

Abstract
High-calorie food leads to nonalcoholic fatty liver disease (NAFLD) through dysregulation of genes involved in lipid metabolism, but the precise mechanism remains unclear. DNA methylation represents one of the mechanisms that contributes to dysregulation of gene expression via interaction with environmental factors. Berberine can alleviate fatty liver in db/db and ob/ob mice. Here, we investigated whether DNA methylation is involved in the pathogenesis of NAFLD induced by a high-fat diet (HFD) and whether berberine improves NAFLD through influencing the methylation status of promoters of key genes. HFD markedly decreased the mRNA levels encoding CPT-1α, MTTP, and LDLR in the liver. In parallel, DNA methylation levels in the MTTP promoter of rats with NAFLD were elevated in the liver. Interestingly, berberine reversed the downregulated expression of these genes and selectively inhibited HFD-induced increase in the methylation of MTTP. Consistently, berberine increased hepatic triglyceride (TG) export and ameliorated HFD-induced fatty liver. Furthermore, a close negative correlation was observed between the MTTP expression and its DNA methylation (at sites −113 and −20). These data indicate that DNA methylation of the MTTP promoter likely contributes to its downregulation during HFD-induced NAFLD and, further, that berberine can partially counteract the HFD-elicited dysregulation of MTTP by reversing the methylation state of its promoter, leading to reduced hepatic fat content.
Keywords: DNA methylation, microsomal triglyceride transfer protein, nonalcoholic fatty liver disease
Nonalcoholic fatty liver disease (NAFLD), characterized by excessive accumulation of triglyceride (TG) in the hepatocytes, affects from 10% to ∼39% of people worldwide (1, 2). It is closely associated with obesity (3), insulin resistance (4), and type 2 diabetes (5). We previously reported that in NAFLD patients without type 2 diabetes, up to 31.4% of individuals meet the criteria of metabolic syndrome, and 43.2% meet the criteria of impaired glucose regulation, of which 14.4% are newly diagnosed with diabetes (6). In a hamster model, preventing intrahepatic lipid accumulation abrogates the development of hepatic insulin resistance; a dose-dependent relationship exists (7). Shulman et al. reported that moderate weight loss normalizes fasting hyperglycemia and improves hepatic insulin sensitivity in patients with poorly controlled type 2 diabetes by reducing hepatic triglyceride content (8). Several prospective studies (4–6, 9) have also shown that NAFLD can predict type 2 diabetes and metabolic syndrome. Thus, reducing hepatic fat accumulation can be an effective strategy to prevent type 2 diabetes. As the pathogenesis of NAFLD remains unclear, no drug is generally accepted, and the only effective treatment is lifestyle intervention, including low-calorie diet, weight loss, and exercise (10). Therefore, it is necessary to understand the pathogenesis of NAFLD to seek a safe and effective drug for reducing hepatic fat accumulation.
Berberine (BBR) is an alkaloid originally isolated from Huanglian (Coptis chinensis). Recent studies have shown that BBR can reduce body weight and improve dyslipidemia and insulin sensitivity in db/db mice (11), hamsters fed a high-fat diet, and patients with type 2 diabetes and dyslipidemia (12, 13). Moreover, BBR reduces serum cholesterol and LDL-cholesterol by elevating hepatic low- density lipoprotein receptor (LDLR) expression through a posttranscriptional mechanism that stabilizes its mRNA (12). Intraperitoneal injection of BBR for three weeks has been shown to alleviate hyperlipidemia and fatty liver in obese db/db and ob/ob mice; BBR is associated with changes in the mRNA levels of genes involved in hepatic and muscular lipid metabolism that enhance fatty acid oxidation and reduce lipogenesis (14). However, because the ob/ob and db/db mice are animal models that contain an inactivating mutation in the leptin or leptin receptor genes, respectively, the pathogenesis of fatty liver in these mice is greatly different from that of human nonalcoholic fatty liver disease. In addition, it has yet to be established whether BBR can improve fatty liver in the wild-type animal model of NAFLD induced by a high-fat diet (HFD).
A set of genes involved in hepatic β-oxidation and lipid export is decreased in the liver of patients with NAFLD (1). Recent studies have shown that modification by DNA methylation of the genes that regulate oxidative phosphorylation is associated with their decreased expression in human skeletal muscle (15, 16) and islets (17) in patients with type 2 diabetes, which increases their susceptibility to insulin resistance. The liver is a central organ in lipid and glucose metabolism, and it remains unclear whether DNA methylation plays a role in dysregulation of these genes in HFD-induced NAFLD and whether BBR can reverse fatty liver by influencing their methylated state.
In the present study, we attempted to investigate 1) whether abnormal expression of key genes involved in lipid metabolism in the liver of Sprague-Dawley (SD) rats with HFD-induced NAFLD is associated with DNA methylation modifications in their promoters; 2) whether BBR-mediated improvement of fatty liver is related to the demethylation within the promoter regions of these genes; and 3) how BBR affects the DNA methylation levels of certain genes.
EXPERIMENTAL PROCEDURES
Animal studies
Healthy male SD rats (5–6 weeks old) weighing 190–210 g were obtained from the Animal Development Center, Chinese Academy of Sciences, Shanghai, and acclimated for 1 week before initiation of the experiment. Rats were given free access to food and water and were maintained on a 12/12-h light/dark cycle. Rats received either a regular rodent chow (normal diet: 62.3% carbohydrate, 12.5% fat, 24.3% protein calories) or a high-fat diet (32.6% carbohydrate, 51.0% fat, 16.4% protein calories) for 24 weeks. Lard was the major constituent of the high-fat diet. After 8 weeks of feeding, rats on the HFD were randomized to receive either BBR (Sigma-Aldrich, Steinheim, UK) at a dose of 200 mg · kg−1·· day−1 (BBR+HFD group) or an equal volume of vehicle (0.5% methylcellulose, HFD group) by gavage for 16 weeks. Rats fed the normal diet received the equal volume of vehicle (0.5% methylcellulose, ND group) as a control group. Body weight and food intake were monitored weekly. Fasting serum insulin (Rat insulin RIA kit, Linco Research, St Charles, MO) and glucose were measured every 4 weeks. At 16 weeks, an intraperitoneal glucose tolerance test (IPGTT) was performed for evaluating insulin sensitivity. After a fasting for 14 h, all rats were euthanized, and their livers were removed and stored in liquid nitrogen for quantitative real-time PCR (qPCR) analysis, hepatic fat content measurement, and DNA methylation analysis. Visceral fat mass, including mesenteric fat pad, epididymal fat pad, and perirenal fat tissue, was weighed. Total blood samples were also collected for measurement of fasting serum total cholesterol (TC), low density lipoprotein cholesterol (LDL-c), and TG levels. Serum TG, TC, and LDL-c were measured using commercially available kits. All experimental procedures involving the use of animals were conducted in conformity with PHS policy and were approved by the Animal Use and Care Committee of Fudan University.
Histological analysis
After the rats were sacrificed, the livers were removed and subsequently fixed in phosphate-buffered 10% formalin. The right lateral lobule of the liver was then divided into 2 sections at the long middle line, one of which was embedded in paraffin blocks and the other in O.C.T. compound. A section from each paraffin block was stained with hematoxylin and eosin (HE) to examine the pathologic structures of the liver and serial cryosections were stained with Sudan III to evaluate lipid droplets.
Liver lipid content
Hepatic lipids were extracted according to the method of Folch et al. (18). The TG content was determined as described previously (19). Briefly, lipid was extracted from frozen liver tissues (30 mg) by homogenization in 1 ml of 2:1 chloroform: methanol, followed by shaking at room temperature for overnight and centrifugation at 3000 rpm for 10 min. Aliquots (400 μl) of the organic-extract lipid suspension were used for the measurement of triglyceride concentrations (TG kit, Sysmex, Japan). Hepatic lipid content was defined as mg of triglyceride per gram of the liver.
Real-time quantitative RT-PCR analysis
Key enzymes of lipid metabolism, which were analyzed by qPCR, were selected as candidate genes for assessment of their mRNA expression levels in the liver of these rats. Those genes that were significantly downregulated in the HFD group and upregulated by BBR treatment were subsequently chosen as targets for further DNA mythelation analysis. Total RNA was isolated from liver tissues using Trizol reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized by reverse transcription using ReverTra Ace (Toyobo, Osaka, Japan). The SYBR Green PCR Master Mix (Toyobo, Osaka, Japan) was used for qPCR with a sequence detection system (ABI PRISM7900, Applied Biosystems, Foster City, CA). The 8 μl reaction mixture contained 1 μl of cDNA and 125 nmol/l of primers. The specific primers used for qPCR are shown in supplementary Table I. The same reaction was performed in triplicate with rat β-actin as an internal control. Fluorescent signals were normalized to an internal reference (ΔRn), and the threshold cycle (Ct) was set within the exponential phase of PCR. The relative gene expression was calculated using the 2−ΔΔCt as described previously (20).
