

Abstract
It is now established that CD1 molecules present lipid antigens to T cells, although it is not clear how the exchange of lipids between membrane compartments and the CD1 binding groove is assisted. We report that mice deficient in prosaposin, the precursor to a family of endosomal lipid transfer proteins (LTP), exhibit specific defects in CD1d-mediated antigen presentation and lack Vα14 NKT cells. In vitro, saposins extracted monomeric lipids from membranes and from CD1, thereby promoting the loading as well as the editing of lipids on CD1. Transient complexes between CD1, lipid, and LTP suggested a “tug-of-war” model in which lipid exchange between CD1 and LTP is on the basis of their respective affinities for lipids. LTPs constitute a previously unknown link between lipid metabolism and immunity and are likely to exert a profound influence on the repertoire of self, tumor, and microbial lipid antigens.
CD1 molecules have evolved a unique hydrophobic binding groove that binds lipid antigens in both the secretory and endosomal compartments for presentation to T lymphocytes (1). In mice, the main population of CD1-restricted T cells, called Vα14 NKT cells, express a semi-invariant Vα14-Jα18/Vβ8 T cell receptor (TCR). These cells exhibit reactivity against CD1d in combination with endogenous ligands (2) that can be mimicked by α-galactosylceramide (αGC) (3). This population is conserved among mammalian species and regulates immune responses (4, 5). Like human CD1b-restricted T cells specific for mycobacterial glycolipids (6), Vα14 NKT cells are dependent on endosomal trafficking of CD1d for natural antigen recognition (7–10). Other endogenous or exogenous antigens do not require endosomal trafficking, however, suggesting that loading may be achieved in distinct cellular compartments depending on the nature of the antigen (9, 11, 12). CD1 endosomal trafficking is tightly controlled by cytoplasmic tail–encoded tyrosine-containing motifs binding adaptor protein 2 and 3 (AP-2 and AP-3) complexes, as well as by association with the invariant chain (Ii) or Ii/major histocompatibility (MHC) class II complexes (13–16).
Because lipids are integral membrane components that might require lipid transfer proteins (LTP) (17) for extraction, we investigated whether various families of LTP might assist antigen presentation. We focused on the Vα14 NKT cell endosomal-dependent system to test the possible involvement of LTPs localized in the endocytic pathway, such as saposins (18) and GM2 activator protein (19). Saposins A, B, C, and D are the four members of a family of small dimeric proteins produced by endosomal proteolytic cleavage of prosaposin (18) that regulate various aspects of lipid metabolism. GM2 activator protein is structurally different from saposins but extracts lipids with the use of similar conformational changes (20). CD1d-αGC tetramers stained Vα14 NKT cells with a CD44high memory phenotype in the thymus and peripheral tissues of wild-type mice, but in SAP−/− littermates (21), these cells were undetectable as in CD1d−/− mice (Fig. 1A). The level and pattern of CD1d expression, however, were conserved in SAP−/− mice (Fig. 1B) (22). SAP−/− mice develop neurological symptoms starting between days 25 and 30 and show retarded growth. Although the thymuses and spleens of the adult animals were relatively small, percentages of naïve (CD44low) and memory (CD44high) CD4+ and CD8+ T cells, B cells, natural killer (NK) cells, and γδ T cells were normal (Fig. 1C) (22). In fetal thymic organ culture, SAP−/− thymic lobes had the same number of cells as those of the wild type but selectively lacked Vα14 NKT cells (Fig. 1D). These findings demonstrate a selective defect in the development of Vα14 NKT cells in the absence of prosaposin.
Fig. 1.

SAP−/− mice selectively lack Vα14 NKT cells. (A) Vα14 NKT cells, identified as positive for CD1d-αGC tetramer (y axis) and negative for empty CD1d tetramer and CD8 (x axis) by flow cytometry of thymocytes, splenocytes, and liver lymphocytes, are indicated in the boxed areas with their corresponding % values. Data representative of four individual adult FVB.SAP−/− mice compared to wild-type (WT) littermates and B6.CD1−/−. (B) Histograms of CD1d surface thymocyte and splenocyte expression in WT and mutant mice. (C) T cell subsets identified by CD4 and CD8 or CD4 and CD44 in the indicated tissues, with corresponding % values as indicated. (D) Vα14 NKT cells in fetal thymuses after 14 days of culture. Data representative of two individual B6.SAP−/− fetal thymuses and their WT littermate controls.
