

Abstract
Long-chain polyunsaturated fatty acids (PUFA) orchestrate immunity and inflammation through their capacity to be converted to potent inflammatory mediators. We assessed associations of FADS gene cluster polymorphisms and fasting serum PUFA concentrations in a fully ascertained, geographically isolated founder population of European descent. Concentrations of 22 PUFAs were determined by gas chromatography, of which ten fatty acids and five ratios defining FADS1 and FADS2 activity were tested for genetic association against 16 single nucleotide polymorphisms (SNP) in 224 individuals. A cluster of SNPs in tight linkage disequilibrium in the FADS1 gene (rs174537, rs174545, rs174546, rs174553, rs174556, rs174561, rs174568, and rs99780) were strongly associated with arachidonic acid (AA) (P = 5.8 × 10−7 – 1.7 × 10−8) among other PUFAs, but the strongest associations were with the ratio measuring FADS1 activity in the ω-6 series (P = 2.11 × 10−13 – 1.8 × 10−20). The minor allele across all SNPs was consistently associated with decreased ω-6 PUFAs, with the exception of dihomo-γ-linoleic acid (DHGLA), where the minor allele was consistently associated with increased levels. Our findings in a geographically isolated population with a homogenous dietary environment suggest that variants in the Δ-5 desaturase enzymatic step likely regulate the efficiency of conversion of medium-chain PUFAs to potentially inflammatory PUFAs, such as AA.
Keywords: fatty acid desaturase, isolated population, genetic association
Several changes in diet over the past century are hypothesized to have been maladaptive, thereby leading to an increase in the incidence of numerous chronic diseases in developed countries (1, 2). Many of these diseases (obesity, diabetes, asthma, allergies, arthritis and chronic joint disease, dementia, and cardiovascular disease) have a substantial inflammatory component (3–6) with notable co-occurrences (7, 8). Perhaps no changes in the modern diet have had a greater impact than the quantitative and qualitative changes in fat consumption. While the percentage of energy from fat (∼35%) is thought to be similar in ancestral and contemporary Western diets, the concentrations and ratios of the types of fat that humans eat (9, 10) have shifted, with marked increases in saturated and omega-6 (ω6) fatty acid consumption. Importantly, these changes have been suggested to alter human health and the incidence of chronic inflammatory diseases (11–13).
Long-chain serum polyunsaturated fatty acids (LC-PUFA; ≥20 carbons), such as arachidonic acid (AA; C20:4ω6), and their metabolic products orchestrate several critical events in immunity and inflammation. PUFAs directly impact normal and pathophysiologic responses through their capacity to be converted to potent bioactive products (such as prostaglandins, thromboxanes, leukotrienes, and lipoxins), to regulate cellular receptors or to modulate the expression of genes that control immune responses (14–18). The MC-PUFAs linoleic acid (LA; C18:2ω6) and α-linolenic acid (ALA; C18:3ω3), which are the precursors of more unsaturated and longer chain ω6 and ω3 PUFAs, respectively, are considered essential because they must be obtained from the diet. Once ingested by mammals, these MC-PUFAs are converted to other more desaturated MC-PUFAs and eventually to LC-PUFAs by the alternate actions of fatty acid desaturases (FADS) and FA elongases (Fig. 1). To date, three members of the FADS gene family (FADS1-3) have been discovered. Their gene products appear to be pivotal in the conversion of MC-PUFAs to LC-PUFAs (Fig. 1). These desaturase genes are localized to a 1.4Mb region on chromosome 11, and they are speculated to have arisen during human evolution through the mechanism of gene duplication, as evidenced by their high degree of sequence identity (62–70%) and almost identical intron/exon organization (19). Candidate gene studies have focused on the contribution of genetic variants in FADS1 and FADS2 to circulating and cellular levels of PUFAs. These studies have relied on a subset of single nucleotide polymorphisms (SNP) that appear to adequately tag the variation in this genomic region (20–24). Additionally, a recent genome-wide association study (GWAS) (25) found little evidence of association for loci outside of 11q12-13. However, the effects of genetic loci can be difficult to fully interpret when a proportionally large source of total phenotypic variation in the quantitative trait distribution comes from genetic heterogeneity and sources other than genetics (i.e., dietary variation in fatty acid intake). Consequently, there is a need to test these associations in cohorts that are not ascertained on the basis of any specific inflammatory disease and for which environments are homogeneous. To address these important issues, we examined the role of FADS in PUFA metabolism in a fully ascertained, geographically isolated (island) population (Tangier Island, Virginia).
Fig. 1.

Biosynthetic pathway of ω3 and ω6 polyunsaturated fatty acids (PUFAs). Fatty acids are obtained from the diet or by de novo FA synthesis (upper left), which builds saturated fatty acids (SFA) by 2-carbon unit increments to the 18-carbon stearic acid (C18:0; i.e., 18 carbons with no double bonds) and takes place in all organisms. Stearoyl coenzyme A desaturase 1(SCD1) initiates fatty acid desaturation and generates the precursors of the ω-7 and ω-9 series fatty acids (upper left). The lack of Δ-12 and Δ-15 desaturase (dashed box) in animals renders linoleic acid (LA) and α-linolenic acid (ALA) essential fatty acids (**) and thus must be obtained from the diet, generally from plant-derived sources. The synthesis of ω-9 (derived from oleic acid, 18:1ω9; series not shown), ω-6 (from LA, 18:2ω6) and ω-3 (from ALA, 18:3ω) PUFAs proceeds in parallel, with the activity of Δ-6 desaturase (FADS2) thought to be the rate-limiting step in PUFA synthesis. In animals and organisms at the base of the food chain, LC-PUFAs are synthesized by alternating actions of elongases (red arrows) and desaturases (blue arrows). In mammals, a variety of enzymes (green arrows) generate numerous bioactive derivatives from ω-6 (DHGLA and AA) and ω-3 (EPA, DHA) PUFAs that have numerous targets and functions throughout the body. With the exception of the series-1 prostaglandins (PG), thromboxanes (TX), and leukotrienes (LT) derived from DHGLA, the ω6-derived lipid mediators (series-2 and -4) tend to have proinflammatory actions. Like the series-1 lipid mediators, the series-3 and -5 metabolites of ω3 PUFAs generally exhibit less inflammatory to anti-inflammatory properties. AA, arachidonic acid; DHGLA, dihomo-gamma-linolenic acid; EPA, eicosapentanoic acid; FADS, fatty acid desaturase.
