


Abstract
DNA dependent protein kinase (DNA-PK) plays a central role in the non-homologous end-joining pathway of DNA double strand break repair. Its catalytic subunit (DNA-PKCS) functions as a serine/threonine protein kinase. We show that DNA-PK forms a stable complex at DNA termini that blocks the action of exonucleases and ligases. The DNA termini become accessible after autophosphorylation of DNA-PKCS, which we demonstrate to require synapsis of DNA ends. Interestingly, the presence of DNA-PK prevents ligation of the two synapsed termini, but allows ligation to another DNA molecule. This alteration of the ligation route is independent of the type of ligase that we used, indicating that the intrinsic architecture of the DNA-PK complex itself is not able to support ligation of the synapsed DNA termini. We present a working model in which DNA-PK creates a stable molecular bridge between two DNA ends that is remodeled after DNA-PK autophosphorylation in such a way that the extreme termini become accessible without disrupting synapsis. We infer that joining of synapsed DNA termini would require an additional protein factor.
INTRODUCTION
DNA double strand breaks (DSBs), which can be introduced by the action of ionizing radiation or mutagenic agents, are repaired either by homologous recombination or non- homologous end-joining (NHEJ). While homologous recombination is considered to be the main DSB repair pathway in lower eukaryotic cells, DSBs in higher eukaryotes, including mammals, appear to be repaired mainly via the NHEJ pathway (1). Defects in end-joining generally result in increased sensitivity to ionizing radiation, as well as immunodeficiency, caused by failure to complete V(D)J recombination (2,3).
In contrast to the homologous recombination repair pathway, NHEJ does not require a DNA template. DNA ends are directly ligated, although processing of the DNA ends prior to ligation is required for a subset of DSBs. For instance, missing 5′ phosphates necessitate addition of a novel phosphate group by polynucleotide kinase (4). Furthermore, the Artemis protein, in complex with the DNA dependent protein kinase (DNA-PK), is probably required for processing of non-matching DNA ends and the opening of DNA hairpin structures, which are formed during V(D)J recombination (5).
The ligase IV/XRCC4 complex and the DNA-PK are the core components of the NHEJ pathway (2), although the involvement of additional factors has been shown by both genetic and biochemical studies (6,7). DNA-PK consists of the Ku70/80 heterodimer and a 470 kDa catalytic subunit (DNA-PKCS), which functions as a serine/threonine protein kinase.
Ku70/80 binds with high affinity to DNA ends, which suggests that it has a role in damage recognition (8). Its recently elucidated three dimensional structure shows that the heterodimer forms a hollow ring around the DNA helix (9), which allows migration along the axis of the DNA molecule to internal positions and even transfer between DNA ends (10,11). In addition, the involvement of Ku70/80 containing protein complexes in end-to-end association of linear DNA molecules and formation of DNA loops has been demonstrated by scanning force microscopy (12–14). Although these structures were observed to be a minority of the protein–DNA complexes, they suggest a role for Ku70/80 or the complete DNA-PK holo-enzyme in mediating the formation of DNA synapsis. In addition, the Ku heterodimer also attracts DNA-PKCS towards the DNA ends and stimulates its kinase activity (8), which is required for efficient DSB rejoining (15).
Several studies have demonstrated a role for DNA-PK in recruitment of the ligase IV/XRCC4 complex towards DNA ends (16–19), which is required for ligation of the DSB by ligase IV. The finding that XRCC4 is a target for the DNA-PKCS protein kinase (20) further supports a model in which both the DNA-PK holo-enzyme and ligase IV/XRCC4 are present at the DNA ends, and have a cooperative role in rejoining of the break.
In addition to a function in attracting ligase IV/XRCC4 towards the DNA break, DNA-PK has recently been found to mediate association of two DNA molecules. This synapsis activated the DNA-PKCS protein kinase (21). Many proteins have been found to be in vitro targets for the DNA-PKCS kinase, although the biological relevance is not clear in most cases. Some interesting candidate target proteins are the tumor suppressor protein p53 and the recently identified Artemis protein. The regulation of Artemis nuclease activity may be a good explanation for the observation that DNA-PKCS deficiency leads to accumulation of DNA hairpins during V(D)J recombination (22).
