

Abstract
Protein clamps are ubiquitous and essential components of DNA metabolic machineries, where they serve as mobile platforms that interact with a large variety of proteins. In this report we identify residues that are required for binding of the β-clamp to DNA polymerase III of Escherichia coli, a polymerase of the Pol C family. We show that the α polymerase subunit of DNA polymerase III interacts with the β-clamp via its extreme seven C-terminal residues, some of which are conserved. Moreover, interaction of Pol III with the clamp takes place at the same site as that of the δ-subunit of the clamp loader, providing the basis for a switch between the clamp loading machinery and the polymerase itself. Escherichia coli DNA polymerases I, II, IV and V (UmuC) interact with β at the same site. Given the limited amounts of clamps in the cell, these results suggest that clamp binding may be competitive and regulated, and that the different polymerases may use the same clamp sequentially during replication and repair.
Keywords: β-clamp/clamp loader/DNA polymerase/DNA repair/PCNA/RFC
Introduction
Protein clamps have been implicated in all processes of DNA metabolism, including replication, transcription, and various pathways of DNA repair (Warbrick, 2000; Stucki et al., 2001). Their biological significance stems, first, from the fact that they are tightly bound to DNA in a topological rather than sequence-specific manner that allows them to slide freely on the nucleic acid (Kong et al., 1992; Stukenberg et al., 1991) and, secondly, from the fact that they can interact with a variety of enzymes to increase their association to DNA. Many proteins that bind to the eukaryotic clamp [proliferating cell antigen (PCNA)] contain a consensus motif (Qxx[I/L]xxFF) that often localizes to the extreme N- or C-terminal regions of the protein (Warbrick, 2000). Polymerases from bacteriophages T4 and RB69 have also been found to interact with their clamps via conserved C-terminal sequences (Berdis et al., 1996; Shamoo and Steitz, 1999). Recently, the Y-family polymerase Pol IV of Escherichia coli was also found to interact with the β-clamp via C-terminal residues (Lenne-Samuel et al., 2002).
In the case of bacterial chromosomal replicases of the Pol C family, exemplified by DNA polymerase III of E.coli, crystal structure information is lacking and no clear map of the clamp–polymerase interaction has been available. The DNA polymerase III core of E.coli consists of three subunits: α (DNA polymerase), ε (3′–5′ proofreading nuclease) and θ. The α DNA polymerase subunit of the Pol III core is responsible for the interaction with β (Stukenberg et al., 1991). Deletion studies of the E.coli α-subunit have suggested that the β interaction site resides in an internal region of the α-protein, spanning residues 542–991 (α is 129 kDa or 1160 amino acids) (Kim and McHenry, 1996). More recently, and based on limited similarity with other β-binding proteins, Dalrymple et al. (2001) proposed a β binding consensus sequence of QLxLF and suggested that β binds at an internal sequence of α at position 918–926. While we cannot exclude a role of internal sequences in the binding to the β-clamp, we have recently performed quantitative binding and functional assays that demonstrate that α binds β mainly through a different site located at the C-terminal 20 residues, and that these residues are required for function of α with β (López de Saro et al., 2003). These C-terminal residues of α are also required for the τ switch, which acts on α to regulate polymerase processivity.
In E.coli it has been shown that the β-clamp can associate with all five known DNA polymerases in the cell (Pol I–V) to increase their processivity of synthesis (Hughes et al., 1991; Bonner et al., 1992; Tang et al., 2000; Wagner et al., 2000; López de Saro and O’Donnell, 2001; Kobayashi et al., 2002). These polymerases belong to four different structural families, namely family A (Pol I), family B (Pol II), family C (Pol III) and the newly discovered Y family (Pol IV and V) (Braithwaite and Ito, 1993; Ohmori et al., 2001; Filée et al., 2002), which have very limited or no apparent sequence similarity between them.
Stimulation of DNA synthesis by the β-clamp is dependent on the topological linkage of the clamp to DNA (Stukenberg et al., 1991). This process involves the transient opening of the protein ring catalyzed by the γ-complex clamp-loader (γ3δδ′χψ) in a reaction driven by ATP hydrolysis (reviewed in O’Donnell et al., 2001). Within the γ-complex, the δ-subunit binds β the tightest and is responsible for opening of the clamp (Turner et al, 1999). In combination with the other γ complex subunits, the primed DNA is placed into the open β-ring and then ATP is hydrolyzed, allowing the ring to close around the DNA. Study of the β-clamp interaction with the δ-subunit of the γ-complex and the α-subunit of the Pol III core shows that they compete for β, and thus likely bind β in the same, or nearly the same, location (Naktinis et al., 1996). During lagging strand synthesis, Pol III core rapidly dissociates from the clamp at the end of each completed Okazaki fragment and hops to a new β-clamp on the next RNA-primed site to be extended (O’Donnell, 1987). The β-clamp is left behind on the completed DNA fragment and is free to function with other β-interactive proteins (Stukenberg et al., 1994). These include proteins involved in Okazaki-fragment maturation (Pol I and ligase), replication-associated DNA repair (Pol I and II), mutation-prone lesion bypass (Pol IV and V), and mismatch repair (MutS).
The elucidation of the molecular structure of the γ-complex (Jeruzalmi et al., 2001), and of the δ-subunit of the γ-complex bound to the β-clamp (Jeruzalmi et al., 2001), was the first step in understanding the molecular details of how a protein interacts with β. The main attachment site of δ to β is mediated by δ-residues Leu73 and Phe74, which are highly conserved among prokaryotic δ-subunits (Jeruzalmi et al., 2001). The site on β to which δ binds is a hydrophobic pocket and residues that define the hydrophobic pocket of β are conserved (Jeruzalmi et al., 2001).
The most defined view of how a DNA polymerase interacts with its clamp is provided by the crystal structure of an 11-residue C-terminal peptide of phage RB69 DNA polymerase (a B-family polymerase) bound to its corresponding clamp, the gp45 protein (Shamoo and Steitz, 1999). The interface mainly consists of a hydrophobic pocket in the gp45 clamp, similar to that displayed by δ binding to β. The cocrystal structure of a C-terminal peptide of the cell regulator, p21WAF1, to the human PCNA clamp is also mediated by a hydrophobic pocket in PCNA (Gulbis et al., 1996). In all three systems the clamps have remarkably similar architecture, consisting of six domains organized on either a dimer (β) or trimer (PCNA, gp45), and interaction with the clamp takes place at equivalent locations between domains of the clamps. These similarities suggest that the basic mechanism by which proteins bind to sliding clamps is conserved across all domains of life.
As a first step towards understanding how clamps are used by multiple proteins during DNA synthesis and repair, one aim of the present study is to map in detail the site of interaction of the E.coli replicative polymerase, the α-subunit of Pol III, with the β-clamp and to examine whether it binds the same site in β as the δ-subunit of the clamp loader. We find that the C-terminal peptide of the Pol III α subunit inhibits clamp loading by the γ-complex and binding to β by the δ-subunit, suggesting that the α·β and δ·β interactions do indeed occur at the same locus on the clamp. This observation provides the structural basis for the internal competition for clamps in the Pol III holoenzyme replicative machinery (Naktinis et al., 1996).