METHODS
Study population
Tangier, Virginia, is a three-by-one mile marshy island situated close to the mouth of the Chesapeake Bay, 12 miles from the nearest mainland port (Crisfield, Maryland). At the time of subject recruitment (1993–2001), the population was composed of approximately 650 individuals, and the isolated nature of the island has led to sustained low levels of inbreeding (26). Briefly, the average inbreeding coefficient (F) for the entire Tangier Island population (dating back to 1722) is 0.009 (range, 0.000–0.156; n = 3,512). While in the early cohorts, close consanguinity contributed to inbreeding, in the later cohorts, as is typical with a relatively isolated island population, there was a conscious avoidance of close mating and the observed inbreeding levels were a result of older lines of more distant relationships. For the majority of the 13 generations on Tangier Island up to the contemporary population, subsistence has been almost exclusively devoted to fishing and crabbing. Due in part to topographical limitations on Tangier Island (i.e., marshy wetlands), kitchen gardens were not common at the time of this study and access to fresh produce was limited to weekly shipments from the mainland and relatively costly to islanders. Ironically, and despite the substantive intake of protein from fish and shellfish, participant observation studies and nutritional surveys collected from 1993 to 2001 implicated an island-wide diet very high in ω6 fatty acids (Barnes et al., unpublished observations).
A total of 453 individuals were recruited, corresponding to 68.2% of the total population residing on the island in 2001. All participants are connected in a single, 13-generational pedigree with an average inbreeding coefficient reflective of an avoidance of close consanguinity (average F = 0.003 per mating summed over an average number of paths connecting each pair = 300) (26, 27). All subjects gave written, informed consent as approved by The Johns Hopkins University Institutional Review Board. Subject characterization included (1) an interviewer-administered standardized questionnaire from which data on age, gender, and smoking exposure were obtained and (2) peripheral fasting blood collection for fatty acid measurements and DNA isolation for genotyping.
Fatty acid levels
Serum was isolated from fasting whole blood samples of 224 individuals (49% of eligible participants) and fatty acids analyzed as described below. A panel of twenty-two ω3 and ω6 fatty acids was quantified by gas chromatography with flame ionization detection (Table 1). Fatty acid methyl esters (FAME) were prepared (28) from duplicate serum samples (100 μl) following as previously described (29). Individual fatty acids are expressed as percent of total fatty acids in a sample.
TABLE 1.
Demographic characteristics and fasting serum FA profile (as % of total FA) in Tangier Island subjects
| Characteristic | Total Sample N = 224 |
|---|---|
| Males, N (%) | 84 (37.7%) |
| Age, years | 46.65 ± 21.15 |
| Myristic acid (C14:0) | 1.5 ± 0.48 |
| Palmitic acid (C16:0) | 26.6 ± 2.56 |
| Palmitoleic acid (C16:1ω7) | 2.49 ± 0.93 |
| Stearic acid (C18:0) | 13.0 ± 3.9 |
| Oleic acid (C18:1ω9) | 22.1 ± 3.30 |
| Vaccenic acid (C18:1ω11c/t) | 2.00 ± 1.12 |
| Linoleic acida (C18:2ω6) | 24.8 ± 4.52 |
| Gamma-linolenic acida (C18:3ω6) | 0.313 ± 0.137 |
| Alpha-linolenic acida (C18:3ω3) | 0.487 ± 0.190 |
| Stearidonic acida (C18:4ω3) | 0.014 ± 0.020 |
| Arachidic acid (C20:0) | 0.241 ± 0.067 |
| Gadoleic acid (C20:1ω9) | 0.136 ± 0.039 |
| Auricolic acid (C20:2, OH) | 0.200 ± 0.050 |
| Dihomo-γ-linolenic acida (C20:3ω6) | 1.0 ± 0.248 |
| Arachidonic acida (C20:4ω6) | 3.6 ± 1.10 |
| Eicosatrienoic acid (C20:3ω3) | 0.039 ± 0.036 |
| Eicosatetraenoic acid (C20:4ω3) | 0.032 ± 0.020 |
| Eicosapentaenoic acida (C20:5ω3) | 0.23 ± 0.110 |
| Docosadienoic acid (C22:2) | 0.0081 ± 0.014 |
| Adrenic acida (C22:4ω6) | 0.14 ± 0.047 |
| Docosapentaenoic acida (C22:5ω3) | 0.2363 ± 0.1007 |
| Docosahexaenoic acida (C22:6ω3) | 0.598 ± 0.270 |
Values are mean ± standard deviation presented for quantitative traits (except gender).
PUFA selected for genetic analysis in this study.
Ten PUFAs were selected for inclusion in the genetic analyses on the basis of their biological relevance in the metabolic pathway outlined in Fig. 1. These included linoleic acid (LA), γ-linolenic acid (GLA), α-linolenic acid (ALA), stearidonic acid (SDA), dihomo-γ-linolenic acid (DHGLA), arachidonic acid (AA), eicosapentanoic acid (EPA), adrenic acid (ADA), docosapentaenoic acid (DPA), and docosahexaenoic acid (DHA) (Table 1). To better elucidate the role of the genetic variants in the FADS gene cluster, we also analyzed derived phenotypes (i.e., ratios derived from specific combinations of PUFAs) that included the precursor and product PUFAs at the pivotal metabolic steps identified in Fig. 1. The ratios included (i) enzymatic activity in the total pathway in ω6 series (TPω6) defined as (AA+ADA)/(LA+GLA+DHGLA); (ii) enzymatic activity in the total pathway in ω3 series (TPω3) defined as (EPA+DPA)/(ALA+SDA+eicosatetraenoic acid [ETEA]); (iii) Fatty acid desaturase 1 (FADS1) activity in the ω6 series (FADS1ω6) defined as (AA+ADA)/DHGLA; (iv) FADS1 activity in the ω3 series (FADS1ω3) defined as (EPA+DPA)/ETEA); and (v) FADS2 activity in the ω6 series (GLA/LA).