Western immunoblot analysis
Protein from rat liver samples was extracted with RIPA buffer [50 mmol/l Tris-HCl (pH 7.4), 1% NP-40, 0.5% sodium deoxycholate, 150 mmol/l NaCl, 0.1% SDS, EDTA, etc.] containing protease and phosphatase inhibitors. Protein concentrations were measured using a BCA-100 Protein Quantitative Analysis Kit. After denatured, protein samples were subjected to SDS-PAGE and blotted onto polyvinylidene difluoride (Millipore) membranes. Nonspecific binding sites were blocked with 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h and then incubated with primary antibodies against MTTP (Santa Cruz Biotechnology, Santa Cruz, CA), CPT-1α (Santa Cruz), SCD-1 (Santa Cruz), LDLR (Abcam) overnight at 4°C. After three washes in Tris-buffered saline containing 0.1% Tween 20, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (anti-mouse or anti-rabbit IgG) for 1 h and visualized by ECL detection (Pierce Biotechnology, Rockford, IL). Quantitation was performed by Fujifilm Las-3000 Luminescent Image Analyzer.
Analysis of DNA methylation within the promoters of target genes in the liver
All reagents were purchased from Sigma (St Louis, MO). Genomic DNA was isolated from the livers of the three groups of rats using the SDS and proteinase K methods, and DNA bisulfite modification was performed as previously described (20). Bisulfite-modified DNA was amplified with primers (see supplementary Table I) designed using the MethPrimer program (http://www.urogene.org/methprimer/index.html). Direct sequencing and classic cloning/sequencing methods were used to detect DNA methylation. Briefly, promoters of target genes were amplified with Taq polymerase (Tiangen Biotech, Beijing, China). Then 2 μl of the PCR products were purified by shrimp alkaline phosphatase-exonuclease I (SAP-ExonI) and subjected to sequencing using a DNA sequencer (ABI PRISM 3730, Applied Biosystems) with Bigdye terminator v3.1 cycle sequencing kit (Applied Biosystems). If there was a single “C” at a cytosine phosphodiester bond guanine (CpG) site, the site was defined as complete methylation; if there was a single “T” at the CpG site, it was considered as no methylation. Overlapping of “C” and “T” was ranked as partial methylation. Classic cloning/sequencing was also used to accurately measure the levels of DNA methylation. The PCR products (n = 4) were cloned into plasmid vectors (pGM-T Cloning kit; Tiangen Biotech, Beijing, China), which were subsequently transformed into Escherichia coli. Plasmid DNA of ten clones derived from each individual hepatic sample was extracted (AxyPrep Plasmid Miniprep Kit, Axygen Bioscience) and sequenced, and the number of methylated sites was determined. The mean methylation level for each rat was calculated as the total number of methylation sites divided by the total number of possible methylation sites in all clones sequenced, and then multiplied by 100. The proportion of methylation for each CpG site was calculated as the number of methylated cytosine divided by total number of samples in each rat.
Serum lipoprotein-associated TG analysis
Equal volumes of serum samples were pooled from rats for three groups in the fasting states at end of 16 weeks of BBR administration. Lipoproteins were separated using fast protein liquid chromatography (FPLC) (21) on a Superose 6 10/300 GL column (GE Healthcare Bio-Sciences AB, Uppasala, Sweden). Samples were chromatographed at a flow rate of 0.5 ml/min, and fractions of 500 µl each were collected and assayed for TG. The contents of apolipoprotein (apo)B100, apoB48, and apoE in the fractions were analyzed by Western immunoblotting. Proteins in the factions were separated by SDS-PAGE using 4–5% or 5–12% gradient gels and transferred to polyvinylidene difluoride (Millipore). The membranes were incubated with primary antibodies against apoB (RayBiotech, Norcross, GA) and apoE (Abcam), followed by donkey anti-rabbit IgG horseradish peroxidase and visualized by ECL detection (Pierce Biotechnology).
In vivo VLDL triglyceride production and serum lipoprotein fractionation
Male SD rats (5–6 weeks old, 190–210 g) fed a HFD (20% carbohydrate, 59% fat, 21% protein calories) were randomized to receive either BBR (Sigma-Aldrich, Steinheim, UK) at a dose of 200 mg · kg−1 · day−1 (BBR+HFD group) or an equal volume of vehicle (0.5% methylcellulose, HFD group) by gavage for 8 weeks, and rats fed regular rodent chow received the vehicle as control (ND group). After BBR treatment, rats were injected with tyloxapol (Sigma-Aldrich) at 600 mg · kg−1 via tail vein after a fasting period of 4 h. Orbital venous samples were taken at 0, 1, 2, and 3 h after tyloxapol injection. After centrifugation, serum was kept at −80°C until use. Serum TG was determined with a commercial kit (Sigma kit TR0100). VLDL triglyceride production was assessed from the triglyceride concentrations. At 24 h after tyloxapol injection, serum was collected and then analyzed for lipoproteins by FPLC.
Cell culture experiments
Buffalo rat liver (BRL) cells were cultured in 6-well plates at 2 × 106 cells per well in DMEM containing 10% FBS in an atmosphere of 10% CO2 at 37°C. After 24 h, the monolayer was washed with PBS and incubated with 1% FBS-DMEM containing 20 µM BBR or 5 µM 5-aza-2'-deoxycytidine (5-Aza-CdR, Sigma). Palmitate (Sigma) was complexed with essentially fatty acid-free BSA (BSA) to generate a stock solution of 11% (w/v) BSA and 10 mM palmitate in serum-free medium, which was diluted into 1% FBS-DMEM for preparation of 0.1 mM palmitate. BRL cells were incubated with 1% FBS-DMEM containing 100 µM palmitate and 20 µM BBR or 5 µM 5-Aza-CdR for 72 h. Cells were washed with PBS and used for RNA extraction and qPCR analysis.
Statistical analysis
All data were presented as mean ± SEM. Significance was assessed among three groups by one-way ANOVA followed by Tukey's multiple comparison test and LSD. To compare food intake, serum glucose, and insulin levels between BBR- and vehicle-treated groups, independent-samples t-test was used, depending on a preliminary F-test for homogeneity of variance. To determine correlations between gene expression levels and DNA methylation degrees, we used Pearson correlation coefficients for normally distributed values or Spearman correlation coefficients when normality was rejected. All P values were two-tailed; P values less than 0.05 were considered statistically significant.
RESULTS
Effects of BBR on body weight, visceral fat, and serum lipid profiles of HFD-fed rats
Rats fed a HFD tended to develop obesity (Fig. 1A). After 11 weeks of BBR treatment, body weights of HFD-fed rats were significantly reduced, reaching similar levels of ND-fed control animals (P < 0.05, Fig. 1B). BBR did not result in dramatic changes in food intake, although slight differences were observed at weeks 10 and 13 (Fig. 1C). Notably, BBR treatment for 16 weeks significantly lowered visceral fat mass by up to 42.8% (P < 0.05) and reduced the serum levels of TC and LDL-c (Table 1), whereas BBR did not significantly affect serum TG levels (HFD group, 1.02 ± 0.10 mmol/l, n = 8; BBR+HFD group, 0.95 ± 0.12 mmol/l, n = 7; P = 0.32) or HDL-c levels (supplementary Table II). Further analysis of the lipid profiles at 4, 8, and 16 weeks after BBR treatment showed that BBR did not exhibit significant effects on serum TC and LDL-c until at 16 weeks (supplementary Table II). These data suggest that BBR exerts antagonizing effects on HFD-induced obesity, and long-term BBR treatment can improve serum TC and LDL-c.
Fig. 1.

Effects of BBR on body weight of rats fed a high-fat diet. Male SD rats at 6 weeks of age received either a regular rodent chow (ND) or a HFD. After 8 weeks of feeding, rats on the HFD were treated for 16 weeks with BBR or vehicle (n = 8 per group). A: Gross appearance of HFD-fed rats at the end of treatment with BBR (BBR+HFD) or vehicle (HFD). B: Changes of body weight were monitored during BBR treatment. C: Effects of BBR on food intake. Values are mean ± SEM. *P < 0.05, **P < 0.01 versus ND-fed control group (ND); #P < 0.05, ##P < 0.01 versus HFD-fed vehicle group (HFD). BBR, berberine; HFD, high-fat diet; ND, normal diet; SD, Sprague-Dawley.
TABLE 1.
General parameters of BBR treatment for 16 weeks and control groups
| Parameter | ND | HFD | BBR+HFD |
|---|---|---|---|
| Visceral fat (g) | 45.1 ± 6.0 | 74.7 ± 8.2a | 42.7 ± 5.8b |
| Serum TC (mmol/l) | 2.17 ± 0.13 | 2.81 ± 0.28a | 2.02 ± 0.21b |
| Serum LDL-c (mmol/l) | 0.54 ± 0.04 | 2.10 ± 0.31a | 1.22 ± 0.19b |
| Serum TG (mmol/l) | 2.01 ± 0.51 | 1.02 ± 0.10a | 0.95 ± 0.12a |
| Serum ALT (IU/l) | 68.0 ± 4.3 | 106.7 ± 15.6a | 69.0 ± 6.4b |
| Serum AST (IU/l) | 194.7 ± 17.3 | 239.9 ± 43.9 | 167.3 ± 13.8 |
Data are presented as mean ± SEM (n = 8). BBR, berberine; HFD, high-fat diet; ND, normal diet; TG, triglyceride; ALT, alanine transaminase; AST, aspartate aminotransferase.