Many NKT cell hybridomas can be activated in vitro to release interleukin-2 (IL-2) by coculture with fresh CD1d-expressing cells, such as thymocytes, which are thought to be the specific antigen-presenting cells (APCs) that positively select Vα14 NKT cells (23). This feature is attributed to the recognition of natural lipid ligands loaded on CD1d in these cells. Unlike wild-type thymocytes, those from SAP−/− animals failed to stimulate the self-reactive Vα14 NKT cell hybridoma DN32.D3. In contrast, the non-Vα14 CD1d-restricted hybridomas TCB11 and 1C8DC1 were able to recognize the SAP−/− thymocytes (Fig. 2A) even in serum-free culture conditions (22). These results, which are consistent with the endosomal trafficking requirement for CD1d to stimulate Vα14 NKT cells (9), suggest that endosomal saposins are selectively involved in the presentation of the Vα14 NKT cell ligands by CD1d.
Fig. 2.

Antigen presentation by SAP−/− APCs. (A) Failure to stimulate CD1d-autoreactive Vα14 NKT hybridomas. Fresh thymocytes from SAP−/− and WT FVB littermates or CD1d−/− mice were used to stimulate CD1d-autoreactive NKT hybridomas expressing a Vα14 invariant TCR α chain (DN32.D3) or other Vα (TCB11 and 1C8DC1). Data representative of four separate sets of mice. (B) Presentation of exogenous glycolipid antigens. Thymocytes, DCs, and splenocytes from WT, SAP−/−, and CD1d−/− mice were pulsed with various concentrations of αGC or Gal-α-1,2 αGal-Cer, as indicated, before coculture with the Vα14 DN32.D3 NKT hybridoma cells.
We examined the presentation of αGC and Galα1,2αGC, which are partially and completely dependent on endosomal functions, respectively (10, 24). Although stimulation of DN32.D3 by SAP−/− thymocytes pulsed with αGC was partially decreased, stimulation by dendritic cells (DCs) was normal (Fig. 2B). In contrast, presentation of Galα1,2αGC, which absolutely requires endosomal processing into αGC, was abolished in all cell types (Fig. 2B) (22). These results demonstrate requirements for saposins in the presentation of exogenous glycolipids.
We next performed two sets of control experiments to exclude the possibility that the lipid presentation defects might be the result of broad nonspecific disruptions of endosomal function in the absence of SAP. SAP−/− APCs were comparable to those of wild type for presentation of intact ovalbumin, hen egg lysozyme, keyhole limpet hemocyanin, and collagen II to various I-Aq– or I-Ab–restricted T cell hybridomas, which require complex processing within an intact endosomallysosomal compartment (25) (fig. S1). Consistent with the preservation of MHC class II and antigen-presenting functions, the Lamp-1+ compartment of SAP−/− cells appeared morphologically normal (Fig. 3), and lysosomal acidification, as assayed by Lysotracker (Materials and Methods) staining, was comparable to that seen in wild type (fig. S1). We also found that mutant mice deficient in α-galactosidase A (26), galactosylceramidase (27), or mucopolysaccharidase (28) had conserved thymic NKT cells and normally presented natural CD1d ligands to NKT cells (fig. S2), indicating that accumulation of lipids or mucopolysaccharides as seen in various lysosomal storage diseases is not accompanied by nonspecific alterations of CD1d-mediated antigen presentation.
Fig. 3.