Genotyping
We selected 16 SNPs in genes previously identified as associated with the above-mentioned PUFAs and ratios (20–22, 24, 25, 30) (Table 2). Tagging approaches (31) show that these 16 SNPs captured 100% of the genetic variation in a 102Kb region on chromosome 11q13.1 harboring the FADS1_FADS2_FADS3_CR11orf cluster in the Utah residents with Northern and Western European ancestry from the CEPH collection (CEU) population in HapMap (http://hapmap.ncbi.nlm.nih.gov/). SNPs were genotyped using the 5′nuclease allelic discrimination Taqman® assay with allelic specific probes on the ABI Prism 7900HT Sequence Detection System (Applies Biosystems, Foster City, CA) according to standard laboratory protocols. Primers and probes were designed and manufactured by Applied Biosystem. Ten percent of the samples were genotyped in duplicate with a100% reproducibility.
TABLE 2.
Characteristics of SNPs analyzed in the C11orf10_FADS1_FADS2_FADS3 gene cluster in the Tangier Island subjects
| SNP | Position | Gene | Position in Gene | Minor Allele | MAF (%) |
|---|---|---|---|---|---|
| rs174537 | 61309256 | C11orf10 | Downstream | T | 29.8 |
| rs174545 | 61325882 | FADS1/C11orf10 | 3′ UTR FADS1 | G | 28.7 |
| rs174546 | 61326406 | FADS1/C11orf10 | 3′ UTR FADS1 | T | 29.4 |
| rs174553 | 61331734 | FADS1 | Intron | G | 29.9 |
| rs174556 | 61337211 | FADS1 | Intron (boundary) | T | 25.6 |
| rs174561 | 61339284 | FADS1 | Intron | C | 26.0 |
| rs174568 | 61350392 | FADS1/FADS2 | Promoter FADS1 | C | 47.3 |
| rs99780 | 61353209 | FADS2 | Intron | T | 29.5 |
| rs174570 | 61353788 | FADS2 | Intron | C | 44.9 |
| rs174575 | 61358579 | FADS2 | Intron | G | 23.8 |
| rs2524299 | 61361358 | FADS2 | Intron | A | 46.5 |
| rs174583 | 61366326 | FADS2 | Intron | T | 30.4 |
| rs498793 | 61381281 | FADS2 | Intron | C | 45.8 |
| rs174611 | 61384457 | FADS2 | Intron | T | 33.0 |
| rs174627 | 61394042 | FADS3 | Downstream | A | 17.4 |
| rs1000778 | 61411881 | FADS3 | Intron | A | 18.7 |
FADS, fatty acid desaturase; MAF, minor allele frequency; SNP, single nucleotide polymorphism.
Statistical analysis
The Kolmogorov-Smirnov test was performed to evaluate normality of the variables, and variables were suitably transformed where necessary to approach normality as is assumed in the tests for association described below. Sensitivity analysis was performed to evaluate the influence of any outliers in distribution; repeating analyses with and without the outlier observations. Correlation between quantitative variables was assessed by the Pearson's correlation test. Quality control assessments for the genotype data included evaluation of the minor allele frequency (MAF) and Mendelian inconsistency checks. Tests for Hardy Weinberg were not performed in these data because there were a small number of true founders (only 19 individuals did not trace their ancestry to the original founders of 1722) in the set of genotyped individuals and isolated/inbred nature of the population violates the core assumption of Hardy Weinberg Equilibrium rules, rendering the test invalid. Linkage disequilibrium (LD) was assessed using D' and r2 within Haploview (32) relying on all individuals in the population (not optimal but necessary given the limited number of genotyped individuals identified as true founders).
Given the complex nature of the pedigree, the single 13-generation-deep pedigree that included >4,000 members was considered at the nuclear family level. Individuals were clustered based on membership in a single sib-ship (i.e., all individuals that shared a parent were given the same cluster number) without the duplication of any individuals (i.e., only siblings were included in the cluster, parents and offspring of the siblings were each included in their own sibling-specific cluster). Tests for association were then performed in a linear regression framework under an additive model at each SNP; coding each SNP on a linear scale of 0/1/2 for 0, 1, or 2 copies of the minor variant allele. The regression models were implemented in the generalized estimating equation (GEE) framework with an exchangeable covariance matrix to correct for sibship clustering. Age and sex were included as covariates in the model. One final model including all significant SNPs, each with its main effects, was also run in an attempt to decipher the SNP with the strongest independent contribution in this region. Locus-specific variation, the phenotypic variance in each PUFA that can be attributed to the specific SNP under consideration, was calculated using the estimated allele frequency, total phenotypic variance, and the three estimated genotypic means and standard deviations (33) assuming no dominance effects.
A total of 15 tests of association were conducted for each of 16 SNPs for a total of 240 statistical tests for association. A Bonferroni approach to maintain a family-wise error rate of 0.05 is stringent given the high correlation between SNPs in this region and the correlation between derived ratios and individual traits that were used for their derivation. Nonetheless, significance was evaluated at both at the nominal level (P < 0.05) as well as the Bonferroni threshold (P < 2.08 × 10−4).
RESULTS
Clinical characteristics of the study subjects (N = 224) are presented in Table 1. The average age was 46.65 ± 21.15 years, and 38% were male. Distribution of age and gender in the subset of Tangier Island residents included in this study is not different in age (44.6 ± 21.8, P = 0.112) or gender (42%, P = 0.114) from that in the fully ascertained sample (N = 453).