P < 0.05 versus ND (normal diet + vehicle group).
P < 0.05 versus HFD (high-fat diet + vehicle group).
BBR alleviates liver steatosis in HFD-fed SD rats
Histological analysis by HE or Sudan Ш staining of the liver sections showed derangement of liver cells and excessive lipid droplets in the hepatocytes of SD rats fed the HFD (Fig. 2A), which were alleviated by BBR treatment. Consistently, treatment of HFD-fed rats with BBR for 16 weeks significantly lowered liver weight by 21% (P < 0.05, Fig. 2B) and decreased hepatic triglyceride contents (by 14%, P < 0.01, Fig. 2C). Serum alanine transaminase (ALT) and aspartate aminotransferase (AST) levels were also reduced by BBR treatment (Table 2). Thus, BBR alleviates HFD-induced liver steatosis.
Fig. 2.

BBR alleviates HFD-induced fatty liver in SD rats. After BBR treatment for 16 weeks, rats were euthanized after a 14 h fast. A: Histological analysis of livers of ND-fed, vehicle-treated rats (ND group) and HFD-fed rats treated with vehicle (HFD group) or BBR (BBR+HFD group). Liver sections were stained with hematoxylin and eosin (upper panel) or with Sudan Ш (lower panel). Photographs are at 200× magnification. B: Effects of BBR on liver weight. C: Effects of BBR on hepatic TG contents. Significance was assessed by one-way ANOVA followed by Tukey's multiple comparison test. Data are mean ± SEM. *P < 0.05, **P < 0.01 versus ND; #P < 0.05, ##P < 0.01 versus HFD. BBR, berberine; HFD, high-fat diet; ND, normal diet; SD, Sprague-Dawley; TG, triglyceride.
TABLE 2.
General parameters of rats treated with BBR or vehicle for 8 weeks
| Parameter | ND | HFD | BBR+HFD |
|---|---|---|---|
| Body weight(g) | 487.4 ± 13.4 | 525.5 ± 10.1 | 472.8 ± 12.4b |
| Visceral fat (g) | 11.2 ± 0.9 | 27.8 ± 3.8a | 13.5 ± 3.8c |
| Liver weight (g) | 17.7 ± 0.8 | 19.3 ± 0.5 | 18.0 ± 1.0 |
| Fast serum TG (mmol/l) | 4.0 ± 0.2 | 7.0 ± 0.7a | 4.2 ± 0.4c |
Data are presented as mean ± SEM (n = 6). BBR, berberine; HFD, high-fat diet; ND, normal diet; TG, triglyceride.
P < 0.01 versus ND (normal diet + vehicle group).
P < 0.05.
P < 0.05 versus HFD (high-fat diet + vehicle group).
BBR improves insulin sensitivity in SD rats with HFD-induced NAFLD
Whereas fasting serum glucose levels were slightly lower in BBR-treated, HFD-fed rats than those of the vehicle-treated group (Fig. 3A), treatment with BBR for 4–16 weeks resulted in sustained reduction in fasting serum insulin levels (Fig. 3B). Analysis by IPGTT showed that the area under curve (AUC) of serum glucose level (P < 0.05, Fig. 3C) was significantly decreased in BBR-treated rats, and notably, the AUC of serum insulin levels in the vehicle-treated, HFD-fed group was significantly higher than that of BBR-treated group (P < 0.05, Fig. 3D). These results demonstrate that BBR improves insulin resistance associated with HFD-induced obesity and fatty liver.
Fig. 3.

BBR lowers serum insulin levels and improves insulin resistance in NAFLD rats. Fasting serum glucose (A) and insulin (B) were measured every 4 weeks during 16-week BBR treatment of HFD-fed rats. Serum glucose (C) and insulin (D) levels were determined during intraperitoneal injection of glucose (2g · kg−1 body weight) in HFD-fed rats at 16 weeks of BBR treatment. Values are mean ± SEM. #P < 0.05, ##P < 0.01 versus HFD group. BBR, berberine; HFD, high-fat diet; NAFLD, nonalcoholic fatty liver disease.
BBR reverses HFD-induced abnormal expression of some key genes associated with lipid metabolism in the liver
Next we tested by qPCR analysis whether BBR affects HFD-induced dysregulation of key genes known to be involved in lipid homeostasis. The relative mRNA levels of CPT-1α (P < 0.05, Fig. 4A), MTTP, and LDLR (P < 0.05, Fig. 4B) were significantly downregulated in the livers of HFD-fed rats relative to the ND control group. BBR treatment significantly reversed the downregulating effects of HFD on the expression of CPT-1α and MTTP (P < 0.05). Meanwhile, BBR upregulated LDLR gene expression, although there is no statistical significant difference between BBR-treated group and HFD control group (P > 0.05). On the other hand, the level of SCD-1 mRNA was increased in rats with NAFLD, which was markedly lowered by BBR treatment (P < 0.05, Fig. 4D). We also found that the PPARγ and GPAT mRNAs were significantly altered by HFD feeding (P < 0.05, Fig. 4C), but these were not affected by BBR treatment. Despite these changes, BBR did not influence the expression of other lipogenic and fatty acid oxidation genes, such as ACC, DGAT1, DGAT2, PPARα, and UCP-2. Consistent with alterations in their mRNA expression levels, Western immunoblot analysis of liver protein extracts also revealed corresponding changes in the protein levels of MTTP, CPT-1α, LDLR, and SCD-1 (Fig. 5A), with CPT-1α, LDLR, and MTTP significantly increased and SCD-1 prominently decreased in BBR-treated, HFD-fed rats (Fig. 5B). Thus, these data suggested that BBR might selectively alter the expression of certain target genes, which prompted us to examine whether the expression of these genes in the liver are influenced by DNA methylation.
Fig. 4.

BBR reverses HFD-induced dysregulation of key genes in lipid metabolism in the liver of SD rats. Real-time quantitative PCR analysis of fatty acid oxidation (A), VLDL assembly and secretion (B), and lipogenesis (C and D) in the livers of SD rats of ND, HFD, and BBR+HFD groups. Relative mRNA amounts of each gene were normalized to that of β-actin. Values are mean ± SEM. *P < 0.05, **P < 0.01 versus ND; #P < 0.05, ##P < 0.01 versus HFD. ACC, acetyl CoA carboxylase; Apo, apolipoprotein; BBR, berberine; CPT-1α, carnitine palmitoyltransferase-1α; DGAT, diacylglycerol O-acyltransferase; HFD, high-fat diet; ND, normal diet; LDLR, low density lipoprotein receptor; MTTP, microsomal triglyceride transfer protein; PPAR, peroxisome proliferator-activated receptor; SCD-1, stearoyl-CoA desaturase-1; SD, Sprague-Dawley; UCP, uncoupling protein.
Fig. 5.

BBR normalizes the liver expression levels of MTTP, CPT-1α, LDLR. and SCD-1 proteins in HFD-fed rats. A: Western immunoblot analysis of the liver protein extracts from ND, HFD, and BBR+HFD group (n = 3 per group). B: Quantitation of the protein levels from the immunoblots after normalization to β-actin as the loading control. BBR, berberine; CPT-1α, carnitine palmitoyltransferase-1α; HFD, high-fat diet; LDLR, low density lipoprotein receptor; MTTP, microsomal triglyceride transfer protein; ND, normal diet; SCD-1, stearoyl-CoA desaturase-1.
Analysis of methylation in the promoter regions of select genes in lipid metabolism
Next we chose MTTP, CPT-1α, and LDLR as target genes for further DNA methylation analysis. As indicated in Fig. 6A, the sequence analyzed for the MTTP promoter (+1 indicates the start site of transcription) contains a CpG island with five potential methylation sites. Genomic sequencing chromatograms for each of the three treatment groups showed that all nonCpG cytosines were replaced by thymine, and that the ratio of C:(C+T) (i.e., methylation) was markedly altered at each CpG site among the three treatment groups of rats (Fig. 6B). Sequences located at −260 to +165 (Fig. 6C) in the CPT-1α promoter region and at −389 to +76 (Fig. 6E) in the LDLR promoter, which contain 37 and 23 potential methylation sites, respectively, were also analyzed for DNA methylation using specific bisulfate-sequencing primers (supplementary Table I). In contrast to the results for the MTTP promoter, direct sequencing indicated that nearly all C's were completely replaced by T in all of the CpG sites within the CPT-1α (Fig. 6D) and LDLR (Fig. 6F) promoter sequences analyzed, suggesting that DNA methylation modifications of the CPT-1α and LDLR promoters were not efficiently upregulated in the livers of the HFD-fed rats.
Fig. 6.