Interestingly, DNA-PKCS can also phosphorylate itself (23,24), which can lead to dissociation of the catalytic subunit from the DNA and inactivation of its kinase activity. DNA-PKCS autophosphorylation takes place in vivo and mutants of DNA-PK that miss one or two of the main autophosphorylation sites are less well able to complement the radiosensitive phenotype of a DNA-PKCS deficient cell line than the wild type protein (25–28).
In this paper, we present evidence that DNA-PKCS requires synapsis of two DNA ends for autophosphorylation. Interestingly, remodeling of DNA-PK complexes bound to DNA ends and subsequent ligation require conditions that allow for DNA-PKCS autophosphorylation, suggesting that DNA-PK regulates end-joining by restricting accessibility to DNA ends until they are synapsed.
MATERIALS AND METHODS
Protein purification
DNA-PK for general use in all assays was purified from human placenta, according to a protocol adapted from Chan et al. (29). After homogenization, ammonium sulphate precipitation, low-pressure purification on DEAE Sepharose, S Sepharose and a second DEAE Sepharose column according to Chan et al., the extract was loaded directly onto a 1 ml Resource Q FPLC column (Amersham Pharmacia). No double-stranded DNA cellulose column was used, in order to obtain a preparation that contained both the DNA-PKCS and the Ku70/80 heterodimer. DNA-PK was bound to Resource Q in buffer B (20 mM Tris pH 7.5, 100 mM KCl, 0.02% Tween-20, 2 mM DTT) and was eluted with a linear gradient from 100 mM to 1 M KCl. Active fractions were eluted at ∼400 mM KCl in the gradient and contained both the DNA-PKCS and the Ku70/80 heterodimer, as confirmed by Coomassie staining and western blot analysis. The final preparation was dialyzed to 25 mM HEPES pH 7.5, 50 mM KCl, 5% glycerol and 2 mM DTT.
The preparation was estimated to be ∼70% pure, containing 100 ng/µl DNA-PKCS and Ku70/80 in a molar ratio of 1:3. Specific activity of the preparation was determined to be 1400 pmol ATP/min/µg with the SignaTECT DNA dependent Protein Kinase Assay System (Promega), according to the manufacturer’s protocol. Total yield of DNA-PKCS from 450 g of placenta was 100 µg.
Highly pure preparations of DNA-PKCS and Ku70/80 (>90% pure), for use in the ligation assay, were obtained from HeLa cells according to Chan et al. (29).
Also for use in the ligation assay, next to the above mentioned preparation, a second DNA-PK preparation from human placenta was made by replacing the final resource Q column by a mono S purification step, using a similar gradient from 100 mM to 1 M KCl.
High mobility group 1 (Hmg1) protein was expressed as a fusion protein with glutathione-S-transferase (GST) in Escherichia coli, using an expression plasmid containing HMG boxes A and B (amino acid residues 1–123). Escherichia coli containing the expression vector was grown at 37°C to OD600 = 0.5 and expression was induced by addition of 0.5 mM IPTG. After 3 h, cells were harvested by centrifugation, lysed in PBS containing 0.1 mg/ml lysozyme and sonicated. The lysate was cleared by centrifugation for 30 min at 12 000 g. Subsequently, the supernatant was loaded onto a glutathione Sepharose 4B (Sigma-Aldrich) column, washed with 5 column vol of PBS and eluted with 15 mM glutathione in 50 mM Tris–HCl, pH 8. GST–HMG containing fractions were pooled and dialyzed against 25 mM Tris–HCl, pH 8, 150 mM KCl, 2 mM DTT, 10% (v/v) glycerol.
DNA-PKCS autophosphorylation
Different concentrations of DNA-PK were incubated with 0.5 nM DNA substrate (either a 250 or a 1000 bp fragment) in the presence of radiolabeled [γ-32P]ATP (Amersham Pharmacia, final activity 0.4 µCi/µl), 25 mM HEPES–KOH pH 7.5, 80 mM KCl, 10 mM MgCl2, 0.1 mM ATP and 0.1 mg/ml BSA. Reaction mixtures were incubated for 30 min at 37°C and the reaction was terminated by addition of SDS–PAGE loading buffer. Reaction mixtures were separated by 6% SDS–PAGE gel electrophoresis. Incorporation of radiolabeled phosphate into the DNA-PKCS band was determined by phospho-imaging. (Image Quant phosphorimager).