A summary of the fasting serum fatty acid profile is presented in Table 1. Palmitic acid constituted the highest proportion of total fatty acids (26.6%, saturated fatty acid) followed by linoleic acid (24.8%, an essential MC-PUFA), oleic acid (22.1%, monounsaturated fatty acid), and stearic acid (13%, saturated fatty acid). There was a significant association between serum fatty acids and age for myristic acid (r2 = 0.225, P = 0.003), palmitic acid (r2 = 0.225, P < 0.001), palmitoleic acid (r2 = 0.229, P < 0.003), oleic acid (r2 = 0.221, P < 0.004), vaccenoic acid (r2 = 0.265, P=0.001), linoleic acid (r2= −0.306., P<0.001), α-linolenic acid (r2 = 0.211, P = 0.006), arachidic acid (r2 = −0.195, P = 0.011), auricolic acid (r2 = −0.158, P = 0.041), eicosatrienoeic acid (r2 = 0.213, P = 0.006), eicosapentenoic acid (r2= 0.216, P = 0.005), and adrenic acid (r2= −0.284, P <0.001). With the exception of palmitic acid (P = 0.005), linoleic acid (P = 0.037), eicosapentanoic acid (P = 0.010), and docosapentaenoic acid (P = 0.004), there were no differences in fatty acid concentrations between genders (data not shown). Ten fatty acids, all PUFAs, were selected for further association analyses (Table 1).
MAFs of the 16 genotyped SNPs ranged from 17.4 to 47.3% (Table 2) and were similar in the subset with PUFA data to the full Tangier sample. Considerable LD was observed for the 16 SNPs with one large block of LD identified according to the algorithm of Gabriel et al. (31), extending 43kb from rs174537 in C11orf10 to rs99780 in intron 1 of FADS2 and including all genotyped SNPs mapping to FADS1 (Fig. 2). A second block of 3kb, extending from rs498793 in intron 6 of FADS2 to rs174611 in intron 7 of FADS2, was also observed (Fig. 2). The values of r2 ranged from 0.93 to 1 in both blocks. These LD patterns are similar to the HapMap CEU reference sample (http://hapmap.ncbi.nlm.nih.gov/).
Fig. 2.

Physical location (A) of the 16 SNPs and the gene structure of the FADS gene cluster are shown (dark blue line = physical location of genotyped SNP; light blue/pink boxes = exon/intron structure with FADS genes). Schematic overview of linkage disequilibrium patterns (B) in the 102 kb region containing the FADS gene cluster (Chr 11 q12-13), as defined by the algorithm of Gabriel et al. (31) using 224 subjects in the analysis (range of LD from high to low displayed as color ranging from dark red to white, respectively). Abbreviations: FADS, fatty acid desaturase; SNP, single nucleotide polymorphism.
Using a heat map of signal intensity (Fig. 3A), the strongest associations were observed for a cluster of SNPs in the FADS1 gene (rs174537, rs174545, rs174546, rs174553, rs174556, rs174561, rs174568, and rs99780) and AA (P values: 1.6 × 10−7, 2.4 × 10−8, 4.05 × 10−8, 1.7 × 10−8, 5.8 × 10−7, 4.14 × 10−8, 5.71 × 10−7 and 7.6 × 10−8, respectively; see also supplementary Table I). All associations met the strict Bonferroni thresholds. Similar associations were observed for GLA, DHGLA, ADA, and DHA, albeit at lower significance thresholds for which only a few survived the Bonferroni correction. Of interest, associations for individual PUFAs, especially AA, were more pronounced when analyzed as ratios, which estimate the biosynthetic capacity of PUFA formation (TPω6, TPω3) and FADS activities (FADS1ω6, FADS1ω3, and FADS2). All three ratios met the strict Bonferroni thresholds for these SNPs, with the strongest association for the ratio that measures FADS1 activity in the ω6 series (FADS1 ω6 defined as AA+ADA/DHGLA, P values: 2.11 × 10−13 – 1.8 × 10−20).
Fig. 3.

A schematic overview of association analyses for 16 SNPs in the FADS gene cluster and PUFAs (N = 10) and PUFA ratios (N = 5) in 224 subjects from the Tangier population. The ratios included are (i) enzymatic activity in the total pathway in ω6 series (TPω6) defined as (AA+ADA)/(LA+GLA+DHGLA); (ii) enzymatic activity in the total pathway in ω3 series (TPω3) defined as (EPA+DPA)/(ALA+SDA+ETEA); (iii) Fatty acid desaturase 1 (FADS1) activity in the ω6 series (FADS1ω6) defined as (AA+ADA)/DHGLA; (iv) FADS1 activity in the ω3 series (FADS1ω3) defined as (EPA+DPA)/ETEA; and (v) FADS2 activity in the ω6 series (GLA/LA). The P values of these association tests are shown in Panel A, and the specific allele associated with an increased mean trait value for each phenotype is shown in Panel B for all tests, where P < 0.05. Color key mapping P values and alleles to specific color codes are displayed. Abbreviations: AA, arachidonic acid; ADA, adrenic acid; ALA, alpha-linolenic acid; DHGLA, dihomo-gamma-linolenic acid; DPA, docosapentaenoic acid; EPA, eicosapentanoic acid; ETEA, eicosatetraenoic acid; FADS, fatty acid desaturase; GLA, gamma-linolenic acid; LA, linoleic acid; PUFA, polyunsaturated fatty acid; SNP, single nucleotide polymorphism.
For all tests of association that were nominally significant (i.e., P < 0.05; Fig. 3, panel A), the specific allele associated with increases in the quantitative phenotype are presented (Fig. 3, panel B). The same allele is consistently associated with increased trait values for each fatty acid, with the exception of DHGLA. Specifically the allele associated with higher concentrations of AA, GLA, EPA, DHA, and ADA are all associated with a lower concentrations of DHGLA across the significant SNPs. Pair-wise correlations between DHGLA and AA, GLA, EPA, DHA, and ADA were all positive (r2 = 0.367, 0.294, 0.131, 0.222 and 0.484, respectively; supplementary Table II).
Fig. 4 presents a closer examination of the allelic effects for the rs174537 genotypes (GG = 51%, GT = 39%, and TT = 10%), the SNP most strongly associated high plasma PUFA concentrations in the recent GWAS (25), and AA, DHGLA, and FADS1 activity in the ω6 series (FADS1ω6) in the Tangier population. The proportion of variance explained by the additive effects of rs174537 was 10% for AA and 6% for DHGLA, but considerably higher (24%) for FADS1ω6. Individuals with the GG genotype (major allele) had significantly higher concentrations of AA (3.9 ± 1.07) compared with individuals with the GT (3.38 ± 1.05) or TT genotype (2.9 ± 0.6; P = 1.66 × 10−7). In contrast, the GG genotype showed lower DHGLA concentrations (1.03 ± 0.2) compared with the GT (1.12 ± 0.2) and the TT genotypes (1.26 ± 0.2; P = 0.0015). This suggests that FADS1 activity, as defined by its product (AA)/substrate (DHGLA) ratio may be a key determinant of PUFA synthetic capacity in humans.