Analysis by direct sequencing of the methylation status of the promoters for MTTP, CPT-1α, and LDLR. Genomic DNA, isolated from the livers, was modified with sodium bisulfite and then amplified. The PCR products were directly sequenced. A: The MTTP promoter sequence with five CpG DNA methylation target sites at positions −174, −165, −141, −113 and −20, and several regulatory elements. B: Representative genomic sequencing chromatograms of the MTTP promoter are shown for rats from the three indicated groups. The upper sequence is the unmodified MTTP promoter sequence. The lower sequence is the modified sequence in ND, HFD, and BBR+HFD groups. The CpG sites are indicated in boxes. C−D: The sequence (C) and representative sequencing chromatograms (D) of the CPT-1α promoter with 37 CpG DNA methylation target sites. E−F: The LDLR promoter with 23 CpG sites (E) and representative genomic sequencing chromatograms (F). BBR, berberine; CPT-1α, carnitine palmitoyltransferase-1α; HFD, high-fat diet; LDLR, low density lipoprotein receptor; MTTP, microsomal triglyceride transfer protein; ND, normal diet.
BBR treatment reduces HFD-elicited increases in the level of DNA methylation in the MTTP promoter region
To examine further whether BBR exerts an inhibitory effect on HFD-dependent methylation changes in the MTTP promoter, we used the classic cloning/sequencing method to quantitatively analyze the methylation levels within the MTTP promoter region in the livers of rats. Sequencing of ten clones of the PCR products from each rat revealed that the mean methylation level of the MTTP promoter was higher in HFD-fed rats than that in the ND-fed control group (37.6 ± 6.3% versus 11.8 ± 0.6%, P < 0.01) (Fig. 7A). When methylation at each CpG site was analyzed individually among the three groups, the degrees of DNA methylation at −174, −113, and −20 CpG sites were significantly increased by HFD feeding (site −174: ND, 9.4 ± 5.9% versus HFD, 48.3 ± 10.7%, P < 0.05; site −113: 9.4 ± 3.1% versus 33.3 ± 2.4%, P < 0.01; site −20: 3.1 ± 3.1% versus 35.6 ± 4.4%, P < 0.01; respectively; Fig. 7B). Importantly, BBR treatment resulted in lower mean methylation levels (HFD, 37.6 ± 6.3% versus BBR+HFD, 16.5 ± 3.4%, P < 0.05) (Fig. 7A) and lower levels of DNA methylation at −174, −113, and −20 CpG sites (site −174: HFD, 48.3 ± 10.7% versus BBR+HFD, 15.0 ± 5.0%, P < 0.05; site −113: 33.3 ± 2.4% versus 7.5 ± 4.9%, P < 0.01; and site −20: 35.6 ± 4.4% versus 15.0 ± 2.9%, P < 0.01, respectively; Fig. 7B) within the MTTP promoter in the liver of HFD-fed rats with NAFLD. Of particular note, an inverse correlation was found between the expression level of MTTP mRNA and the degrees of DNA methylation at −113 and −20 CpG sites (site −113, r = −0.636, P = 0.026, Fig. 7C; site −20, r = −0.726, P = 0.008, Fig. 7D). On the other hand, no significant correlation was observed between the MTTP mRNA level and the DNA methylation at −174 CpG site (r = −0.481, P = 0.1135) or the mean DNA methylation degree (r = −0.434, P = 0.158). These data suggest that BBR may influence the expression of genes involved in lipid metabolism by affecting the methylation modifications of their promoters, which contributes to its alleviating actions in reducing HFD-induced hepatic lipid overload.
Fig. 7.

BBR inhibits HFD-induced upregulation of DNA methylation in the MTTP promoter. A: Percentage of DNA methylation was determined from subcloned PCR products in the pGM-T vector (10 clones from each rat, n = 4 per group). Shown is the mean methylation level. B: Methylation degree (%) of each indicated cytosine phosphodiester bond guanine (CpG) site was determined for rats of the three indicated groups, calculated as the ratio of the number of methylated cytosine to the total number of samples. C–D: Negative correlations between the MTTP mRNA expression level and the degree of DNA methylation at −113 (C) and −20 (D) CpG sites in the liver. Values are mean ± SEM. *P < 0.05, **P < 0.01 versus ND; #P < 0.05, ##P < 0.01 versus HFD. BBR, berberine; HFD, high-fat diet; MTTP, microsomal triglyceride transfer protein; ND, normal diet.
Direct effects of BBR on the expression of MTTP in cultured rat liver cells
To confirm further that BBR affects MTTP transcription through DNA methylation inhibition, we investigated whether BBR could directly upregulate the expression of MTTP compared with 5-aza-2'-deoxycytidine (5-aza-CdR, a potent demethylating agent) in a rat liver cell line, BRL cells. Treatment of BRL cells with 20 µM BBR or 5 µM 5-aza-CdR caused similarly time-dependent increases in the mRNA expression of MTTP as determined by qPCR (Fig. 8A). In addition, incubation of BRL cells with palmitate (100 μM) resulted in reduced MTTP mRNA abundance, and treatment by 20 µM BBR or 5 μM 5-aza-2'-deoxycytidine effectively reversed the suppressive effect of palmitate on MTTP mRNA expression (Fig. 8B). These data indicate that BBR is able to upregulate MTTP expression in a cell-autonomous manner, likely through affecting its methylation status.
Fig. 8.
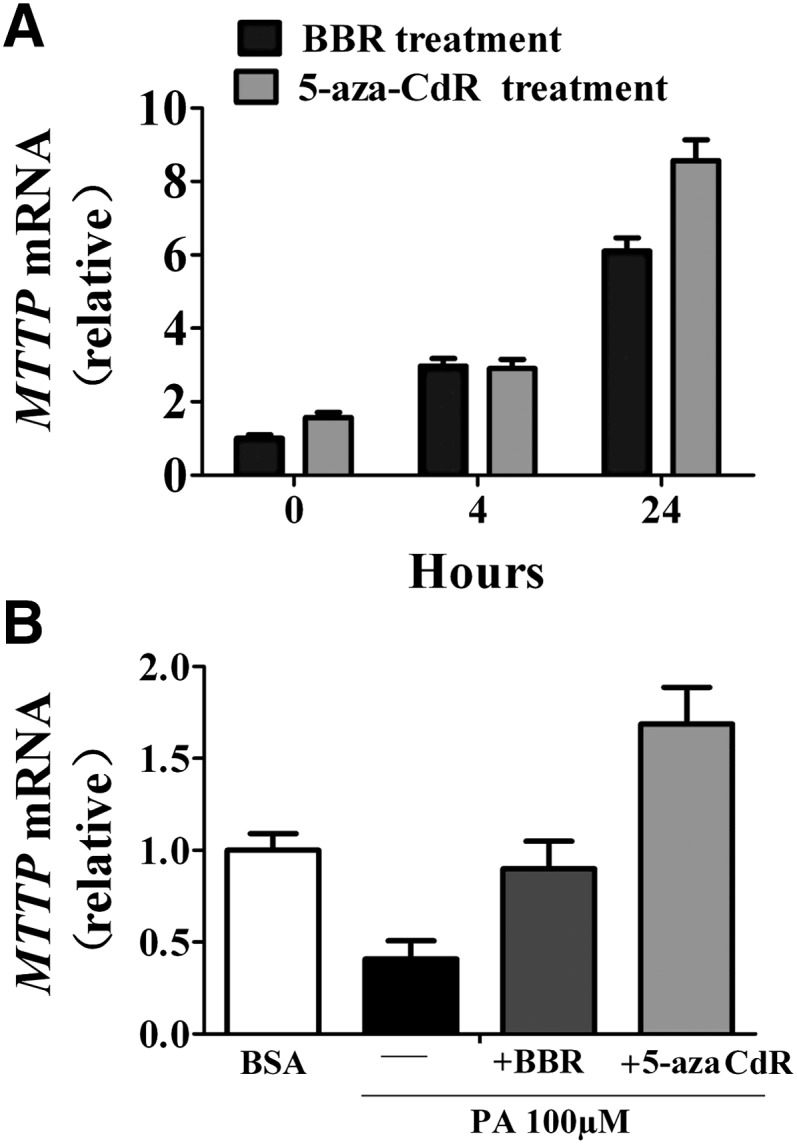
Direct effect of BBR and 5-aza-CdR on MTTP mRNA expression. A: The expression levels of MTTP mRNA were determined by qPCR from BRL rat liver cells treated with BBR (20 µM) or 5-aza-CdR (5 µM) for 4 or 24 h. B: Analysis of the relative MTTP mRNA levels in BRL cells treated with 5 µM 5-aza-CdR or 20 µM BBR for 72 h in the presence 100 µM PA. BBR, berberine; BRL, buffalo rat liver; MTTP, microsomal triglyceride transfer protein; PA, palmitate.
BBR does not affect hepatic DNA methyl transferases but reduces serum homocysteine levels
Next we tested whether BBR affected MTTP expression through regulating DNA methyl transferases. No significant differences were detected in the mRNA and protein levels of DNA methyltransferase (DNMT)1 and DNMT3b from the liver of HFD-fed rats upon BBR treatment (supplementary Fig. I). Moreover, BBR treatment did not alter the total hepatic DNMT enzyme activity (P > 0.05, supplementary Table III). On the other hand, in rats with HFD-induced fatty liver, the serum level of homocysteine was strikingly higher than that in ND group (P < 0.01, supplementary Table III). Interestingly, BBR treatment significantly reduced serum homocysteine to the level of the ND group, whereas no significant differences were found in the hepatic contents of S-adenosylmethionine (SAM), S-adenosylhomocyteine (SAH), or the SAM/SAH ratio between BBR-treated, HFD-fed rats and control rats fed the HFD or ND (P > 0.05, supplementary Table III). These data imply that BBR might suppress DNA methylation through its effect on the pool of methyl donors that remain to be further defined.