The autophosphorylation assay has been performed at least four times, yielding reproducible results.
In vitro ligation reactions
Ligation reactions were performed in the presence of a 965 bp overall radiolabeled DNA substrate, made by PCR on pDVG137 using primers DAR5 and NEB1224 (sequence details available upon request), yielding a 1.9 kb fragment. This fragment was gel purified and cohesive ends were introduced by SphI digestion, followed by gel purification of the 965 bp fragment. The reaction system contained DNA-PK at a concentration of ∼10 ng/µl. Ligation reactions were initiated by addition of 3 U T4 DNA ligase (Promega). After incubation at 37°C for 30 min, reaction mixtures were treated overnight with 0.5 µg/µl proteinase K in the presence of 0.1% SDS at 50°C. Subsequently, proteins were removed by phenol/chloroform extraction, after which the ligation products were separated by gel electrophoresis in a 1.3% agarose gel in TBE buffer, containing 0.5 µg/ml ethidiumbromide. Reaction products were visualized by phospho-imaging.
When using E.coli DNA ligase, DNA-PK and the DNA substrate were incubated for 30 min at 37°C under the reaction conditions described above, after which ligation buffer (New England Biolabs) and 5 U E.coli DNA ligase (New England Biolabs) were added. The ligation reaction took place at 16°C for 90 min.
When wortmannin was included in the ligation reaction mixtures, it was added as a stock solution in DMSO to a final concentration of 10 µM wortmannin and 5% DMSO in the reaction mixtures.
Exonuclease digestions
Exonuclease digestion reactions were performed under identical buffer conditions as were used for the ligation reactions. DNA-PK (10 ng/µl) was incubated with 0.5 nM radioactively labeled DNA substrate for 30 min at 37°C before exonuclease was added. Nucleases were added at concentrations that were just sufficient for complete digestion of an unprotected DNA substrate.
For Bal31 digestion, CaCl2 was added to a final concentration of 12.5 mM and digestion was performed by incubation for 5 min at room temperature. Each reaction mixture contained 5 × 10–3 U of Bal31 (Life Technologies). The reactions were stopped by addition of 10 mM EGTA, 0.1% SDS and 0.5 ng/µl proteinase K. After incubation at 50°C for >2 h, DNA was purified by phenol/chloroform extraction and ethanol precipitation. The DNA ends were separated from the DNA substrate by restriction endonuclease digestion with either Eco47III or XhoI, which both digest close to either DNA end. Digestion products were separated by polyacrylamide gel electrophoresis on a 12% denaturing gel in TBE buffer and were visualized by phospho-imaging.
Exonuclease V (Amersham Bioscience) was added to the reaction mixtures to a final concentration of 5 × 10–3 U and incubation was performed for 10 min at 37°C. The reactions were stopped by addition of 0.1% SDS. Digestion products were separated by electrophoresis on a 1% agarose gel in TBE buffer and were visualized by phospho-imaging. DNA substrate was a 5′ labeled 1.2 kb fragment with 5′ protruding ends.
Footprinting experiments
Protein/DNA complexes were assembled under the same reaction conditions as were used in the ligation assays. DNA substrate was a 5′ radioactively labeled 874 bp fragment, made by PCR on pECFP-C1 (Clontech), using DG159 and 5′ radioactively labeled DG156 as primers (sequence details available upon request), followed by gel purification of the 874 bp fragment. After incubation of this DNA substrate with DNA-PK, DNAse I was added to a final concentration of 250 ng/ml and digestion was allowed to proceed for 15 min at room temperature. The digestion reactions were terminated by addition of 10 mM EDTA and 0.1% SDS, and DNA was purified by proteinase K treatment, phenol/chloroform extraction and ethanol precipitation. One volume of loading buffer (95% deionized formamide, 20 mM EDTA) was added and incubation took place for 10 min at 95°C. Electrophoresis was performed on a 12% denaturing polyacrylamide gel in TBE buffer, followed by phospho-imaging.