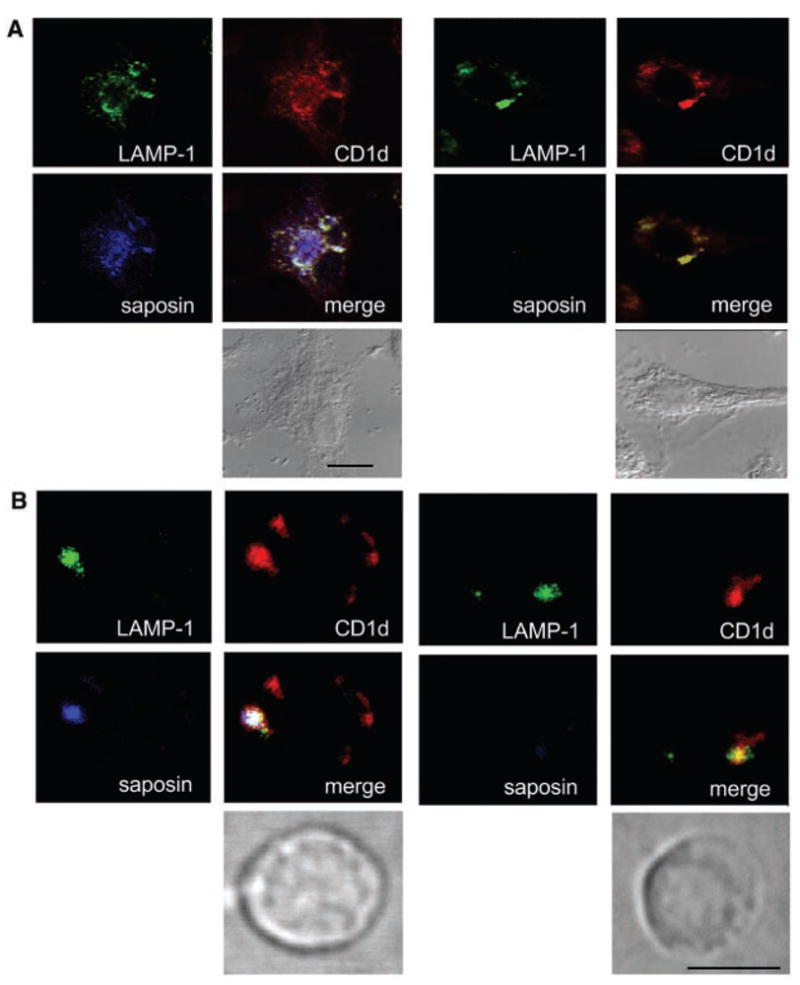
Colocalization of CD1d, saposin, and Lamp1. (A) Tumor necrosis factor–α–activated DC stained with antibodies against CD1d, saposin, and Lamp1, as indicated. Direct transmission image is shown underneath to delineate the outer cell membrane. Left, WT; right, SAP−/−. (B) Same experiment as in (A) with fresh thymocytes. Scale bars indicate 10 μm.
Both CD1d and saposins have been reported to preferentially localize in late endosomal/lysosomal compartments (15, 29). We found that in DCs, as well as in thymocytes, saposin and CD1d colocalized in the Lamp-1+ late endosome/lysosome (Fig. 3), These results demonstrate colocalization of CD1d and saposin in the subcellular compartment where glycolipid processing and loading occur.
Because no evidence exists that CD1d itself can function as a LTP, we directly tested whether saposins might be responsible for the transfer of lipids onto CD1d. To examine this, we devised a cell-free system in which lipid exchange between, on one hand, purified complexes of CD1d bound to the acidic trisialoganglioside GT1b (CD1-GT) and, on the other hand, liposomes made of phosphatidylserine (PS) could be measured by native isoelectrofocusing (IEF) (30). In the absence of LTP, PS could not be loaded. However, each of the four saposins readily promoted exchange (Fig. 4A). Incubation of purified CD1-GT with individual saposins alone did not promote the removal of GT1b, suggesting that, in this particular experimental condition, saposins could extract lipids from membranes but not from CD1d. The loading of PS was verified by repurifying CD1d and analyzing the lipid extract by mass spectrometry (22). In the case of PS, dose-effect studies and kinetic analysis determined that saposin A was the most effective transfer protein. By comparison, C displayed intermediate activity, and B and D were relatively inefficient (fig. S3). Importantly, the titration experiments revealed that saposins were not enzymatic in their activity, because they needed to be present at an equimolar ratio with CD1d to carry their transfer function. This property clearly differentiates saposins, in their lipid-editing function, from the peptide editor for MHC class II, HLA-DM (31). It is consistent with the nonenzymatic mechanism of saposin function in the hydrolysis of cerebrosides (32). The fine specificity of saposins was further examined by comparing the transfer of sulfatide (SFT) onto CD1d (fig. S3). In this case, saposins A and C were the most efficient, saposin D had no activity, and B was intermediate. All saposins were most active at 37°C and highly pH-dependent, with maximal activity at pH values below 5.5 (fig. S3). However, saposin A and C, but not B and D, had substantial function at pH = 6.0, suggesting possible different functions of saposins in endosomal subcompartments and during the maturation process of dendritic cells.
Fig. 4.