BBR treatment for 16 weeks increased serum VLDL-associated TG levels
MTTP is required for the secretion of VLDL from the liver, and the amount of triglycerides in VLDL fractions is strikingly decreased in the liver of tissue-specific MTTP knockout mice (22). Given our observation that BBR treatment resulted in upregulated hepatic MTTP expression and increased MTTP protein abundance in HFD-fed rats, we examined whether BBR could increase the TG content in VLDL fractions. Indeed, measurement of serum lipoprotein-associated TG (Fig. 9A) by FPLC showed that BBR treatment for 16 weeks significantly increased VLDL-TG levels, by ∼26% according to the area under curve (AUC) (Fig. 9B) compared with vehicle-treated animals fed a HFD. On the other hand, HFD feeding in the absence of BBR treatment led to decreased serum VLDL-TG levels by ∼41% compared with ND control group (Fig. 9A, B). Moreover, Western immunoblot analysis showed higher levels of apoB100 and apoB48 in VLDL fractions from the BBR-treated group than in those from the control group (Fig. 9C), whereas no apparent alterations were observed for apoE protein from these VLDL fractions. These data indicate that BBR treatment caused an increase in the triglyceride-rich VLDL particles, which was decreased upon long-term HFD feeding, consistent with the observed changes of hepatic MTTP expression levels.
Fig. 9.
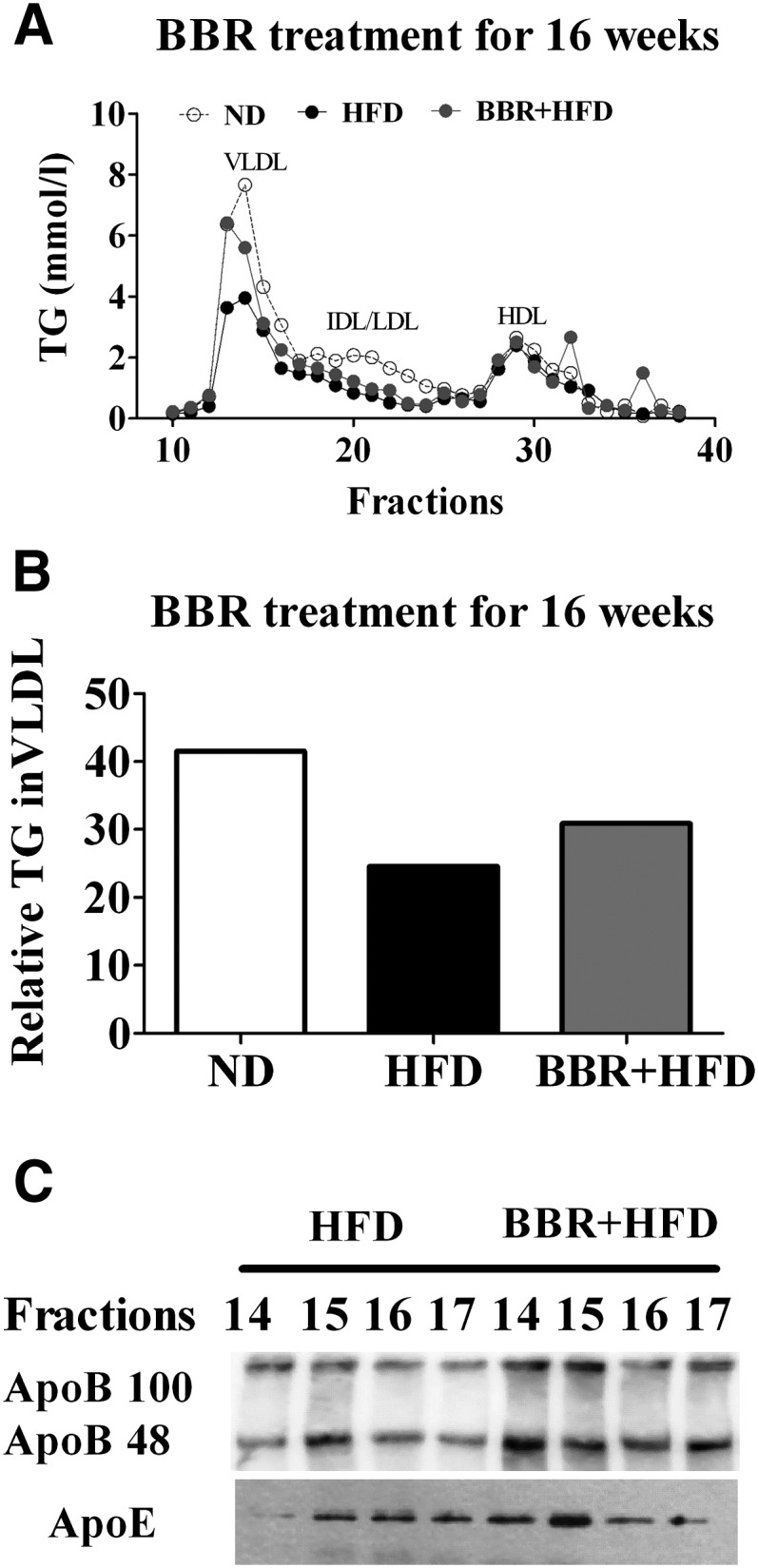
Effects of BBR on the plasma apoB-containing lipoproteins. A: Analysis of lipoprotein profiles from ND-fed control group or HFD-fed rats treated by BBR or vehicle for 16 weeks. Pooled serum samples (n = 6 per group) were fractionated by FPLC, and TG from each fraction was measured. Fractions corresponding to VLDL, IDL/LDL, and HDL are indicated. B: Relative TG contents in VLDL fractions were calculated using AUCs of the FPLC fractions in (A). C: Western immunoblot analysis of apoB100/48 and apoE protein levels in the indicated VLDL fractions (14–17). Apo, apolipoprotein; AUC, area under curve; BBR, berberine; FPLC, fast protein liquid chromatography; HFD, high-fat diet; MTTP, microsomal triglyceride transfer protein; ND, normal diet; TG, triglyceride.
Effect of BBR treatment for 8 weeks on hepatic VLDL production
To directly test the effect of BBR treatment on VLDL production from the liver, we measured serum TG and fractionated VLDL-TG from BBR-treated SD rats following intravenous injection of tyloxapol, which can prevent the metabolism and removal of plasma lipoprotein by inhibiting lipoprotein lipase activity (23). Rats that were fed HFD and simultaneously treated with BBR for 8 weeks exhibited decreased body weight and serum TG by 18% and 41%, respectively (Table 2). Notably, more increasingly elevated serum TG levels were observed in BBR-treated, HFD-fed rats (by ∼55%, ∼195%, and ∼257% at 1, 2, and 3 h) relative to the vehicle-treated control group (by ∼40%, ∼132%, and ∼160% at 1, 2, and 3 h) after tyloxapol administration (Fig. 10A), suggesting the tendency of increased hepatic TG production upon BBR treatment. While we observed considerably higher amounts of TG in the VLDL fractions in HFD-fed rats than in those of ND-fed control animals (Fig. 10B, C), markedly increased TG levels (∼2-fold) in VLDL particles were observed in BBR-treated, HFD-fed rats compared with the vehicle-treated group (Fig. 10B, C). These results further indicated that BBR treatment led to increased secretion of VLDL-TG from the liver.
Fig. 10.