Fig. 4.
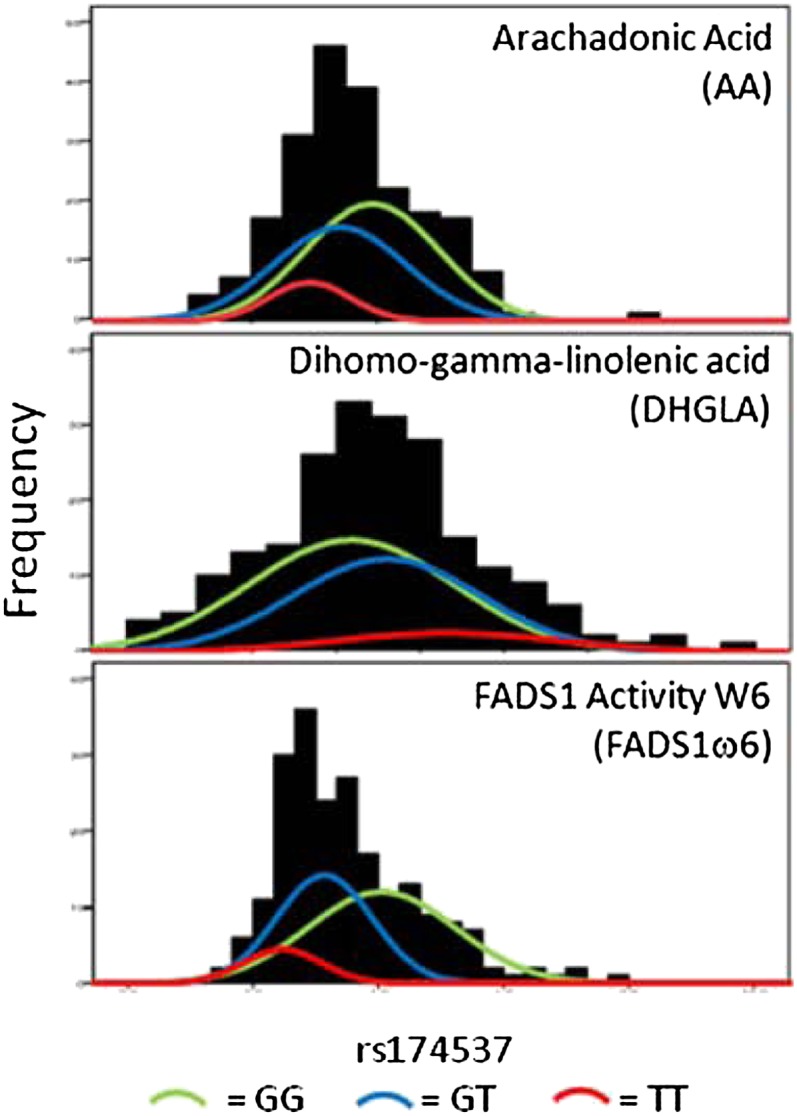
Phenotypic distributions of AA (top panel, product of FADS1), DHGLA (middle panel, substrate of FADS1), and FADS1ω6 (bottom panel, FADS1 activity in the ω6 series) by the three genotypes at SNP rs174537) in 224 subjects from the Tangier population. In each panel (1), a frequency distribution of the full distribution of the phenotype (N = 224) is shown in black boxes (2); the histogram of the distribution in the subjects with the GG genotype (N = 107) in a green line (3); the histogram of the distribution in the subjects with the GT heterozygote genotype (N = 83) in a blue line; and (4) the histogram of the distribution in the subjects with the TT homozygote genotype (N = 21) in a red line. Abbreviations: AA, arachidonic acid; DHGLA, dihomo-gamma-linolenic acid; FADS, fatty acid desaturase; SNP, single nucleotide polymorphism.
DISCUSSION
Specific genetic variants have been hypothesized to alter the capacity of individuals to convert dietary MC-PUFAs to LC-PUFAs. This is particularly relevant given the high levels of MC-ω6 PUFAs (e.g., 14–20 LA g/day) commonly obtained in the typical Western diet. High concentrations of LC-PUFAs, such as AA (derived from the conversion of LA to AA), may subsequently influence levels of proinflammatory eicosanoids, which in turn appear to be associated with elevated markers of low-level systemic inflammation, such as CRP, and increase the risk for diseases, such as atherosclerosis (34–36). To date, this hypothesis has been studied in heterogeneous human populations for which there is also a high intersubject variability in dietary concentrations of MC- and LC-PUFAs and in populations typically ascertained on the basis of specific proinflammatory clinical conditions.
One of the most difficult aspects of understanding the ω6/ω3 desaturation/elongation pathway (Fig. 1) in humans is the fact that different dietary fatty acids (from heterogeneous diets) can impact several points the biosynthetic pathway. It is estimated that the daily consumption of PUFAs by humans eating a Western diet is 14–20 g LA, 0.5–1.5g ALA, 50–200 mg AA, and 50–200 mg EPA/DHA (37, 38). The quantities and ratios of these dietary PUFAs depends in large part on (1) the quantities of and types of cooking and vegetable oils used and (2) the dairy and meat products consumed. This dietary heterogeneity adds complexity to the interpretation of in vivo PUFA data at multiple levels. For example, ω3 PUFAs compete with ω6 PUFAs at several points in the biosynthetic pathway (Fig. 1). In addition, some PUFAs serve as enzymatic substrates for steps early in the pathway, while others serve as product inhibitors for the same enzymatic steps. An isolated population with genetic and environmental homogeneity provides an excellent model for the identification of genetic variants. This has been demonstrated in other isolated populations, such as the Hutterites, Finish, and Amish, for multiple complex disorders like bipolar disease, allergic asthma, and familial combined hyperlipidemia, among others (39). Although the Tangier Island population is isolated and inbred, our previous work has demonstrated that inbreeding levels are reflective of an avoidance of close consanguinity (26), rendering Tangier Island generalizable to other European-ancestry populations as reflected in similar LD patterns and allele frequencies to publicly available databases (Fig. 2; R. A. Mathias, unpublished observations). Collectively, this unique geographically isolated island population presents a remarkable opportunity to analyze the effect of these FADS gene variants on PUFA levels in a setting that may limit the confounding between genetics and the environmental (dietary) factors discussed above.