LTP-mediated lipid transfer to or extraction from CD1d. (A) Saposins transfer lipids from liposomes onto CD1d molecules. CD1-GT (2 μM) incubated at pH = 5.0 with 1 mM PS (prepared as 400 nm liposomes) and 15 μM saposin. Lipid loading is visualized by native IEF. The migration of the CD1d band toward the cathode indicates loading with PS. (B) Association of saposin A with CD1d. CD1-GT, CD1-SFT, and CD1-αGC complexes were purified to homogeneity by anion exchange, incubated at equimolar ratio with saposin A (4 μM) for 1 hour at room temperature, and analyzed by native IEF. After transfer, blots were hybridized with the CD1 antibody 20H2 and a rabbit anti-saposin A antiserum. Lane 1, saposin alone; lane 2, saposin plus CD1-GT; lane 3, saposin plus CD1d-αGC; and lane 4, saposin plus CD1-SFT. The presence of CD1-saposin complexes is marked by the appearance of bands stained by both anti–saposin A and anti-CD1d (arrows). Controls including saposin A alone (lane 5), saposin plus GT1b (lane 6), saposin plus αGC (lane 7), saposin plus sulfatide (lane 8), and saposin plus H-2Kb (lane 9) were hybridized with the anti-saposin serum. Note that the anti-saposin serum reacts to different levels when incubated with various lipids. (C) GM2A unloads glycolipids bound to CD1d. Purified CD1-GT complexes (2.5 μM) were incubated with increasing concentrations of GM2A. Coomassie Blue staining reveals the appearance of a neutral band stained by anti-CD1 and corresponding to the migration of empty CD1d. GM2A (not seen on Coomassie Blue staining) appears in the Western blot by itself (top of gel) and in an intermediate band that corresponds to the GM2A-GT1b complex. (Far right) Anti-GM2A Western blot of GM2A by itself (lane 1) and GM2A incubated with 2.5 μM GT1b (lane 2). In the composite overlay, CD1 species are in green, whereas GM2A species are in blue.
To better understand the nature of molecular interactions leading to lipid exchange, we attempted to visualize a potential physical interaction between saposins and CD1d. In order to do so, anion exchange–purified CD1-GT, CD1-SFT, and CD1-αGC complexes were incubated with an equimolar amount of saposin A at room temperature for 1 hour (Fig. 4B). Saposin A ran to the anodic front because of its low pKa (4.9) and stained very poorly with Coomassie Blue on native IEF. When CD1-GT or CD1-SFT complexes were incubated with saposin A, a band comigrating with CD1d was detected by a serum against saposin A (anti-saposin) as well as by an antibody against CD1d (anti-CD1) in Western blot (Fig. 4B). No complex formation could be observed between saposin A and classical MHC class I molecules (H-2Kb) or albumin. The absence of complex formation with CD1-αGC strongly suggested that the interaction was highly lipid head group– dependent, and it also correlated with the inability of saposin A to transfer αGC onto CD1d (see below). We note that the association between CD1-GT and saposin A was not accompanied by any notice-able unloading of GT1b from CD1d, which would have appeared as neutral bands of empty CD1d.
We considered the possibility that other LTPs might reveal that capacity to extract lipids bound to CD1 molecules. Indeed, we found that the endosomal GM2 activator protein (GM2A) (33) could remove the GT1b from CD1-GT. Purified CD1-GT complexes were incubated in the absence of exogenous lipids with increasing concentrations of recombinant GM2A (Fig. 4C). The appearance of a lower band corresponding to neutral, empty CD1d demonstrated the loss of GT1b on both the Coomassie Blue–stained gel and the anti-CD1 Western blot. Concomitantly, GM2A-GT complexes were revealed by a corresponding change in IEF on the anti-GM2A Western blot. Importantly, in contrast with CD1–GT–saposin A, we did not detect CD1-GT-GM2A trimolecular complexes. These findings support an affinity-based “tug-of-war” model of lipid exchange between CD1d and LTP. Transient complexes would not be detected when unloading is rapid, whereas slow or inefficient unloading would result in more stable complexes.