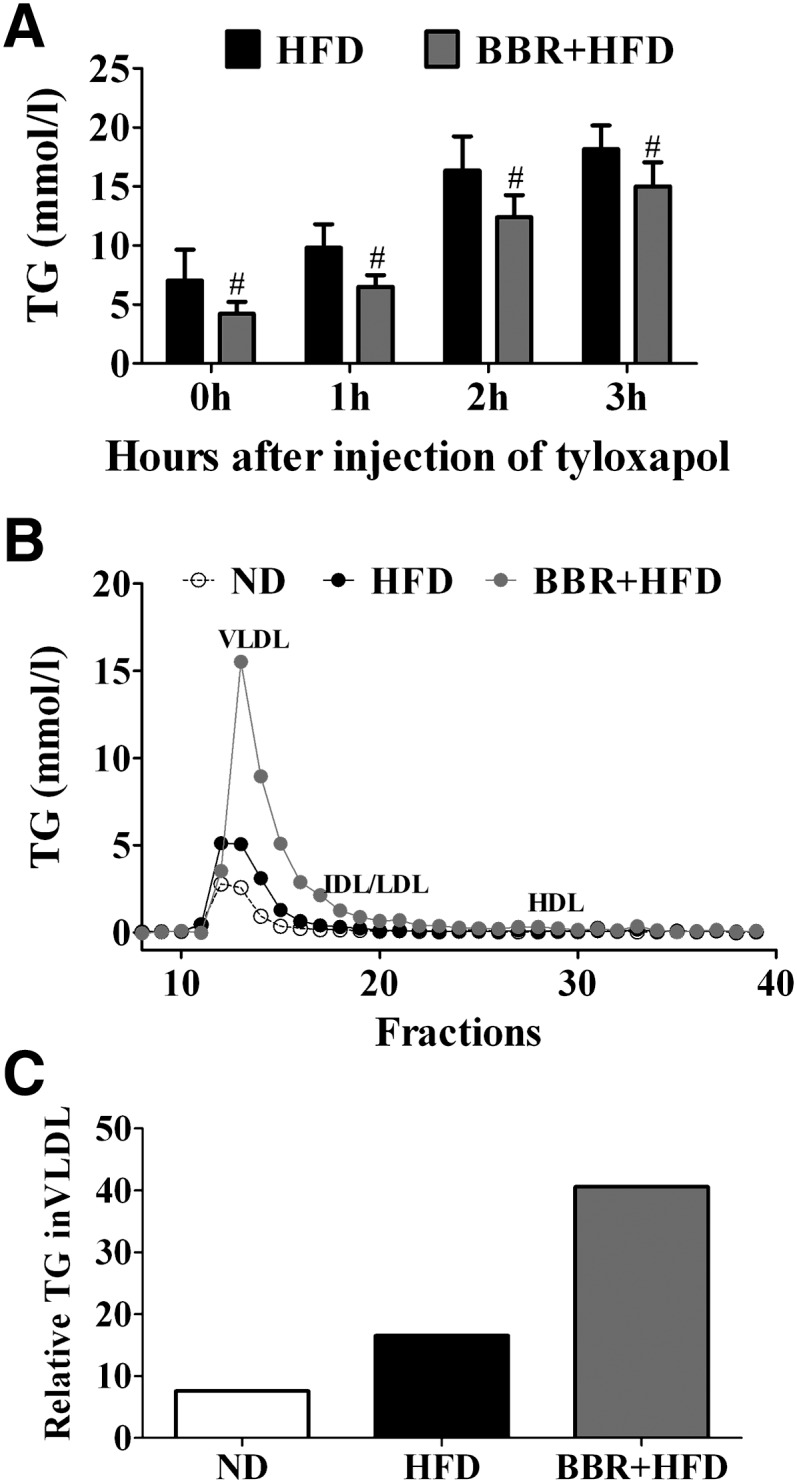
Effects of BBR on the hepatic TG production in HFD-fed rats. Male SD rats were fed ND or a HFD, with the HFD-fed animals simultaneously treated with vehicle or BBR for 8 weeks (n = 6 per group). Rats were then treated via tail vein injection with 600 mg/kg tyloxapol after a 4 h fast. A: Serum TG was measured at 0, 1, 2, and 3 h after tyloxapol injection for the indicated groups. Data are shown as mean ± SEM. #P < 0.05 versus HFD. B: Analysis of lipoprotein profiles from the indicated groups. Pooled serum samples (n = 6 per group) were fractionated by FPLC, and TG in each fraction was measured. Fractions corresponding to VLDL, IDL/LDL, and HDL are indicated. C: Relative TG contents in the VLDL fractions were calculated using AUCs from the FPLC fractions in (B). AUC, area under curve; BBR, berberine; FPLC, fast protein liquid chromatography; HFD, high-fat diet; ND, normal diet; SD, Sprague-Dawley; TG, triglyceride.
DISCUSSION
In this study we have shown that BBR could alleviate HFD-induced fatty liver and reversed the abnormal expression of MTTP, CPT-1α, SCD-1, and LDLR in SD rats with HFD-induced NAFLD. BBR treatment increased TG-rich VLDL particles from the liver of HFD-fed rats. Importantly, our results demonstrated that DNA methylation of the MTTP promoter was selectively increased in the liver of SD rats with NAFLD and that BBR was able to reverse HFD-elicited hypermethylation of the MTTP promoter.
Recently, BBR has been shown to reduce body weight and improve dyslipidemia in obese db/db mice (11, 14), HFD-fed hamsters, and patients with type 2 diabetes and dyslipidemia (12, 13). Here we found that BBR displayed similarly beneficial effects on rats with NAFLD induced by HFD. BBR significantly decreased liver weight and alleviated fatty liver, which is consistent with the reported effects of BBR on db/db mice (14). In accordance with the alleviations observed in histological analyses of BBR-treated rats, liver function was also improved by BBR. The IPGTT analysis showed that BBR also significantly improved insulin resistance in the HFD-induced NAFLD SD rat model. These results support the notion that BBR effectively reduces body weight and hepatic fat content, thereby improving insulin sensitivity (8).
The precise mechanisms by which HFD causes the development of NAFLD or BBR improves fatty liver remain largely unclear. Defects in lipid metabolism pathways, including de novo lipogenesis, fatty acid oxidation, hepatic fatty acid influx, and/or fat export in the form of VLDL, are closely linked to hepatic steatosis (1). Our results revealed dramatically reduced expression level of MTTP in the liver of HFD-fed rats. In 1984, Wetterau et al. found that an intracellular protein accelerates the transfer of triglyceride and cholesteryl ester, which was subsequently named microsomal triglyceride transfer protein (MTTP). Later studies have demonstrated that MTTP is necessary for the assembly and secretion of apoB-containing lipoproteins (e.g., VLDL and LDL), whereby lipids are normally exported from the liver (24). A common polymorphism exists within the MTTP promoter, a G to T transition at 493 nucleotides upstream of the initiation start site in different populations (25–28). The G allele, which is responsible for a decrease in the MTTP gene transcription, has been shown to be prone to developing increased intrahepatic triglycerides content (25, 28). In ob/ob mice, downregulation of MTTP gene expression facilitates intracellular fat accumulation in hepatocytes, increasing the susceptibility to hepatic steatosis (29). Therefore, a decrease in the MTTP mRNA may play a critical role in the pathogenesis of obesity-associated NAFLD. Interestingly, we observed that BBR treatment led to upregulated expression of MTTP mRNA. Further, the amount of triglyceride and apoB in the VLDL fractions were increased in SD rats upon BBR treatment for 16 weeks, indicating that BBR-upregulated hepatic MTTP expression led to increased MTTP function. Consistent with this, we found that BBR increased the amount of TG in VLDL particles, particularly in HFD-fed rats treated with tyloxapol (21), which can decrease TG hydrolysis by inhibiting lipoprotein lipase (LPL). Moreover, we found that BBR could also increase LDLR protein levels in the liver, which is in line with previously reported studies (12, 30). Hepatic LDLR is known to regulate human plasma LDL-c homeostasis and a high level of hepatic LDLR mRNA is associated with improved clearance of plasma LDL-c. Thus, BBR might result in more clearance of serum LDL-c through its action on LDLR. These results suggest that MTTP is an important target of BBR in reducing hepatic triglyceride content, while more physiological targets are most likely to exist in mediating the effects of BBR on lipid homeostasis.
In addition, the present study revealed decreased expression of CPT-1α, a key rate-limiting enzyme in mitochondrial β-oxidation, in the liver of rats with HFD-induced NAFLD, which is consistent with the results reported in patients with NAFLD (1). HFD-induced elevation in the expression of SCD-1, the main enzyme responsible for the conversion of saturated fatty acyl-CoAs, stearoyl-CoA (18:0) and palmitoyl-CoA (16:0), to their respective monounsaturated fatty acyl-CoAs, oleyl-CoA (18:1) and palmitoleyl-CoA (16:1) (31), was also observed in the liver of SD rats with NAFLD. Lack of SCD-1 expression in mice has been shown to result in reduced tissue lipid content and protect against both diet-induced and genetically induced obesity (32). Therefore, HFD-elicited upregulation of SCD-1 as well as repressed expression of CPT-1α (33) could suppress fatty acid β-oxidation in the mitochondria and thus facilitate the development of NAFLD. More importantly, we observed that BBR could reverse the HFD-induced dysregulation of these genes and improve fatty liver. A previous study (34) demonstrated that overexpression of CPT-1α was accompanied by a 69% reduction in hepatic TG accumulation in obese SD rats. Thus, the regulatory effects of BBR on the expression of MTTP, CPT-1α, and SCD-1 in the liver could contribute to its reducing effects on hepatic TG in rats with NAFLD.