Another goal of the present study is to determine whether the other E.coli polymerases also interact with β in the same locus as α and δ, and if the interaction is mediated by C-terminal residues. We find that DNA polymerase II (Pol II), despite having no sequence similarity with the Pol III α-subunit, also interacts with the β-clamp in the same location, via the C-terminal end of the protein. Pol IV also interacts with β via C-terminal residues, consistent with a previous study (Lenne-Samuel et al., 2002), and we find that it binds β in the same locale as α and Pol II. Furthermore, we find that the sites of interaction between the clamp and DNA polymerases I and V are the same as those of Pol II and Pol III, although these polymerases do not appear to utilize C-terminal residues for this interaction. Overall, we find that all E.coli polymerases interact at the same point on the clamp, namely the hydrophobic pocket to which the δ-subunit binds. The implications of these findings with respect to how these enzymes traffic on the β-clamp are discussed.
Results
Characterization of an α-subunit C-terminal peptide interaction with the β-clamp
We have recently demonstrated that the C-terminal 20 residues of the α-subunit are required for functional interaction with β (López de Saro et al., 2003). To define the critical residues that contribute to the α–β interaction, we designed overlapping N-biotinylated peptides spanning the C-terminal region of α and assayed them for interaction with the β-clamp by attaching them to streptavidin-coated microplates and then probing them for ability to bind 32P-β. Radiolabeled β used in these studies contained a six-residue C-terminal extension that can be phosphorylated with protein kinase A and [32P]γ-ATP (Stukenberg et al., 1994; Kelman et al., 1995). We found that peptides corresponding to the C-terminal 20 amino acids interact with the β-clamp, as did a peptide lacking the last residue, but deletion of two residues from the C-terminus abolished the interaction (Figure 1A). Hence, the penultimate F-residue of the α-peptide is required for interaction with β.

Fig. 1. The extreme C-terminus of the α-subunit of DNA polymerase III interacts with the β-clamp. (A) Sequences probed by peptide analysis are shown as stippled boxes in the DnaE protein scheme, but only the analysis of the C-terminal peptides is shown. N-terminal biotinylated peptides were immobilized on streptavidin-coated 96-well plates and probed with 32P-β. It is estimated that 30 nmol of peptide is retained in each well. Peptide sequences used in the microplate assay on the right are shown as lines under the sequence of the C-terminus. Results of the assays are shown to the right. (B) Native polyacrylamide electrophoresis was used to separate a complex of 32P-β with biotinylated α1141–1160 bound to streptavidin. Lane 1 contained only 32P-β; the α1141–1160 peptide (220 nM) was added in lanes 3 and 4, and streptavidin (2.2 µM) in lanes 2 and 4, as indicated. (C) Gel shift assay of the radiolabeled β-clamp. Native polyacrylamide electrophoresis was performed to separate free 32P-β from 32P-β·core complexes as described in Materials and methods. Pol III core was added in lanes 2–6 and the complex was challenged with either BSA (lane 2), non-labeled E.coli β-clamp (lane 3), human PCNA (lane 4), phage T4 gp45 (lane 5) or α1141–1160 peptide (lane 6), as indicated above the gel. (D) DNA synthesis is inhibited by α C-terminal peptides. Reactions were performed using primed M13mp18 ssDNA, β, core and γ complex in the presence of 100 µM of each peptide as described in Materials and methods.
Next, we used a protein shift assay (López de Saro and O’Donnell, 2001) to determine whether the C-terminal α-peptide (α1141–1160) complexed to streptavidin in solution is sufficient to bind to 32P-β and to produce a mobility shift in a native polyacrylamide gel. The results showed that streptavidin does indeed produce a slower-mobility complex, presumably consisting of streptavidin· biotin-peptide bound to 32P-β (Figure 1B, lane 4). We also observed that Pol III core retards the mobility of 32P-β in a native gel (Figure 1C, compare lanes 1 and 2). If the polymerase·β interaction requires the α C-terminal sequences, the α1141–1160 peptide may be expected to disrupt this interaction, thus displacing the 32P-β from core polymerase, and the results showed that this is in fact the case (Figure 2C, lane 6). To demonstrate specificity of this interaction further, heterologous clamps were tested for displacement of the Pol III core·32P-β interaction. The addition of excess yeast PCNA clamp or phage T4 gp45 clamp did not displace core from 32P-β (Figure 1C, lanes 4 and 5, respectively). Nor did the α-peptide bind to human 32P-PCNA or inhibit the binding of 32P-PCNA to the cell-cycle regulator p21WAFI (data not shown). As expected from their ability to compete with DNA polymerase for β, the β-binding peptides α1140–1159 and α1141–1160 were inhibitory to Pol III core when added to a replication reaction using primed M13mp18 ssDNA as template; core requires β for synthesis of this substrate (Figure 1D).

Fig. 2. Alanine scan analysis of α C-terminal peptide binding to β. (A) Peptides were pre-bound to streptavidin-coated microtiter plates and probed with 32P-β as in Figure 1A. A clear spot indicates a critical residue for 32P-β binding. (B) Peptides were used to compete Pol III core off the 32P-β·core complex in the native PAGE mobility shift assay, as in Figure 1C. (C) Purified δ protein (20 pmol) was fixed to microtiter plates and probed with 32P-β in the presence of the indicated peptide. Retention of 32P-β in the well indicates that a critical residue has been changed to Ala.
Similar binding assays as described above were performed on peptides spanning the N-terminal region of α and the region of the protein from residue 812 to 991 (gray boxes in the key of Figure 1A), with negative results (data not shown). Since the interaction could require a specific conformation not provided by 20-mer peptides, our results do not exclude the possibility that additional sites of contact between α and the β-clamp could be present in this area, as suggested by the studies of Kim and McHenry (1996). On the other hand, our results do not detect the interaction observed by Dalrymple et al. (2001), in which a sequence corresponding to region 918–926 of α (sequence IGQADMFGV), fused onto a reporter protein, resulted in an interaction with the β-clamp using a yeast two-hybrid assay. Peptides spanning this region do not show binding to the β-clamp using the assays and conditions described here (data not shown).
Alanine-scan analysis of the α C-terminal peptide
To identify residues at the C-terminus of the α-subunit of Pol III that are critical for interaction with β, we performed an alanine scan analysis of the α1141–1160 peptide. These peptides were used in three different assays: (i) direct binding of 32P-β to biotinylated peptide immobilized in wells of a microtiter plate (Figure 2A); (ii) ability to displace Pol III core from a 32P-β·core complex observed in a native polyacrylamide gel (Figure 2B); and (iii) ability to displace 32P-β from δ-subunit immobilized in wells of a microtiter plate (Figure 2C). The δ–β displacement assay is based on the previous finding that binding of α and δ to β is sterically exclusive, and that the same single amino-acid changes on β that abolish interaction with α also abolish interaction with δ, suggesting that α and δ share a common site on β (Naktinis et al., 1996). This conclusion is supported here by the finding that peptide α1141–1160 competes 32P-β off the δ subunit (Figure 2C).