The current study took a biochemical approach, utilizing both fatty acid profiles (ten ω6 and ω3 PUFAs) and PUFA ratios (five separate ratios), to evaluate the potential functional consequences of genetic variations in the FADS gene cluster. This approach allowed us to estimate the biochemical efficiency of FADS1 and FADS2 enzyme activities. One of the greatest surprises from these data is that the FADS1 (Δ5-desaturase) and not FADS2 (Δ6-desaturase) step of PUFA biosynthesis appears to be the most highly genetically regulated step in human PUFA biosynthesis. Historically, most studies in this field have pointed to the FADS2 (Δ-6 desaturase) step as being the critical rate-limiting step to the formation of LC-PUFAs, such as AA, EPA, and DHA (40, 41). However, many of the studies to date examining rates of PUFA biosynthesis through the various enzymatic steps have been done in rodent models on low-fat diets (typically 5% of calories) (42, 43). Studies designed to evaluate the ability of humans to metabolize LA and ALA to LC-PUFAs have shown variable results and are complicated by a background dietary fat content higher than that used in animal studies (reviewed by Brenna et al. (44). As described above from the tests for association and patterns of linkage disequilibrium, the strongest associations in our study are at FADS1 step, and the role of this FADS1 pathway is supported by the inverse relationship in allelic effect between DHGLA and AA levels.
The direction of the effect of the SNPs with respect to fatty acid levels observed in our study is similar to that conserved across different studies, wherein the minor (variant) allele in all selected SNPs was significantly associated with lower concentrations of LC-PUFAs (most notably, AA and EPA) but higher concentrations of LA and ALA (30). In our study, the patterns of association with increased trait values for each PUFA are consistent, with the exception of DHGLA (i.e., the alleles associated with higher concentrations of AA, GLA, EPA, DHA, and ADA are all associated with a lower concentrations of DHGLA). This is not a statistical artifact as would have occurred if the phenotypic correlation between DHGLA and these fatty acids were all negative. All observed pair-wise correlations between DHGLA and AA, GLA, EPA, DHA, and ADA (supplementary Table II) were positive, making this difference in risk allele particularly interesting. In fact, tests of association on the ratio that measures FADS1 activity in the ω6 series (FADS1 ω6) had the most significant P value observed in our study (P = 10−13–10−20) as it best defines the effects of these SNP loci on AA and DHGLA (where the allelic effects are opposite on the substrate and product for FADS1 activity). All things being equal (i.e., the same amount of MC-PUFA entering the ω6 PUFA biosynthetic pathway), concentrations of the metabolic precursor of FADS1, DHGLA, would be expected to be lower as it is more rapidly converted to AA. In fact, while the proportion of variance in the quantitative trait that is explained by the additive effects at rs174537 is 10% for AA and 6% for DHGLA, it is considerably higher, 24%, for FADS1 activity in the ω6 series when relevant PUFAs are factored into the equation.
Recent functional analyses indicated that a polymorphism located in the promoter of FADS2 (rs968567) influences the transcription of this gene (45). The physical proximity of the 16 SNPs and LD patterns in our data suggests that the strength of association across these SNPs is a function of the high degree of correlation between them. Therefore, it is difficult to identify the SNP that best accounts for the variation in the PUFA phenotypes. While there are a few SNPs with strong association signals that fall outside the defined LD block that includes FADS1, the significance of their effects disappeared when they were included in a single regression model that adjusted for effects of SNPs within the LD block (data not shown). Furthermore, while they are not within the LD block, they do appear to have high correlations with the SNPs within the block, and for these reasons, we suggest that most of the genetic contribution from this chromosomal region to FAs in the Tangier Island population appears to come from SNPs in FADS1.
In summary, we observe a strong relationship between the FADS gene cluster and fatty acid precursors of inflammatory mediators in a genetically and environmentally homogeneous population of European descent. Importantly, these data point to the FADS1 (Δ5-desaturase) enzymatic step as the key step in humans where genetic variance among individuals may regulate the efficiency of the conversion of the high concentrations of MC-PUFAs, such as LA, found in Western diets to potentially inflammatory PUFAs, such as AA.
Supplementary Material
Acknowledgments
The authors thank the families from Tangier Island, Virginia, for their wholehearted cooperation and participation in this study, especially Inez Pruitt, Jean Crockett, and Cindy Parks for their local support. We thank Mark Liu, Eva Ehrlich, Maria Stockton, Renate Nickel, Lun Xiu, Kathryn Held, Sharon Patterson, April Zambelli-Weiner, Cassandra Foster, Chris Cheadle, Alan Berger, Tonya Watkins, and Pat Oldewurtel for technical support.
Footnotes
Abbreviations:
- AA
- arachidonic acid
- ADA
- adrenic acid
- ALA
- alpha-linolenic acid
- CEU
- Utah residents with Northern and Western European ancestry from the CEPH collection in HapMap
- DHA
- docosahexaenoic acid
- DHGLA
- dihomo-gamma-linolenic acid
- DPA
- docosapentaenoic acid
- EPA
- eicosapentanoic acid
- FADS
- fatty acid desaturase
- FAME
- fatty acid methyl ester
- GEE
- generalized estimating equation
- GLA
- gamma-linolenic acid
- GWAS
- genome-wide association study
- LA
- linoleic acid
- LC
- long chain
- LD
- linkage disequilibrium
- MAF
- minor allele frequency
- MC
- medium chain
- SDA
- stearidonic acid
- SNP
- single nucleotide polymorphism
This work was supported by Asthma and Allergy Foundation of America Young Investigator's Award (K.C.B.), National Institutes of Health Grants U01 HL-066583 (K.C.B.) and P50 AT-0027820 (F.H.C.), and the Mary Beryl Patch Turnbull Scholar Program (K.C.B.). F.H.C. has published books with Rodale and Simon and Schuster and is a founder and consultant to GeneSmart Health, Inc., which may be partially related to his research. His potential conflicts of interest have been disclosed to Wake Forest University Health Sciences and to outside sponsors and are institutionally managed. No other authors have a conflict of interest.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of two tables.