How does high-fat diet feeding cause abnormal expression of these genes and how can BBR reverse their dysregulated expression? DNA methylation is usually involved in the interaction between genes and environment factors. Cytosine residues occurring in CG dinucleotides are targets for DNA methylation, and gene expression is usually downregulated when DNA methylation modification occurs at its promoter. Ling et al. reported that DNA methylation of NDUFB6 (15), COX7A1 (16), and PPARGC1A (17) is involved in the pathogenesis of insulin resistance and type 2 diabetes, suggesting that DNA methylation may play a role in regulating the expression of these genes in the diseases influenced by environmental and genetic factors. Promoters of MTTP, LDLR, and CPT-1α genes are rich in CpG sites and susceptible to DNA methylation modifications. This prompted us to hypothesize that DNA methylation may be involved in altering hepatic MTTP, CPT-1α, and LDLR expression in SD rats with HFD-induced NAFLD and that BBR may counteract HFD-induced dysregulation of these genes through promoting demethylation of their promoters. Indeed, our results showed that the mean methylation levels of MTTP promoter and three out of five CpG sites (−174, −113, and −20) were increased in the liver of HFD-fed rats. Even more interestingly, BBR was able to decrease the mean methylation level as well as methylation at the three CpG sites (the greatest by up to 30%) in the MTTP promoter region. Furthermore, the levels of DNA methylation of two CpG sites (−113 and −20) were inversely correlated with the expression level of MTTP mRNA in the liver of rats of the three treatment groups. These results suggest that HFD resulted in DNA hypermethylation of the MTTP promoter region, causing a decrease in its mRNA expression level. The relatively lower abundance of MTTP protein might block the assembly and secretion of VLDL, resulting in defective fat export from the liver. BBR treatment led to decreased level of DNA methylation in the MTTP promoter region and thus upregulated the expression of MTTP, which contributed to triglyceride export from the liver and improved fatty liver. Thus, the methylation modifications within the sequence located at −113 to −20 of MTTP promoter region may play a prominent role in regulation of its transcription, which includes several positive elements, such as HNF1, LRH-1, HNF4, and DR1 (35). Whether DNA methylation of these CpG sites directly blocks the binding of transcription factors to these response elements within the MTTP promoter or changes the molecular conformation of the MTTP promoter has yet to be further investigated. On the other hand, no methylation modifications were found in the potential CpG sites within the CPT-1α and LDLR promoter sequences analyzed. Epigenetic regulatory mechanisms include histone modifications and microRNAs besides DNA methylation, which are also emerging as important players in polygenic human diseases and are influenced by a complex interaction of environmental, dietary, and exercise factors (36). Therefore, it remains to be understood whether the expression of CPT-1α and LDLR might be regulated by other mechanisms, such as histone modification or microRNAs.
In contrast to our results from the present study, elevated MTTP mRNA levels have been reported in the liver from OLETF rats (37) and Syrian Golden hamsters (38) when fed HFD for 28 days. Quesada et al. (39) also reported increased MTTP expression (by ∼1.5-fold) in the liver of Wistar rats fed HFD for 13 weeks. While we found HFD feeding for 24 weeks led to severe hepatic steatosis in SD rats, the amount of VLDL-TG was increased in the early stage of HFD feeding (at 8 weeks) as opposed to a decline of VLDL-TG in the late stage of NAFLD (at 24 weeks). Although it is likely that the regulation of hepatic MTTP expression may respond differently to short-term versus long-term HFD intake, this inconsistency might also result from the different genetic background of animals or differences in the high-fat diets used in these different experimental settings.
In keeping with a direct effect of BBR on the expression of MTTP by influencing DNA methylation, we observed that treatment of rat liver BRL cells with both BBR and 5-aza-CdR could lead to time-dependent increases in the MTTP mRNA expression. Moreover, both could reverse the suppressive effect of palmitate on MTTP expression. Given the fact that 5-aza-CdR can strongly inhibit DNA methyltransferase activity and induce the expression of many genes through reversing gene-specific hypermethylation (40, 41), BBR might regulate MTTP gene expression through a similar mechanism, i.e., DNA demethylation. In our attempt to explore the mechanism(s) by which BBR alters the DNA methylation level of MTTP promoter, we found that the serum homocysteine level was markedly elevated in SD rats with HFD-induced NALFD, which was accompanied by a significant hypermethylation of MTTP promoter in the liver. Moreover, treatment with BBR lowered the serum homocysteine content as well as the methylation level of MTTP promoter. On the other hand, we observed that BBR neither significantly altered hepatic SAM or SAH concentrations nor affected the total DNMT enzyme activities. Although it remains unclear how BBR treatment led to lower serum homocysteine levels or how this might relate to hypomethylation of certain gene promoters, these results suggest that BBR may exert important effects on the pool of these species involved in the transfer of methyl group, thereby altering the methylation status of select genes that include those associated with lipid metabolism. While the exact mechanisms underlying BBR's actions remain to be elucidated, it is possible that BBR may also exert its demethylation effects on the promoters of other genes involved in modulation of DNA methylation (e.g., DNA demethylases).
In summary, the present study has demonstrated that BBR reduces hepatic fat content in rats with HFD-induced NAFLD by increasing TG export from hepatocytes, at least partially through decreasing the methylation of the MTTP promoter. Our findings not only illustrate that BBR represents a promising agent for the treatment of NAFLD but also provide novel insights into the importance of epigenetic factors such as DNA methylation in the pathogenesis of NAFLD induced by excessive intake of a high-calorie diet.
Supplementary Material
Acknowledgments
The authors thank Yong Liu (The Institute for Nutritional Sciences, Chinese Academy of Sciences, China) for critical reading of this manuscript; J.Z. Gu and L. Liang (Shanghai Experimental Animal Center, China Academy of Sciences, China) for their help with animal studies; and Qiqun Tang and Xi Li (The Institute of Biomedical Sciences, Fudan University, China) for their advice.
Footnotes
Abbreviations:
- ACC
- acetyl CoA carboxylase
- ALT
- alanine transaminase
- Apo
- apolipoprotein
- AST
- aspartate aminotransferase
- BBR
- berberine
- BRL
- buffalo rat liver
- CpG
- cytosine phosphodiester bond guanine
- CPT-1α
- carnitine palmitoyltransferase-1α
- DGAT
- diacylglycerol O-acyltransferase
- DNMT
- DNA methyltransferase
- FPLC
- fast protein liquid chromatography
- GPAT
- glycerol-3-phospate acyltransferase
- HFD
- high-fat diet
- LDLR
- low-density lipoprotein receptor
- MTTP
- microsomal triglyceride transfer protein
- NAFLD
- nonalcoholic fatty liver disease
- ND
- normal diet
- PPAR
- peroxisome proliferator-activated receptor
- SAH
- S-adenosylhomocyteine
- SAM
- S-adenosylmethionine
- SCD-1
- stearoyl-CoA desaturase-1
- 5-Aza-CdR
- 5-aza-2'-deoxycytidine
- SD
- Sprague-Dawley
- TC
- total cholesterol
- TG
- triglyceride
- UCP
- uncoupling protein
This work was supported by Science and Technology Commission of Shanghai Municipality Grants 07JC1401107 and 08dj1400601 (X.G.) and by National Natural Science Foundation of China Grant 30900500 (M.L.).
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of one fi gure and three tables.