While most alanine mutants of the α1141–1160 peptide retain full activity for binding 32P-β, mutation of residues Q1154, L1157 and F1159 to alanine clearly diminished the activity of the peptides in all three assays. Interestingly, peptide D1160A showed no retention of 32P-β on the microtiter plate (Figure 3A), but was almost as effective as wild-type peptide in competition assays (Figure 3B and C). Since deletion of residue D1160 (peptide α 1140–1159) in the experiment shown in Figure 1A did not impair binding to 32P-β, we suggest that the presence of alanine at the position of D1160 lowers the affinity of peptide for the β-clamp. The large concentration of peptide used in the competition assays of Figure 1B and C may overcome the deficiency.

Fig. 3. An α-subunit truncated in the C-terminal seven residues loses affinity and function with the β-clamp. (A) Fluorescently labeled β-clamp (βOG) at residue 333 was used in KD measurements with wild-type α (squares) and αΔC7 (circles). (B) KD measurements were obtained over a 0–230 mM range of NaCl. (C) Wild-type core (squares) and core reconstituted using αΔC7 (circles) were assayed for processive DNA synthesis on singly primed M13mp18 ssDNA in the presence of the β-clamp and γ-complex.
Deletion of the C-terminal seven residues of α greatly reduces interaction with the β-clamp
In light of the experiments described above, we deleted the seven C-terminal residues of α that contain the three critical β-interactive residues identified in the alanine scanning experiments. This C-terminal deletion will be referred to here as αΔ7. We then labeled the β-clamp with the fluorescent 488 Oregon Green (OG) probe at the single exposed Cys333, and used fluorescence to determine the affinity of the labeled β-clamp for wild-type α and for αΔC7. Cys333 is on the opposite face of β from where α binds, and OG-labeled βOG was as active as wild-type β in DNA synthesis activity assays with γ-complex and core (data not shown). Titration of α and αΔ7 into βOG reveals that αΔC7 has a 10-fold reduced affinity for the β-clamp (Figure 3A; KD of 1.16 µM versus 0.11 µM at 100 mM NaCl). The residual binding of αΔC7 for the β-clamp could be due to additional interaction points between the two proteins, perhaps in regions distinct from the C-terminal region. Alternatively, C-terminal residues immediately upstream could participate in binding β via protein backbone contacts with the clamp, as in the p21·PCNA complex (Gulbis et al., 1996). This possibility is supported by our earlier study of αΔ20 (deletion of the C-terminal 20 residues), which binds β 100-fold weaker than wild-type α (López de Saro et al., 2003). In this case, the upstream residues that contribute peptide backbone contacts to β would not be detected by the alanine scanning experiments described above. This residual affinity of αΔC7 for the β-clamp was clearly salt dependent (Figure 3B), suggesting that the contribution of the last seven residues has a strong hydrophobic component, consistent with the requirement of L1157 and F1159 in the interaction of α with β.
DNA synthesis assays performed with αΔC7 reconstituted with the ε and θ subunits to form Pol III (αΔ7) core showed that its activity in a β-clamp-dependent replication assay is considerably lower than that of wild-type core (Figure 3C). This result supports the conclusion that the extreme C-terminal seven residues of α are important to functional interaction with β. Both core complexes displayed similar activities in a β-independent extension assay using gapped DNA as a substrate (not shown), indicating that αΔC7 is not impaired in its catalytic activity.
Pol III core interacts with β at the same locus as the δ subunit of the clamp loader
During lagging strand synthesis, the clamp loader (γ-complex) loads β-clamps onto multiple RNA primers. The KD of γ-complex for β in solution is 8 nM, ∼30-fold lower than that of the Pol III core·β complex (250 nM). However, when β is on DNA this affinity is reversed, resulting in a KD value for the Pol III core·β of <5 nM (Naktinis et al., 1996) and a reduced affinity of the γ-complex for β (Turner et al., 1999; Ason et al., 2000). This DNA-modulated competition between clamp loader and polymerase for β generates a molecular switch that assures an ordered sequence of events during the assembly of the replicase at a primed site (Ason et al., 2003). A previous study on the site of interaction of δ subunit (of γ-complex) with β, and α subunit (of Pol III core) with β, demonstrated that both proteins interact with the clamp near the C-terminus, which is near the hydrophobic pocket between β domains II and III (Naktinis et al., 1996). Here, as shown in Figure 2C, we find that the 20-mer peptide derived from the C-terminus of α displaces δ from 32P-β. This result indicated that the α and δ proteins bind the same site on β, rather than competing by steric occlusion, in which two large proteins bind different sites on β, but are too close for both α and δ to bind β at the same time.
The ability of the α C-terminal peptide to compete δ from β suggests that this peptide may inhibit the clamp loading reaction. Indeed, the peptide inhibits loading of the β-clamp on DNA by γ-complex, but a related peptide mutated in a single residue (Q1154A) does not (Figure 4). These results further support the conclusion that the site of interaction of δ and α with β is the same, namely the hydrophobic pocket between domains II and III of the β ring, which is near the β C-terminus.

Fig. 4. The α C-terminal peptide inhibits β-clamp loading by the γ-complex. (A) In the presence of ATP, the γ-complex (γ3δδ′χψ) loads 32P-β onto primed M13mp18 ssDNA, resulting in a complex with a molecular mass that elutes earlier than free 32P-β in gel filtration. (B) The plot shows the gel filtration profiles of reactions without peptide added (squares), or with a peptide that does not interact with 32P-β (Q1154A) (circles), or with wild-type α1141–1160 peptide that binds and inhibits the clamp loading reaction (diamonds).
Conserved residues at the C-terminus of Pol II, Pol III and Pol IV interact at the same site of β
The α C-terminal residues identified by alanine scanning as being essential for interaction with β appear to be present in the C-terminal sequences of Pol II and Pol IV, although the spacing is somewhat different (Figure 5A). A peptide corresponding to the C-terminus of Pol IV has been shown recently to interact with β directly, and a C-terminal deletion of this Y-family polymerase no longer functions with β (Lenne-Samuel et al., 2002). The experiment shown in Figure 5B (upper panel) demonstrates that immobilized peptides derived from the C-terminus of either Pol II or Pol IV interact with 32P-β and retain 32P-β in ELISA plates. If these Pol II and Pol IV peptides bind β at the same site, they should be capable of competing 32P-β off δ protein. The experiment shown in Figure 5B (lower panel) illustrates that this prediction is valid. Immobilized δ retains 32P-β in the well of a microtiter plate, and the addition of C-terminal peptide derived from α, Pol II or Pol IV displaces 32P-β from the immobilized δ. Further, the Pol II and Pol IV proteins also displace Pol III core from the Pol III core·β complex in the native PAGE assay, as shown in Figure 5C. The 20-mer peptides derived from the C-terminus of Pol II and Pol IV also inhibit β-dependent replication by Pol III core (data not shown).