REFERENCES
- 1.Cordain L., Eaton S. B., Sebastian A., Mann N., Lindeberg S., Watkins B. A., O'Keefe J. H., Brand-Miller J. 2005. Origins and evolution of the Western diet: health implications for the 21st century. Am. J. Clin. Nutr. 81: 341–354. [DOI] [PubMed] [Google Scholar]
- 2.Robson A. A. 2009. Preventing diet induced disease: bioavailable nutrient-rich, low-energy-dense diets. Nutr. Health. 20: 135–166. [DOI] [PubMed] [Google Scholar]
- 3.Calder P. C., Albers R., Antoine J. M., Blum S., Bourdet-Sicard R., Ferns G. A., Folkerts G., Friedmann P. S., Frost G. S., Guarner F., et al. 2009. Inflammatory disease processes and interactions with nutrition. Br. J. Nutr. 101(Suppl 1): S1–S45. [DOI] [PubMed] [Google Scholar]
- 4.Micha R., Mozaffarian D. 2008. Trans fatty acids: effects on cardiometabolic health and implications for policy. Prostaglandins Leukot. Essent. Fatty Acids. 79: 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt M. I., Duncan B. B. 2003. Diabesity: an inflammatory metabolic condition. Clin. Chem. Lab. Med. 41: 1120–1130. [DOI] [PubMed] [Google Scholar]
- 6.Wellen K. E., Hotamisligil G. S. 2005. Inflammation, stress, and diabetes. J. Clin. Invest. 115: 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iribarren C., Tolstykh I. V., Eisner M. D. 2004. Are patients with asthma at increased risk of coronary heart disease? Int. J. Epidemiol. 33: 743–748. [DOI] [PubMed] [Google Scholar]
- 8.Onufrak S. J., Abramson J. L., Austin H. D., Holguin F., McClellan W. M., Vaccarino L. V. 2008. Relation of adult-onset asthma to coronary heart disease and stroke. Am. J. Cardiol. 101: 1247–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corne J. M., Holgate S. T. 1997. Mechanisms of virus induced exacerbations of asthma. Thorax. 52: 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eaton S. B. 2006. The ancestral human diet: what was it and should it be a paradigm for contemporary nutrition? Proc. Nutr. Soc. 65: 1–6. [DOI] [PubMed] [Google Scholar]
- 11.Astrup A., Dyerberg J., Selleck M., Stender S. 2008. Nutrition transition and its relationship to the development of obesity and related chronic diseases. Obes. Rev. 9(Suppl 1): 48–52. [DOI] [PubMed] [Google Scholar]
- 12.Chess D. J., Stanley W. C. 2008. Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc. Res. 79: 269–278. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert P. A., Khokhar S. 2008. Changing dietary habits of ethnic groups in Europe and implications for health. Nutr. Rev. 66: 203–215. [DOI] [PubMed] [Google Scholar]
- 14.Gottrand F. 2008. Long-chain polyunsaturated fatty acids influence the immune system of infants. J. Nutr. 138: 1807S–1812S. [DOI] [PubMed] [Google Scholar]
- 15.Crawford M. A., Costeloe K., Ghebremeskel K., Phylactos A., Skirvin L., Stacey F. 1997. Are deficits of arachidonic and docosahexaenoic acids responsible for the neural and vascular complications of preterm babies? Am. J. Clin. Nutr. 66: 1032S–1041S. [DOI] [PubMed] [Google Scholar]
- 16.Nettleton J. A., Katz R. 2005. n-3 long-chain polyunsaturated fatty acids in type 2 diabetes: a review. J. Am. Diet. Assoc. 105: 428–440. [DOI] [PubMed] [Google Scholar]
- 17.Simopoulos A. P. 2002. Omega-3 fatty acids in inflammation and autoimmune diseases. J. Am. Coll. Nutr. 21: 495–505. [DOI] [PubMed] [Google Scholar]
- 18.Simopoulos A. P. 2008. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. (Maywood). 233: 674–688. [DOI] [PubMed] [Google Scholar]
- 19.Marquardt A., Stohr H., White K., Weber B. H. 2000. cDNA cloning, genomic structure, and chromosomal localization of three members of the human fatty acid desaturase family. Genomics. 66: 175–183. [DOI] [PubMed] [Google Scholar]
- 20.Xie L., Innis S. M. 2008. Genetic variants of the FADS1 FADS2 gene cluster are associated with altered (n-6) and (n-3) essential fatty acids in plasma and erythrocyte phospholipids in women during pregnancy and in breast milk during lactation. J. Nutr. 138: 2222–2228. [DOI] [PubMed] [Google Scholar]
- 21.Schaeffer L., Gohlke H., Muller M., Heid I. M., Palmer L. J., Kompauer I., Demmelmair H., Illig T., Koletzko B., Heinrich J. 2006. Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum. Mol. Genet. 15: 1745–1756. [DOI] [PubMed] [Google Scholar]
- 22.Rzehak P., Heinrich J., Klopp N., Schaeffer L., Hoff S., Wolfram G., Illig T., Linseisen J. 2009. Evidence for an association between genetic variants of the fatty acid desaturase 1 fatty acid desaturase 2 (FADS1 FADS2) gene cluster and the fatty acid composition of erythrocyte membranes. Br. J. Nutr. 101: 20–26. [DOI] [PubMed] [Google Scholar]