REFERENCES
- 1.Kohjima M., Enjoji M., Higuchi N., Kato M., Kotoh K., Yoshimoto T., Fujino T., Yada M., Yada R., Harada N., et al. 2007. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 20: 351–358. [PubMed] [Google Scholar]
- 2.Adams L. A., Angulo P., Lindor K. D. 2005. Nonalcoholic fatty liver disease. CMAJ. 172: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leung D. H., Williams K., Fraley J. K., Klish W. J. 2009. Age- and ethnic-specific elevation of ALT among obese children at risk for nonalcoholic steatohepatitis (NASH): implications for screening. Clin. Pediatr. (Phila.). 48: 50–57. [DOI] [PubMed] [Google Scholar]
- 4.Hanley A. J., Williams K., Festa A., Wagenknecht L. E., D'Agostino R. B., Jr, Kempf J., Zinman B., Haffner S. M. 2004. Elevations in markers of liver injury and risk of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes. 53: 2623–2632. [DOI] [PubMed] [Google Scholar]
- 5.Vozarova B., Stefan N., Lindsay R. S., Saremi A., Pratley R. E., Bogardus C., Tataranni P. A. 2002. High alanine aminotransferase is associated with decreased hepatic insulin sensitivity and predicts the development of type 2 diabetes. Diabetes. 51: 1889–1895. [DOI] [PubMed] [Google Scholar]
- 6.Yan H-M., Gao X., Liu M., Gu Q., Zhang B., Li X., Gao J., Zhao N. Q. 2006. Study of the association between non-alcoholic fatty liver disease and metabolic syndrome. Chin. J. Diabetes. 14: 326–328. [Google Scholar]
- 7.Samuel V. T., Liu Z. X., Qu X., Elder B. D., Bilz S., Befroy D., Romanelli A. J., Shulman G. I. 2004. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 279: 32345–32353. [DOI] [PubMed] [Google Scholar]
- 8.Petersen K. F., Dufour S., Befroy D., Lehrke M., Hendler R. E., Shulman G. I. 2005. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 54: 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schindhelm R. K., Dekker J. M., Nijpels G., Bouter L. M., Stehouwer C. D., Heine R. J., Diamant M. 2007. Alanine aminotransferase predicts coronary heart disease events: a 10-year follow-up of the Hoorn Study. Atherosclerosis. 191: 391–396. [DOI] [PubMed] [Google Scholar]
- 10.Postic C., Girard J. 2008. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Invest. 118: 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee Y. S., Kim W. S., Kim K. H., Yoon M. J., Cho H. J., Shen Y., Ye J. M., Lee C. H., Oh W. K., Kim C. T., et al. 2006. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes. 55: 2256–2264. [DOI] [PubMed] [Google Scholar]
- 12.Kong W., Wei J., Abidi P., Lin M., Inaba S., Li C., Wang Y., Wang Z., Si S., Pan H., et al. 2004. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat. Med. 10: 1344–1351. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y., Li X., Zou D., Liu W., Yang J., Zhu N., Huo L., Wang M., Hong J., Wu P., et al. 2008. Treatment of type 2 diabetes and dyslipidemia with the natural plant alkaloid berberine. J. Clin. Endocrinol. Metab. 93: 2559–2565. [DOI] [PubMed] [Google Scholar]
- 14.Kim W. S., Lee Y. S., Cha S. H., Jeong H. W., Choe S. S., Lee M. R., Oh G. T., Park H. S., Lee K. U., Lane M. D., et al. 2009. Berberine improves lipid dysregulation in obesity by controlling central and peripheral AMPK activity. Am. J. Physiol. Endocrinol. Metab. 296: E812–E819. [DOI] [PubMed] [Google Scholar]
- 15.Ling C., Poulsen P., Simonsson S., Ronn T., Holmkvist J., Almgren P., Hagert P., Nilsson E., Mabey A. G., Nilsson P., et al. 2007. Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle. J. Clin. Invest. 117: 3427–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ronn T., Poulsen P., Hansson O., Holmkvist J., Almgren P., Nilsson P., Tuomi T., Isomaa B., Groop L., Vaag A., et al. 2008. Age influences DNA methylation and gene expression of COX7A1 in human skeletal muscle. Diabetologia. 51: 1159–1168. [DOI] [PubMed] [Google Scholar]
- 17.Ling C., Del Guerra S., Lupi R., Ronn T., Granhall C., Luthman H., Masiello P., Marchetti P., Groop L., Del Prato S. 2008. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia. 51: 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Folch J., Lees M., Sloane Stanley G. H. 1957. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226: 497–509. [PubMed] [Google Scholar]
- 19.Cnop M., Hannaert J. C., Hoorens A., Eizirik D. L., Pipeleers D. G. 2001. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes. 50: 1771–1777. [DOI] [PubMed] [Google Scholar]
- 20.Jiang M. H., Fei J., Lan M. S., Lu Z. P., Liu M., Fan W. W., Gao X., Lu D. R. 2008. Hypermethylation of hepatic Gck promoter in ageing rats contributes to diabetogenic potential. Diabetologia. 51: 1525–1533. [DOI] [PubMed] [Google Scholar]
- 21.Millar J. S., Cromley D. A., McCoy M. G., Rader D. J., Billheimer J. T. 2005. Determining hepatic triglyceride production in mice: comparison of poloxamer 407 with Triton WR-1339. J. Lipid Res. 46: 2023–2028. [DOI] [PubMed] [Google Scholar]
- 22.Raabe M., Veniant M. M., Sullivan M. A., Zlot C. H., Bjorkegren J., Nielsen L. B., Wong J. S., Hamilton R. L., Young S. G. 1999. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J. Clin. Invest. 103: 1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyake J. H., Doung X. D., Strauss W., Moore G. L., Castellani L. W., Curtiss L. K., Taylor J. M., Davis R. A. 2001. Increased production of apolipoprotein B-containing lipoproteins in the absence of hyperlipidemia in transgenic mice expressing cholesterol 7 alpha-hydroxylase. J. Biol. Chem. 276: 23304–23311. [DOI] [PubMed] [Google Scholar]
- 24.Wetterau J. R., Lin M. C., Jamil H. 1997. Microsomal triglyceride transfer protein. Biochim. Biophys. Acta. 1345: 136–150. [DOI] [PubMed] [Google Scholar]
- 25.Bernard S., Touzet S., Personne I., Lapras V., Bondon P. J., Berthezene F., Moulin P. 2000. Association between microsomal triglyceride transfer protein gene polymorphism and the biological features of liver steatosis in patients with type II diabetes. Diabetologia. 43: 995–999. [DOI] [PubMed] [Google Scholar]
- 26.Merriman R. B., Aouizerat B. E., Bass N. M. 2006. Genetic influences in nonalcoholic fatty liver disease. J. Clin. Gastroenterol. 40 (Suppl 1): S30–S33. [DOI] [PubMed] [Google Scholar]
- 27.Namikawa C., Shu-Ping Z., Vyselaar J. R., Nozaki Y., Nemoto Y., Ono M., Akisawa N., Saibara T., Hiroi M., Enzan H., et al. 2004. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J. Hepatol. 40: 781–786. [DOI] [PubMed] [Google Scholar]
- 28.Gambino R., Cassader M., Pagano G., Durazzo M., Musso G. 2007. Polymorphism in microsomal triglyceride transfer protein: a link between liver disease and atherogenic postprandial lipid profile in NASH? Hepatology. 45: 1097–1107. [DOI] [PubMed] [Google Scholar]
- 29.Stefano J. T., de Oliveira C. P., Correa-Giannella M. L., de Lima V. M., de Sa S. V., de Oliveira E. P., de Mello E. S., Giannella-Neto D., Alves V. A., Carrilho F. J. 2007. Nonalcoholic steatohepatitis (NASH) in ob/ob mice treated with yo jyo hen shi ko (YHK): effects on peroxisome proliferator-activated receptors (PPARs) and microsomal triglyceride transfer protein (MTP). Dig. Dis. Sci. 52: 3448–3454. [DOI] [PubMed] [Google Scholar]
- 30.Abidi P., Zhou Y., Jiang J. D., Liu J. W. 2005. Extracellular signal-regulated kinase-dependent stabilization of hepatic low-density lipoprotein receptor mRNA by herbal medicine berberine. Arterioscler. Thromb. Vasc. Biol. 25: 2170–2176. [DOI] [PubMed] [Google Scholar]
- 31.Gutierrez-Juarez R., Pocai A., Mulas C., Ono H., Bhanot S., Monia B. P., Rossetti L. 2006. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J. Clin. Invest. 116: 1686–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mar-Heyming R., Miyazaki M., Weissglas-Volkov D., Kolaitis N. A., Sadaat N., Plaisier C., Pajukanta P., Cantor R. M., de Bruin T. W., Ntambi J. M., et al. 2008. Association of stearoyl-CoA desaturase 1 activity with familial combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 28: 1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobrzyn P., Dobrzyn A., Miyazaki M., Cohen P., Asilmaz E., Hardie D. G., Friedman J. M., Ntambi J. M. 2004. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc. Natl. Acad. Sci. USA. 101: 6409–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stefanovic-Racic M., Perdomo G., Mantell B. S., Sipula I. J., Brown N. F., O'Doherty R. M. 2008. A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. Am. J. Physiol. Endocrinol. Metab. 294: E969–E977. [DOI] [PubMed] [Google Scholar]
- 35.Hussain M. M., Rava P., Pan X., Dai K., Dougan S. K., Iqbal J., Lazare F., Khatun I. 2008. Microsomal triglyceride transfer protein in plasma and cellular lipid metabolism. Curr. Opin. Lipidol. 19: 277–284. [DOI] [PubMed] [Google Scholar]
- 36.Knutson S. K., Chyla B. J., Amann J. M., Bhaskara S., Huppert S. S., Hiebert S. W. 2008. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 27: 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuriyama H. S., Yamashita I., Shimomura T., Funahashi M., Ishigami K., Aragane K., Miyaoka T., Nakamura K., Takemura Z., Man, et al. 1998. Enhanced expression of hepatic acyl-coenzyme A synthetas and microsomal triglyceride transfer protein messenger RNAs in the obese and hypertriglyceridemic rat with visceral fat accumulation. Hepatology. 27: 557–562. [DOI] [PubMed] [Google Scholar]
- 38.Bennett A. J., Billett M. A., Salter A. M., White D. A. 1995. Regulation of hamster hepatic microsomal triglyceride transfer protein mRNA levels by dietary fats. Biochem. Biophys. Res. Commun. 212: 473–478. [DOI] [PubMed] [Google Scholar]
- 39.Quesada H., del Bas J. M., Pajuelo D., Diaz S., Fernandez-Larrea J., Pinent M., Arola L., Salvado M. J., Blade C. 2009. Grape seed proanthocyanidins correct dyslipidemia associated with a high-fat diet in rats and repress genes controlling lipogenesis and VLDL assembling in liver. Int. J. Obes. 33: 1007–1012. [DOI] [PubMed] [Google Scholar]
- 40.Liang G., Gonzales F. A., Jones P. A., Orntoft T. F., Thykjaer T. 2002. Analysis of gene induction in human fibroblasts and bladder cancer cells exposed to the methylation inhibitor 5-aza-2'-deoxycytidine. Cancer Res. 62: 961–966. [PubMed] [Google Scholar]
- 41.Momparler R. L., Ayoub J. 2001. Potential of 5-aza-2'-deoxycytidine (Decitabine) a potent inhibitor of DNA methylation for therapy of advanced non-small cell lung cancer. Lung Cancer. 34: S111–S115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.