Fig. 5. Pol II and Pol IV interact with the β-clamp via extreme C-terminal sequences. (A) C-terminal sequences from Pol III, Pol II and Pol IV. Conserved residues that correspond to residues in E.coli α needed to bind β highlighted. (B) Synthetic biotinylated peptides derived from the C-terminus E.coli Pol II and Pol IV bind 32P-β as determined using streptavidin-coated microtiter plates (upper panel), and by ability to disrupt the δ·32P-β complex (lower panel). (C) C-terminal peptides from Pol II and Pol IV displace Pol III core from 32P-β·core complex in a native PAGE assay (as in Figure 1C). (D) The native PAGE assay was used to challenge the 32P-β·core complex with C-terminal Pol II peptide (Pol II764–783) (left panel) or the 32P-β·Pol II complex with a C-terminal α peptide (α1141–1160) (right panel). Peptide was added in reactions 3–10 as follows: 500, 500, 250, 125, 62.5, 31.2, 15.6 and 7.8 µM, respectively. Concentrations of Pol III core (left panel) and Pol II (right panel) were 500 nM and 925 nM, respectively. (E) Fluorescence KD measurements of the interaction between Pol III core (upper left panel) or Pol II to βOG as described in the Materials and methods. The lower panels show the interaction of wild-type β with rhodamine-labeled peptides corresponding to the C-terminal 20 residues of α (left panel) and Pol II (right).
The alignment of the C-terminal tails of various polymerases reveals slight differences in the presence and/or spacing of the residues that are important for α to bind β (see Figure 5A). To assess the importance of these variations to the relative affinity to β, we compared the Pol II and Pol III C-terminal peptides for their effectiveness in disrupting polymerase·clamp complexes. In Figure 5D (upper panel) we titrated the 20-mer peptide of Pol II into the 32P-β·Pol III core complex, and in the lower panel, the C-terminal Pol III α 20-mer peptide was titrated into 32P-β·Pol II complex. The results demonstrate that the peptides have similar efficiencies in disrupting the polymerase–clamp complex.
We next determined the apparent KD of Pol III core·β (51 ± 9 nM) and Pol II·β (120 ± 23 nM) using βOG (Figure 5E, upper panels). To compare the relative affinity of the C-terminal 20 residues of both polymerases for β, we used synthetic peptides labeled at their N-terminus with rhodamine (TAMRA); the apparent KD values calculated from titration measurements were 3.2 µM for the α peptide and 3.5 µM for the Pol II peptide, revealing that despite the differences in sequence, both peptides bind to the clamp with very similar affinities (Figure 5E, lower panels). Calculation of the free energy of the polymerase·β interaction indicates that ∼75% of the interaction energy between Pol III and β resides in the C-terminal 20 residues (ΔG = 9.94 kcal/mol for Pol III·β, ΔG = 7.50 kcal/mol for Pol III peptide·β) as does 79% of the interaction energy between Pol II and β (ΔG = 9.45 kcal/mol for Pol II·β, ΔG = 7.44 kcal/mol for Pol II peptide·β).
DNA polymerases I and V (UmuC) also interact with β in the same locus as Pol III
Our previous studies have shown that the β-clamp can stimulate the Klenow fragment of DNA polymerase I in synthesis on SSB-coated, singly primed M13mp18 ssDNA (López de Saro and O’Donnell, 2001). However, the C-terminus of the Klenow fragment lacks a sequence containing the β-binding residues of the Pol III α-subunit. Consistent with this, we detected no binding to the β-clamp of a C-terminal 20-residue peptide derived from the Klenow sequence (data not shown). However, the Pol III α subunit peptide (α 1141–1160) did inhibit β-stimulated DNA synthesis by Pol I Klenow fragment (Figure 6A). In this experiment the β-clamp was loaded onto the primed DNA before the peptide and Pol I (Klenow fragment) were added. Hence, the observed inhibition is likely the result of disruption of the Pol I·β contact by the peptide. Further, 32P-β is retained in wells by immobilized Klenow fragment (Figure 6B), suggesting that the site on β that is bound by the α C-terminal peptide and the Pol I Klenow fragment is the same, and that both polymerases interact with the clamp in a competitive manner.
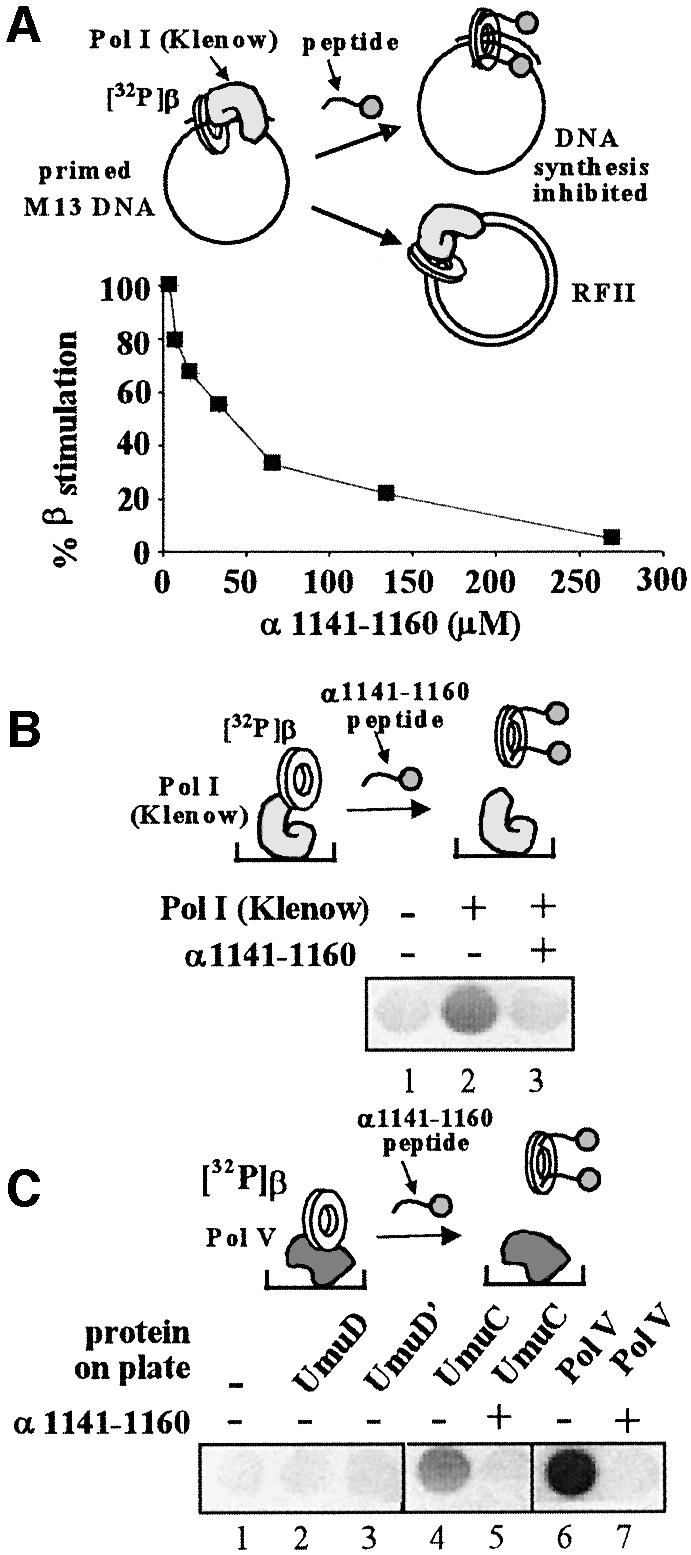
Fig. 6. Pol I (Klenow fragment) and Pol V also bind the β-clamp in the same locus as δ, Pol III, Pol II and Pol IV. (A) Stimulation of DNA synthesis by the β-clamp is inhibited by peptide α1141–1160. The plot shows the result of a titration of the α1141–1160 peptide into a β-stimulated Pol I Klenow fragment DNA replication assay on primed M13mp18 ssDNA. β was loaded on the primed template prior to the addition of Pol I. (B) Pol I Klenow fragment (50 pmol) was fixed in microtiter plates and assayed for binding to 32P-β in the presence or absence of α1141–1160 peptide (100 µM). (C) UmuD, UmuD′, UmuC or Pol V (UmuD′2UmuC) (50 pmol each) were fixed in a microtiter plate and probed with 32P-β. In reactions 5 and 7, 100 µM α1141–1160 peptide was added.