- 23.Plaisier C. L., Horvath S., Huertas-Vazquez A., Cruz-Bautista I., Herrera M. F., Tusie-Luna T., Aguilar-Salinas C., Pajukanta P. 2009. A systems genetics approach implicates USF1, FADS3, and other causal candidate genes for familial combined hyperlipidemia. PLoS Genet. 5: e1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinelli N., Girelli D., Malerba G., Guarini P., Illig T., Trabetti E., Sandri M., Friso S., Pizzolo F., Schaeffer L., et al. 2008. FADS genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am. J. Clin. Nutr. 88: 941–949. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka T., Shen J., Abecasis G. R., Kisialiou A., Ordovas J. M., Guralnik J. M., Singleton A., Bandinelli S., Cherubini A., Arnett D., et al. 2009. Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI Study. PLoS Genet. 5: e1000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathias R. A., Bickel C. A., Beaty T. H., Petersen G. M., Hetmanski J. B., Liang K. Y., Barnes K. C. 2000. A study of contemporary levels and temporal trends in inbreeding in the Tangier Island, Virginia, population using pedigree data and isonymy. Am. J. Phys. Anthropol. 112: 29–38. [DOI] [PubMed] [Google Scholar]
- 27.Mathias R. A., Beaty T. H., Bailey-Wilson J. E., Bickel C., Stockton M. L., Barnes K. C. 2005. Inheritance of total serum IgE in the isolated Tangier Island population from Virginia: complexities associated with genealogical depth of pedigrees in segregation analyses. Hum. Hered. 59: 228–238. [DOI] [PubMed] [Google Scholar]
- 28.Metcalfe L. D., Schmitz A. A., Pelka J. R. 1966. Rapid preparation of fatty acid esters from lipids for gas chromatographic analysis. Anal. Chem. 38: 514–515. [Google Scholar]
- 29.Weaver K. L., Ivester P., Seeds M., Case L. D., Arm J. P., Chilton F. H. 2009. Effect of dietary fatty acids on inflammatory gene expression in healthy humans. J. Biol. Chem. 284: 15400–15407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lattka E., Illig T., Koletzko B., Heinrich J. 2010. Genetic variants of the FADS1 FADS2 gene cluster as related to essential fatty acid metabolism. Curr. Opin. Lipidol. 21: 64–69. [DOI] [PubMed] [Google Scholar]
- 31.Gabriel S. B., Schaffner S. F., Nguyen H., Moore J. M., Roy J., Blumenstiel B., Higgins J., DeFelice M., Lochner A., Faggart M., et al. 2002. The structure of haplotype blocks in the human genome. Science. 296: 2225–2229. [DOI] [PubMed] [Google Scholar]
- 32.Barrett J. C., Fry B., Maller J., Daly M. J. 2005. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 21: 263–265. [DOI] [PubMed] [Google Scholar]
- 33.Falconer D. S., Mackay T. F. C. 1996. Introduction to Quantitative Genetics. 4th edition Pearson Education Limited, Essex, England. [Google Scholar]
- 34.Cesari M., Penninx B. W., Newman A. B., Kritchevsky S. B., Nicklas B. J., Sutton-Tyrrell K., Rubin S. M., Ding J., Simonsick E. M., Harris T. B., et al. 2003. Inflammatory markers and onset of cardiovascular events: results from the Health ABC study. Circulation. 108: 2317–2322. [DOI] [PubMed] [Google Scholar]
- 35.Chilton F. H., Rudel L. L., Parks J. S., Arm J. P., Seeds M. C. 2008. Mechanisms by which botanical lipids affect inflammatory disorders. Am. J. Clin. Nutr. 87: 498S–503S. [DOI] [PubMed] [Google Scholar]
- 36.Poudel-Tandukar K., Nanri A., Matsushita Y., Sasaki S., Ohta M., Sato M., Mizoue T. 2009. Dietary intakes of alpha-linolenic and linoleic acids are inversely associated with serum C-reactive protein levels among Japanese men. Nutr. Res. 29: 363–370. [DOI] [PubMed] [Google Scholar]
- 37.Meyer B. J., Mann N. J., Lewis J. L., Milligan G. C., Sinclair A. J., Howe P. R. 2003. Dietary intakes and food sources of omega-6 and omega-3 polyunsaturated fatty acids. Lipids. 38: 391–398. [DOI] [PubMed] [Google Scholar]
- 38.Astorg P., Arnault N., Czernichow S., Noisette N., Galan P., Hercberg S. 2004. Dietary intakes and food sources of n-6 and n-3 PUFA in French adult men and women. Lipids. 39: 527–535. [DOI] [PubMed] [Google Scholar]
- 39.Varilo T., Peltonen L. 2004. Isolates and their potential use in complex gene mapping efforts. Curr. Opin. Genet. Dev. 14: 316–323. [DOI] [PubMed] [Google Scholar]
- 40.Guillou H., Zadravec D., Martin P. G., Jacobsson A. 2010. The key roles of elongases and desaturases in mammalian fatty acid metabolism: insights from transgenic mice. Prog Lipid Res. 49: 186–199. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura M. T., Nara T. Y. 2003. Essential fatty acid synthesis and its regulation in mammals. Prostaglandins Leukot. Essent. Fatty Acids. 68: 145–150. [DOI] [PubMed] [Google Scholar]
- 42.Cho H. P., Nakamura M. T., Clarke S. D. 1999. Cloning, expression, and nutritional regulation of the mammalian Delta-6 desaturase. J. Biol. Chem. 274: 471–477. [DOI] [PubMed] [Google Scholar]
- 43.Christiansen E. N., Lund J. S., Rortveit T., Rustan A. C. 1991. Effect of dietary n-3 and n-6 fatty acids on fatty acid desaturation in rat liver. Biochim. Biophys. Acta. 1082: 57–62. [DOI] [PubMed] [Google Scholar]
- 44.Brenna J. T., Salem N., Jr., Sinclair A. J., Cunnane S. C. 2009. alpha-Linolenic acid supplementation and conversion to n-3 long-chain polyunsaturated fatty acids in humans. Prostaglandins Leukot. Essent. Fatty Acids. 80: 85–91. [DOI] [PubMed] [Google Scholar]
- 45.Lattka E., Eggers S., Moeller G., Heim K., Weber M., Mehta D., Prokisch H., Illig T., Adamski J. 2010. A common FADS2 promoter polymorphism increases promoter activity and facilitates binding of transcription factor ELK1. J. Lipid Res. 51: 182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.