None of the saposins could readily transfer αGC or Galα1,2αGC onto CD1d in vitro (22), whereas GM2A efficiently assisted the loading of αGC onto CD1d (fig. S4), explaining why presentation of αGC by SAP−/− cells was subnormal (Fig. 2). Neither saposins nor GM2A assisted the loading of Galα1,2αGC (22). However, because Galα1,2αGC needs processing into αGC by α-glycosidase A, a saposin-dependent enzyme (34), defective presentation of Galα1,2αGC by SAP−/− cells (Fig. 2) is likely a result of defective processing. Together, our findings suggest that defective presentation of glycolipid antigens by SAP−/− cells may result from single or combined defects in lipid processing, loading, and unloading. Further, because of the individual specificities of LTPs, it is likely that different LTPs cooperate to generate and edit the glycolipid repertoire presented by CD1 molecules. Also, we can assume that LTP dependency for loading will vary for various lipid families.
In conclusion, it appears that endosomal LTPs, such as the saposin family and GM2A, fill a major gap between CD1-mediated antigen presentation and lipid metabolism. Saposins are involved in the generation and/or loading of the natural thymic antigens that determine selection of Vα14 NKT cells. Our biochemical studies suggest an affinity-based tug-of-war model of lipid exchange between LTPs and CD1 molecules (Fig. 5). LTPs extract lipids from membranes and CD1d by interacting specifically with their polar heads. On the basis of their relative affinities for the polar head and the fatty acid portion of a given lipid, respectively, LTP and CD1d form transient complexes that lead to lipid exchange and determine the repertoire of glycolipids presented by CD1d molecules.
Fig. 5.
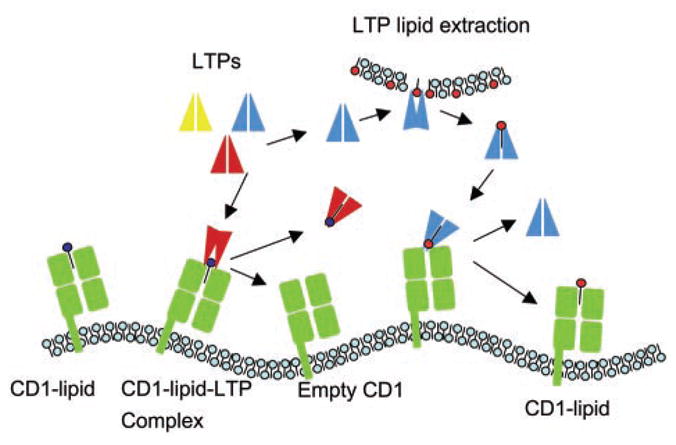
Schematic representation of the hypothetical mechanism of lipid editing by LTPs. LTPs are presented in two conformations reflecting the structural changes associated with lipid extraction (20).
In vivo, such lipid-editing function is likely to have an impact on CD1-restricted T cells similar to that of HLA-DM editing of MHC class II– bound peptides on the selection and expansion of CD4− T cells. Studies of the differential distribution and lipid specificity of individual LTPs, which allow for considerable flexibility in regulating the presentation of lipid antigens, will be critical to improve the rational design of lipid vaccines and adjuvants.
Acknowledgments
We thank R. Brown, E. Berger, K. Sandhoff, and B. Jabri for helpful suggestions and E. Rosloniec and A. Rudensky for I-Ab– and I-Aq–restricted T cell hybridomas. Supported by NIH grants (PO1 AI53725 to P.S., A.B., and L.T. and AI50867 and AI38339 to A.B.) and the Cancer Research Institute (D.Z. and Y.S.).