Finally, we turned our attention to Pol V of E.coli, a heterotrimeric polymerase of the Y-family with the subunit structure UmuD′2UmuC (Woodgate et al., 1989; Bruck et al., 1996) that is involved in translesion synthesis (Tang et al., 2000). UmuD′ (115 amino acids) is produced by RecA-induced proteolytic removal of an N-terminal peptide from UmuD (139 amino acids). We examined 20-mer peptides derived from the N- and C-terminal sequences of UmuD and UmuC for binding to the β-clamp, with negative results. We then fixed the full UmuD, UmuD′, UmuC and Pol V complex (UmuD′2UmuC) to ELISA plates and probed the immobilized protein with 32P-β. As shown in Figure 6C, only UmuC and Pol V complex showed an interaction with 32P-β, and these interactions were disrupted by the addition of the Pol III α C-terminal peptide (α 1141–1160). These results indicate that the main connection between Pol V and β is mediated by the single UmuC subunit, and that it binds to β in the same location as Pol III and the other DNA polymerases. Consistent with this, Dalrymple et al. (2001) have reported, on the basis of sequence similarity, a putative binding site on UmuC for the β-clamp that is situated ∼62 residues from the C-terminus, and more recently it has been shown that removal of these residues abolishes the lesion bypass ability of Pol V (Becherel et al., 2002). This sequence (QLNLF) resembles the motifs present in the other E.coli DNA polymerases. An interaction between UmuD and β has been reported using crosslinking assays (Sutton et al., 1999). Despite identification of a UmuC-β interaction, we cannot exclude the possibility that UmuD or UmuD′ may interact with β in a fashion undetected under the conditions of this report.
Discussion
Protein clamps play a central role in DNA metabolism, either as processivity factors for DNA polymerases or to orchestrate complex multienzymatic reactions. In this report we performed a detailed analysis of a β-clamp interaction motif in the main replicase, a C-family DNA polymerase. Peptide mapping revealed critical residues at the extreme C-terminus of the α subunit needed to function with β, consistent with our previous finding that this region is critical for the sequential binding and release of the clamp during Okazaki-fragment synthesis. We find that the α subunit of Pol III interacts with the β-clamp at the same locus as the δ-subunit of the γ-complex, providing the basis for the competitive switch between the loading machinery and the polymerase. We have shown that this competitive switch can occur for all five E.coli polymerases interacting with the β-clamp at a common locus.
A peptide tail in Pol II, III and IV interact with the β-clamp
This report demonstrates that E.coli Pol II, III and IV interact with the β-clamp via the C-terminal tail of these polymerases. It has been hypothesized that attachment via a flexible C-terminal extension could allow for the clamp and polymerase to rotate with respect to one another, perhaps aiding the 3′ terminus of DNA to switch from the polymerization to the exonuclease site (Shamoo and Steitz, 1999). Some degree of flexibility would also be advantageous if two different polymerases were to bind the same clamp concurrently, as some models have proposed. Since the β-clamp is a homodimer, two hydrophobic pockets could potentially be available for binding two proteins. The arrangement of the subunits places both sites on the same face of the ring, which may create steric constraints that preclude binding of more than one protein at a time. No conclusive evidence has been presented in favor of or against this possibility, but the alternative model, one involving sequential binding by different proteins, is also possible. In this case, different proteins would compete for attachment to the clamp, and traffic flow on β could be regulated by other factors, such as DNA structure or post-translational modifications of the clamp.
Competition of proteins for the same clamp has been documented in the case of the γ-complex and the Pol III core, which switch their affinity for β-clamps sequentially during the first steps in processive DNA synthesis (illustrated in Figure 7A). The switch is regulated by whether DNA is inside the β-ring or not (Stukenberg et al., 1994). When β is in solution (i.e. not on DNA), γ-complex binds β tighter than core, but when DNA goes through β, core develops a much greater affinity for β and takes the ring over from the clamp loader (Ason et al., 2003). A second switch, also dependent on DNA structure, occurs at the end of processive Okazaki-fragment synthesis. In this case Pol III core is released from the β-clamp only when synthesis is complete, allowing core to rapidly recycle to a β-clamp at a new primed site for the synthesis of the next Okazaki fragment (O’Donnell, 1987; Stukenberg et al., 1991). In a recent study we have demonstrated that the C-terminus of the α-subunit is also a target for binding by the τ-subunit of the clamp loader (López de Saro et al., 2003). The τ-subunit senses DNA structure, and when it lacks ssDNA template, τ binds the C-terminus of Pol III core and displaces it from β and DNA as illustrated in Figure 7A (Leu et al., 2003). This ‘processivity switch’ is mediated by direct competition between a DNA-sensing protein (τ) and the β-clamp for a common site on the Pol III α-subunit and reveals an elegant mechanism by which polymerase trafficking on clamps can be connected to the different structures of the DNA substrate and product.

Fig. 7. Trafficking of various proteins on β-clamps at different stages of replication and repair. (A) The γ-complex loads β-clamps on DNA, which are then used for processive elongation by Pol III core. These proteins trade places on β. The affinity of these proteins for β is modulated by whether β is on or off DNA, thus regulating this traffic flow as described in the text. (B) Pol I and ligase are thought to interact sequentially during removal of the RNA primer and sealing of the nick during Okazaki fragment synthesis. β-clamps interact with repair proteins which may utilize β for genome surveillance directly after Okazaki fragment maturation. (C) Lesion bypass polymerases IV and V are thought to replace Pol III at sites of replication blocks or damage. Pol III may take over with β again after bypass is complete. (D) Model of the crystal structure of δ-subunit bound to the β-clamp. Domains of the β-dimer are labeled I, II and III. δ interacts with β via two residues of an exposed loop (Leu73 and Phe74) that bind a hydrophobic pocket between domains II and III of β. (E) Location of the sites of interaction on β of the DNA polymerases and the δ-subunit.
Analogous competition mechanisms for clamps regulated by DNA structure may be discovered for other clamp-related reactions. For example, the clamp left by Pol III core at the completed Okazaki fragment may be sequentially used by Pol I and DNA ligase for RNA primer removal and sealing of the nick, respectively (see key to Figure 7B). Perhaps Pol I binds β tightest when RNA is present, but loses affinity for β when the processing job is complete. Ligase may interact most favorably with β at the nicked site created by Pol I action. Besides polymerases and ligase, MutS has also been shown to bind β (López de Saro and O’Donnell, 2001). Hence, either before or following Okazaki-fragment processing, β-clamps left on DNA may serve as markers of newly synthesized DNA for genome surveillance by repair proteins or other DNA modifying proteins before being recycled from DNA for use at other locations.
Traffic flow on β during bypass of a DNA lesion
The SOS response in E.coli includes the induction of three polymerases; Pol II, Pol IV and Pol V (Goodman, 2002). It has been suggested that these polymerases are called into action to bypass DNA damage during chromosomal replication, and that their diversity reflects the variety of damaged DNA that can be encountered (Goodman and Tippin, 2000; Sutton and Walker, 2001; Wagner et al, 2002). The finding that they interact with the β-clamp on the same locus as Pol III core suggests that they compete with core for β at sites of DNA damage (see Figure 7C). The levels of Pol V (UmuC subunit) in the cell rise to ∼200 copies per cell upon induction (Woodgate and Ennis, 1991), ∼10-fold higher than the intracellular level of Pol III core, which is present at a concentration of ∼20 copies per cell (Baker and Kornberg, 1992).
A combination of DNA structural elements or signals from the stalled replication fork could be responsible for the selection of the polymerase that resides with β at a 3′ terminus at any given time. However, the fact that the repair polymerases and Pol III core compete for the same site on the clamp suggests that control of polymerase selection may be exercised at the level of their affinity for the clamp. In the case of Pol V, action with β requires RecA, which is also induced in the SOS response (Pham et al., 2001, 2002). Pol III core, on the other hand, is inhibited by RecA. Hence, one may expect that Pol III core, stalled at a DNA lesion, may be displaced by RecA, and at the same time the presence of RecA and β would recruit Pol V (Figure 7C) (Pham et al., 2001). After lesion bypass and disassembly of the RecA filament (due to inherent instability of RecA), Pol V would lose affinity in the absence of RecA and dissociate from β (Pham et al., 2001). Pol III core could then reassociate with β to resume chain extension.
Similarity within the clamp-binding motifs
Despite the lack of sequence similarity between Pol II, III and IV of E.coli, they have evolved similar C-terminal sequence motifs that bind to β. The sequence motif consists of the pattern QxxL(x)F. This sequence is somewhat reminiscent of the PCNA consensus binding motif (Qxx[I/L]xxFF) (Warbrick, 2000) and the β binding motif (QLxLF) suggested by Dalrymple et al. (2001). This is even more remarkable in view of the fact that β and PCNA share no sequence similarity, despite having a common fold. Clamp-binding motifs could either be ancient and reflect a common ancestor, or may have occurred as a result of convergent evolution.
The finding that the clamp loader and all the polymerases present in the E.coli cell interact with the β-clamp at the same location suggests that this site may be an attractive target for the development of DNA replication inhibitors. This is especially true considering that many proteins interact with the same hydrophobic pocket on β. Overcoming drug binding to this pocket would likely impair its binding to several proteins at once, thus making the appearance of antibiotic-resistant variants less likely.
Materials and methods
Materials
HPLC-purified, N-terminal biotinylated 20-mer peptides corresponding to N- and C-terminal sequences of Pol III α, Pol II and Pol IV were purchased from Bio-synthesis, Inc.; N-terminal biotinylated 20-mer peptides corresponding to internal regions of the α-protein and the Ala-scanning peptide series shown in Figure 2 were purchased from Chiron Technologies; they were synthesized using Multipin peptide technology, which leaves a diketopiperidine linker at the C-terminus of the peptide. Streptavidin-coated, 96-well microtiter plates were from Costar, and non-streptavidin plates (MaxiSorp) were from Nalge Nunc, Inc. Streptavidin was from Sigma Co. Labeled nucleotides were from Dupont, New England. Nuclear and unlabeled nucleotides were from Pharmacia-LKB.
Proteins were purified as described previously: α, ε, θ (Studwell and O’Donnell, 1990) and β (Kong et al., 1992). 32P-β was labeled using γ-32P ATP as described previously (Kelman et al., 1995). Pol III core was reconstituted from isolated subunits, then purified from unbound proteins as described (Studwell-Vaughn and O’Donnell, 1991).
Microplate assays
Microplate assays were performed as described previously by Yuzhakov et al. (1999). N-terminally biotinylated peptides (100 pmol) were added to streptavidin-coated 96-well plates in 30 µl PBST buffer [0.01 M phosphate-buffered saline pH 7.2 (PHS) supplemented with 0.1% v/v Tween 20] and incubated for 1 h at 23°C. They were then washed three times by the addition and removal of 100 µl PBST buffer. 32P-β was added (90 nM) directly to wells in a volume of 30 µl and incubated for 1 h, after which each well was washed three times with 50 µl PBST buffer and the plates were analyzed using a PhosphorImager (Molecular Dynamics).
MaxiSorp plates were used in 96-well plate assays in which other proteins were immobilized. Proteins (100 pmol of either δ, Pol I, Pol IV, UmuC, UmuD or UmuD′) were added directly to the plate in 30 µl of water and the plates were dried at 23°C. Plates were then blocked with 5% non-fat dry milk in PBST for 1 h and washed. 32P-β (90 nM in 30 µl PBST buffer) was then added and the plates analyzed using a PhosphorImager.
Native gel mobility shift assays
Native gel electrophoresis assays using 32P-β were performed as described by López de Saro and O’Donnell (2001). Reactions (15 µl) contained 20 mM Tris–HCl (pH 7.5), 0.1 mM EDTA, 4% glycerol, 50 µg/ml bovine serum albumin (BSA), 100 mM NaCl, 5 mM DTT, 200 nM Pol III core (as indicated) and 90 nM 32P-β (150 d.p.m./fmol). When present, either of the following components were added: 4 µg E.coli β, 4 µg human PCNA, 4 µg T4gp45 or 25 pmol α1141–1161 peptide. Reactions were incubated at 37°C for 5 min before loading 4 µl onto a 4% native polyacrylamide gel. Electrophoresis was performed using 40 mM Tris-acetate (pH 8.0), 1 mM EDTA, at 4°C and developed at 17 mA for 90 min. Gels were dried and detection of 32P-β was performed using a PhosphorImager.
Replication assays
DNA synthesis assays were performed using M13mp18 phage circular ssDNA (1.7 nM) primed with a DNA 30-mer. Reactions included 0.42 µM ssDNA-binding protein (as tetramer), 2 nM γ-complex (γ3δδ′χψ) 30 nM β (as dimer), and either 25 nM α, Pol III core or Pol I Klenow fragment, as indicated, in 25 µl 20 mM Tris–HCl (pH 7.5), 0.1 mM EDTA, 4% glycerol, 40 µg/ml BSA, 5 mM DTT, 8 mM MgCl2, 0.5 mM ATP, 60 µM each of dGTP, dATP and dCTP, and 20 µM [α-32P]dTTP. When present, peptide was added at the concentrations indicated prior to shifting to 37°C. Reactions were incubated for 30 s (Pol III) or 30 min (Pol I Klenow fragment) at 37°C and stopped with 25 µl of 1% SDS, 50 mM EDTA, then analyzed for nucleotide incorporation by spotting onto DE81 filter papers as described previously (Studwell and O’Donnell, 1991).