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/1092009/DC1
Materials and Methods
Figs. S1 to S4
References and Notes
- 1.Moody DB, Porcelli SA. Nat Rev Immunol. 2003;3:11. doi: 10.1038/nri979. [DOI] [PubMed] [Google Scholar]
- 2.Bendelac A, et al. Science. 1995;268:863. doi: 10.1126/science.7538697. [DOI] [PubMed] [Google Scholar]
- 3.Kawano T, et al. Science. 1997;278:1626. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 4.Godfrey DI, Hammond KJ, Poulton LD, Smyth MJ, Baxter AG. Immunol Today. 2000;21:573. doi: 10.1016/s0167-5699(00)01735-7. [DOI] [PubMed] [Google Scholar]
- 5.Bendelac A, Bonneville M, Kearney JF. Nat Rev Immunol. 2001;1:177. doi: 10.1038/35105052. [DOI] [PubMed] [Google Scholar]
- 6.Moody DB, et al. J Exp Med. 2000;192:965. doi: 10.1084/jem.192.7.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugita M, et al. Science. 1996;273:349. doi: 10.1126/science.273.5273.349. [DOI] [PubMed] [Google Scholar]
- 8.Jackman RM, et al. Immunity. 1998;8:341. doi: 10.1016/s1074-7613(00)80539-7. [DOI] [PubMed] [Google Scholar]
- 9.Chiu YH, et al. J Exp Med. 1999;189:103. doi: 10.1084/jem.189.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiu YH, et al. Nat Immunol. 2002;3:55. doi: 10.1038/ni740. [DOI] [PubMed] [Google Scholar]
- 11.Shamshiev A, et al. Immunity. 2000;13:255. doi: 10.1016/s1074-7613(00)00025-x. [DOI] [PubMed] [Google Scholar]
- 12.Moody DB, et al. Nat Immunol. 2002;3:435. doi: 10.1038/ni780. [DOI] [PubMed] [Google Scholar]
- 13.Sugita M, et al. Immunity. 2002;16:697. doi: 10.1016/s1074-7613(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 14.Briken V, Jackman RM, Dasgupta S, Hoening S, Porcelli SA. EMBO J. 2002;21:825. doi: 10.1093/emboj/21.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jayawardena-Wolf J, Benlagha K, Chiu YH, Mehr R, Bendelac A. Immunity. 2001;15:897. doi: 10.1016/s1074-7613(01)00240-0. [DOI] [PubMed] [Google Scholar]
- 16.Kang SJ, Cresswell P. EMBO J. 2002;21:1650. doi: 10.1093/emboj/21.7.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rueckert DG, Schmidt K. Chem Phys Lipids. 1990;56:1. doi: 10.1016/0009-3084(90)90083-4. [DOI] [PubMed] [Google Scholar]
- 18.Sandhoff K, Kolter T, Van Echten-Deckert G. Ann NY Acad Sci. 1998;845:139. doi: 10.1111/j.1749-6632.1998.tb09667.x. [DOI] [PubMed] [Google Scholar]
- 19.Wright CS, Zhao Q, Rastinejad F. J Mol Biol. 2003;331:951. doi: 10.1016/s0022-2836(03)00794-0. [DOI] [PubMed] [Google Scholar]
- 20.Ahn VE, Faull KF, Whitelegge JP, Fluharty AL, Prive GG. Proc Natl Acad Sci USA. 2003;100:38. doi: 10.1073/pnas.0136947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujita N, et al. Hum Mol Genet. 1996;5:711. doi: 10.1093/hmg/5.6.711. [DOI] [PubMed] [Google Scholar]
- 22.Zhou D, et al. unpublished observations. [Google Scholar]
- 23.Bendelac A, Rivera MN, Park SH, Roark JH. Annu Rev Immunol. 1997;15:535. doi: 10.1146/annurev.immunol.15.1.535. [DOI] [PubMed] [Google Scholar]
- 24.Prigozy TI, et al. Science. 2001;291:664. doi: 10.1126/science.291.5504.664. [DOI] [PubMed] [Google Scholar]
- 25.Harding CV, Collins DS, Slot JW, Geuze HJ, Unanue ER. Cell. 1991;64:393. doi: 10.1016/0092-8674(91)90647-h. [DOI] [PubMed] [Google Scholar]
- 26.Ohshima T, et al. Proc Natl Acad Sci USA. 1997;94:2540. doi: 10.1073/pnas.94.6.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakai N, et al. J Neurochem. 1996;66:1118. doi: 10.1046/j.1471-4159.1996.66031118.x. [DOI] [PubMed] [Google Scholar]
- 28.Vogler C, et al. Pediatr Res. 2001;49:342. doi: 10.1203/00006450-200103000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Q, Morales CR. J Biol Chem. 2000;275:24829. doi: 10.1074/jbc.M003497200. [DOI] [PubMed] [Google Scholar]
- 30.Cantu C, III, Benlagha K, Savage PB, Bendelac A, Teyton L. J Immunol. 2003;170:4673. doi: 10.4049/jimmunol.170.9.4673. [DOI] [PubMed] [Google Scholar]
- 31.Denzin LK, Cresswell P. Cell. 1995;82:155. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 32.Qi X, Leonova T, Grabowski GA. J Biol Chem. 1994;269:16746. [PubMed] [Google Scholar]
- 33.Wu YY, et al. J Biol Chem. 1994;269:16276. [PubMed] [Google Scholar]
- 34.Kase R, et al. FEBS Lett. 1996;393:74. doi: 10.1016/0014-5793(96)00863-0. [DOI] [PubMed] [Google Scholar]