Clamp loading assay
Clamp loading assays of 32P-β onto singly primed M13mp18 ssDNA by γ-complex were performed as described for replication reactions, but in the absence of DNA polymerase or dNTPs. Reaction volumes were 100 µl of 20 mM Tris–HCl (pH 7.5), 0.1 mM EDTA, 4% glycerol 40 µg/ml BSA, 5 mM DTT, 8 mM MgCl2, 0.5 mM ATP and incubation was for 5 min at 37°C. Reactions were then applied to a 5 ml BioGel A15M column (BioRad) pre-equilibrated with 20 mM Tris–HCl (pH 7.5), 0.1 mM EDTA, 4% glycerol, 40 µg/ml BSA, 5 mM DTT, 8 mM MgCl2 and 100 mM NaCl. Chromatography was performed at 22°C and fractions of 180 µl were collected and analyzed by liquid scintillation counting.
KD values using fluorescent βOG
The β-subunit can be uniquely labeled at Cys333 using maleimide derivatives (Griep and McHenry, 1988). β (3 mg) was labeled using OG 488 maleimide (Molecular Probes) in 1 ml of 50 mM potassium phosphate (pH 7.5) and 100 mM NaCl. The OG maleimide (90 nmol) was dissolved in 100 µl DMSO, then 80 µl was added to β with gentle stirring at 4°C, followed by overnight incubation at 4°C in the dark. βOG was separated from unreacted reagent on a 50 ml column of BioGel 6 and contained ∼0.91 molecules of OG per β monomer as determined from protein absorbance at 280 nM (ε280 = 14 890/M/cm) and OG at 490 nm (ε491–76 000/M/cm).
Titration of wild-type α or core αΔC7 into βOG was performed as follows. Reactions contained 50 nM β2OG and 1.5 µM α in 60 µl 20 mM Tris–HCl (pH 7.5), 0.5 mM EDTA, 1 mM DTT and 0–250 mM NaCl (as indicated) on ice. Titrations of Pol III core or Pol II were performed in a similar buffer supplemented with 100 mM NaCl. Reactions were shifted to 22°C for 15 min then 51 µl was placed in a 3 X 3 mm cuvette. Excitation was at 490 nm and emission was monitored from 500–600 nm in a PTI Quantamaster spectrofluorimeter. Fluorescence emission at 517 nm was used for analysis. Additional titrations of wild-type α into high concentrations of β2OG (250 nM and 500 nM) showed a α:β2 =1:1 stoichiometry. As a consequence, data points were fit according to the model, A + B → AB using the Origin software (Microcal, Inc.). Reactions using rhodamine (TAMRA)-labeled peptides contained 1 µM peptide and β-subunit as indicated. Excitation was at 490 nm, and the emission was monitored from 500–600 nm. Fluorescence emission at 517 nm was used for analysis as above.
Acknowledgments
Acknowledgements
We are grateful to David Jeruzalmi (Harvard University) for Figure 7D. This work was supported by a grant from the National Institutes of Health (NIH; Bethesda, MD, USA) (GM38839) and the Howard Hughes Medical Institute (to M.O’D.), and NIH grants GM21422 and GM42554 (to M.F.G.).
References
- Ason B., Hingorani M.M., Beechem,J.M., O’Donnell,M., Goodman,M.F. and Bloom,L.B. (2000) A model for Escherichia coli DNA polymerase III holoenzyme assembly at primer/template ends. DNA triggers a change in binding specificity of the gamma complex clamp loader. J. Biol. Chem., 275, 3006–3015. [DOI] [PubMed] [Google Scholar]
- Ason B., Handayani,R., Williams,C.R., Bertram,J.G., Hingorani,M.M., O’Donnell,M., Goodman,M.F. and Bloom,L.B. (2003) Mechanism of loading the Escherichia coli DNA polymerase III β sliding clamp on DNA. Bona fide primer/template preferentially trigger the γ complex to hydrolyze ATP and load the clamp. J. Biol. Chem., 278, 10033–10040. [DOI] [PubMed] [Google Scholar]
- Baker T. and Kornberg,A. (1992) DNA Replication. Second edition. W.H. Freeman and Co, New York. [Google Scholar]
- Becherel O.J., Fuchs,R.P.P. and Wagner,J. (2002) Pivotal role of the β-clamp in translesion DNA synthesis and mutagenesis in E.coli cells. DNA Repair, 1, 703–708. [DOI] [PubMed] [Google Scholar]
- Berdis A.J., Soumillion,P. and Benkovic,S.J. (1996) The carboxyl terminus of the bacteriophage T4 DNA polymerase is required for holoenzyme complex formation. Proc. Natl Acad. Sci. USA, 93, 12822–12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner C.A., Stukenberg,P.T., Rajagopalan,M., Eritja,R., O’Donnell,M., McEntee,K., Echols,H. and Goodman,M.F. (1992) Processive DNA synthesis by DNA polymerase II mediated by DNA polymerase III accessory subunits. J. Biol. Chem., 267, 11431–11438. [PubMed] [Google Scholar]
- Braithwaite D.K. and Ito,J. (1993) Compilation, alignment and phylogenetic relationships of DNA polymerases. Nucleic Acids Res., 21, 787–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck I., Woodgate,R., McEntee,K. Goodman,M.F. (1996) Purification of a soluble UmuD′C complex from Escherichia coli.J. Biol. Chem., 271, 10767–10774. [DOI] [PubMed] [Google Scholar]
- Dalrymple B.P., Kongsuwan,K., Wijffels,G., Dixon,N.E. and Jennings,P.A. (2001) A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc. Natl Acad. Sci. USA, 98, 11627–11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filée J., Forterre,P., Sen-Lin,T. and Laurent,J. (2002) Evolution of DNA polymerase families: evidences for multiple gene exchange between cellular and viral proteins. J. Mol. Evol., 54, 763–773. [DOI] [PubMed] [Google Scholar]
- Goodman M. (2002) Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem., 71, 17–50. [DOI] [PubMed] [Google Scholar]
- Goodman M. and Tippin,B. (2000) Slopier copier DNA polymerases involved in DNA repair. Curr. Opin. Genet. Dev., 10, 162–168. [DOI] [PubMed] [Google Scholar]
- Griep A. and McHenry,C. (1988) The dimer of the β subunit of Escherichia coli DNA polymerase III holoenzyme is dissociated into monomers upon binding Mg2+. Biochemistry, 27, 5210–5215. [DOI] [PubMed] [Google Scholar]
- Gulbis J.M., Kelman,Z., Hurwitz,J., O’Donnell,M. and Kuriyan,J. (1996) Structure of the C-terminal region of p21WAF1/CIP1 complexed with human PCNA. Cell, 87, 297–306. [DOI] [PubMed] [Google Scholar]
- Hughes A.J., Bryan,S.K., Chen,H., Moses,R.E. and McHenry,C.S. (1991) Escherichia coli DNA polymerase II is stimulated by DNA polymerase III holoenzyme auxiliary subunits. J. Biol. Chem., 266, 4568–4573. [PubMed] [Google Scholar]
- Jeruzalmi D., Yurieva,O., Zhao,Y., Young,M., Stewart,J., Hingorani,M., O’Donnell,M. and Kuriyan,J. (2001) Mechanism of processivity clamp opening by the delta subunit wrench of the clamp loader complex of E.coli DNA polymerase III. Cell, 106, 417–428. [PubMed] [Google Scholar]
- Kelman Z., Naktinis,V. and O’Donnell,M. (1995) Radiolabeling of proteins for biochemical studies. Methods Enzymol., 262, 430–442. [DOI] [PubMed] [Google Scholar]
- Kelman Z., Zuo,S., Arroyo,M.P., Wang,T.S.-F. and Hurwitz,J. (1999) The C-terminal region of Schizosaccharomyces pombe proliferating cell nuclear antigen is essential for DNA polymerase activity. Proc. Natl Acad. Sci. USA, 96, 9515–9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.R. and McHenry,C.S. (1996) Identification of the β-binding domain of the α subunit of Escherichia coli Polymerase III holoenzyme. J. Biol. Chem., 271, 20699–20704. [DOI] [PubMed] [Google Scholar]
- Kobayashi S., Valentine,M.R., Pham,P., O’Donnell,M. and Goodman,M.F. (2002) Fidelity of Escherichia coli DNA Polymerase IV. Preferential generation of small deletion mutations by dNTP-stabilized misalignment. J. Biol. Chem., 277, 34198–34207. [DOI] [PubMed] [Google Scholar]
- Kong X.-P., Onrust,R., O’Donnell,M. and Kuriyan,J. (1992) Three dimensional structure of the β subunit of Escherichia coli DNA polymerase III Holoenzyme: a sliding DNA-clamp. Cell, 69, 425–437. [DOI] [PubMed] [Google Scholar]
- Lenne-Samuel N., Wagner,J., Etienne,H. and Fuchs,R.P.P. (2002) The processivity factor β controls DNA polymerase IV traffic during spontaneous mutagenesis and translesion synthesis in vivo. EMBO Rep., 3, 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu F., Georgescu,R. and O’Donnell,M. (2003) Mechanism of the E.coli τ processivity switch during lagging-strand synthesis. Mol. Cell, 11, 315–327. [DOI] [PubMed] [Google Scholar]
- López de Saro F.J. and O’Donnell,M. (2001) Interaction of the β sliding clamp with MutS, ligase and DNA polymerase I. Proc. Natl Acad. Sci. USA, 98, 8376–8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López de Saro F.J., Georgescu,R.E. and O’Donnell,M. (2003) A peptide switch regulates DNA polymerase processivity. Proc. Natl Acad. Sci. USA, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naktinis V., Turner,J. and O’Donnell,M. (1996) A molecular switch in a replication machine defined by an internal competition for protein rings. Cell, 84, 137–145. [DOI] [PubMed] [Google Scholar]
- O’Donnell M.E. (1987) Accessory proteins bind a primed template and mediate rapid cycling of DNA polymerase III holoenzyme from Escherichia coli. J. Biol. Chem., 262, 16558–16565. [PubMed] [Google Scholar]
- O’Donnell M., Jeruzalmi,D. and Kuriyan,J. (2001) Clamp loader structure predicts the architecture of DNA polymerase III holoenzyme and RFC. Curr. Biol., 11, R935–R946. [DOI] [PubMed] [Google Scholar]
- Ohmori H. et al. (2001) The Y-family of DNA polymerases. Mol. Cell, 8, 7–8. [DOI] [PubMed] [Google Scholar]
- Pham P., Bertram,J.G., O’Donnell,M., Woodgate,R. and Goodman,M.F. (2001) A model for SOS-lesion-targeted mutations in Escherichia coli. Nature, 409, 366–370. [DOI] [PubMed] [Google Scholar]
- Pham P., Seitz,E.M., Saveliev,S., Shen,X., Woodgate,R., Cox,M.M. and Goodman,M.F. (2002) Two distinct modes of RecA action are required for DNA polymerase V-catalyzed translesion synthesis. Proc. Natl Acad. Sci. USA., 99, 11061–11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamoo Y. and Steitz,T.A. (1999) Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell, 99, 155–166. [DOI] [PubMed] [Google Scholar]
- Stucki M., Stagljar,I., Jónsson,Z.O. and Hübscher,U. (2001) A coordinated interplay: proteins with multiple functions in DNA replication, DNA repair, cell cyle/checkpoint control and transcription. Prog. Nucleic Acids Res., 65, 261. [DOI] [PubMed] [Google Scholar]
- Stuckenberg P.T., Studwell-Vaughan,P.S. and O’Donnell,M. (1991) Mechanism of the sliding β-clamp of DNA polymerase III holoenzyme. J. Biol. Chem., 266, 11328–11334. [PubMed] [Google Scholar]
- Stuckenberg P.T., Turner,J. and O’Donnell,M. (1994) An explanation for lagging strand replication: polymerase hopping among DNA sliding clamps. Cell, 78, 877–887. [DOI] [PubMed] [Google Scholar]
- Studwell P.S. and O’Donnell,M. (1990) Processive replication is contingent on the exonuclease subunit of DNA polymerase III holoenzyme. J. Biol. Chem., 265, 1171–1178. [PubMed] [Google Scholar]
- Studwell P.S. and O’Donnell,M. (1991) Constitution of the twin polymerase of DNA polymerase III holoenzyme. J. Biol. Chem., 266, 19833–19841. [PubMed] [Google Scholar]
- Studwell-Vaughan P.S. and O’Donnell,M. (1991) Constitution of the twin polymerase of DNA polymerase III holoenzyme. J. Biol. Chem., 266, 19833–19841. [PubMed] [Google Scholar]
- Sutton M.D. and Walker,G.C. (2001) Managing DNA polymerases: coordinating DNA replication, DNA repair and DNA recombination. Proc. Natl Acad. Sci. USA, 98, 8342–8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton M.D., Opperman,T. and Walker,G.C. (1999) The Escherichia coli SOS mutagenesis proteins UmuD and UmuD′ interact physically with the replicative DNA polymerase. Proc. Natl Acad. Sci. USA, 96, 12373–12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M., Pham,P., Shen,X., Taylor,J.-S., O’Donnell,M., Woodgate,R. and Goodman,M.F. (2000) Roles of E.coli DNA polymerases IV and V in lesion-targeted and untargeted mutagenesis. Nature, 404, 1014–1018. [DOI] [PubMed] [Google Scholar]
- Turner J., Hingorani,M.M., Kelman,Z. and O’Donnell,M. (1999). The internal workings of a DNA polymerase clamp-loading machine. EMBO J., 18, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J., Fujii,S., Gruz,P., Nohmi,T. and Fuchs,R.P.P. (2000) The β clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep., 1, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J., Etienne,H., Janel-Bintz,R. and Fuchs,R.P.P. (2002) Genetics of mutagenesis in E.coli: various combinations of translesion polymerases (PolII, IV and V) deal with lesion/sequence context diversity. DNA Repair, 1, 159–167. [DOI] [PubMed] [Google Scholar]
- Warbrick E. (2000) The puzzle of PCNA’s many partners. BioEssays, 22, 997–1006. [DOI] [PubMed] [Google Scholar]
- Woodgate R. and Ennis,D.G. (1991) Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol. Gen. Genet., 229, 10–16. [DOI] [PubMed] [Google Scholar]
- Woodgate R., Rajagopalan,M., Lu,C. and Echols,H. (1989) UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD’. Proc. Natl Acad. Sci. USA, 86, 7301–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzhakov A., Kelman,Z. and O’Donnell,M. (1999) Trading places on DNA: a three point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell, 96, 153–163. [DOI] [PubMed] [Google Scholar]