RESULTS
DNA-PK forms protein–DNA complexes at the DNA ends
We studied the behavior of DNA-PK on DNA ends, especially in situations where these DNA ends are brought in close proximity. We first confirmed the presence of protein–DNA complexes at the DNA termini by testing the susceptibility of a 5′ radioactively labeled DNA substrate for exonuclease digestion in the presence or absence of purified DNA-PK (Fig. 1A). As expected, DNA-PK rendered the DNA substrate unsusceptible to digestion by exonuclease V, whereas the naked substrate was completely degraded (compare lanes 2 and 4), indicating that DNA associated proteins block access of the exonuclease to both ends of the DNA. Similar results were obtained by using Bal31 exonuclease (data not shown), suggesting that DNA associated proteins reside at the DNA termini, sterically inhibiting exonuclease.
Figure 1.
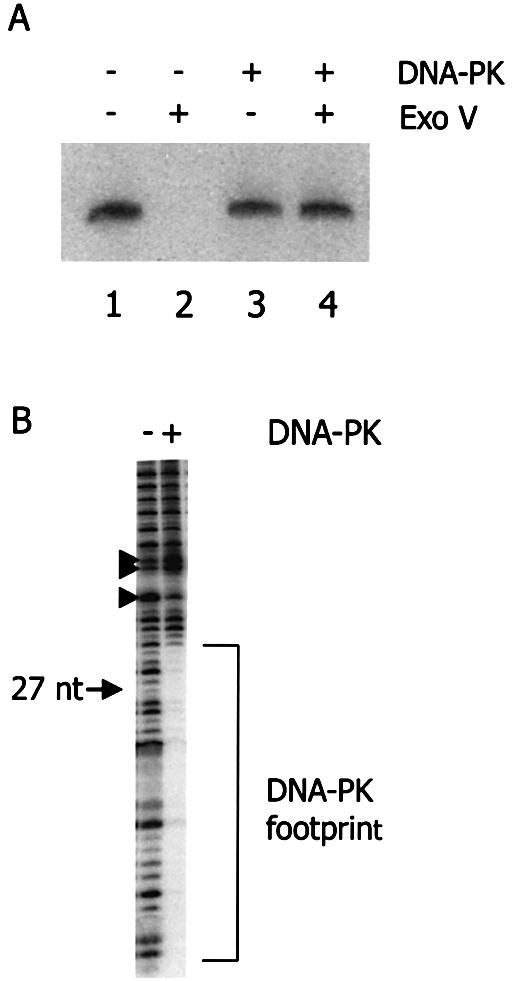
DNA-PK associates with DNA termini. (A) Analysis by nuclease protection assay. A 5′ radioactively labeled 1.2 kb DNA substrate was first incubated with (lanes 3 and 4) or without (lanes 1 and 2) DNA-PK. Subsequently, susceptibility of the DNA–protein complex for degradation by exonuclease V was tested (lanes 2 and 4). (B) DNAse I footprint of DNA-PK, associated with the terminus of a DNA substrate. An 874 bp DNA substrate with one 5′ radioactively labeled strand was first incubated with (lane 2) or without (lane 1) DNA-PK. Subsequently, the reaction mixtures were incubated with DNase I. The position of a 27 nt marker fragment has been indicated on the left.
Protein–DNA complexes were studied in more detail by DNase footprinting (Fig. 1B). This clearly revealed the presence of protein complexes at the DNA termini, which span a specific region of a few dozen nucleotides. By comparing with a 27 nt fragment, obtained by NheI digestion of the DNA substrate, we determined that DNA-PK covers 31–33 nt of the DNA terminus, which is similar to what has been reported before (30). In addition to this area of DNase protection, a clear difference in DNase sensitivity can also be observed at ∼38 and 43 nt from the end (arrowheads in Fig. 1B), suggesting that this part of the DNA may also be in contact with DNA-PK.
Effect of ATP on DNA-PK complexes
Conditions that allow for DNA-PK phosphorylation have been reported to alter the composition of DNA end-bound complexes (23). In order to study the effect of ATP addition on DNA end accessibility, we tested susceptibility of the DNA termini for Bal31 exonuclease in both the presence and the absence of ATP (Fig. 2A). An overall 32P-labeled DNA substrate was incubated with purified DNA-PK, followed by a short Bal31 digestion. Subsequently, the DNA ends were separated from the substrate by digestion with restriction endonuclease, which allowed visualization of Bal31 digestion of the DNA termini.
Figure 2.

ATP renders the DNA termini with complexed DNA-PK more susceptible to nucleolytic degradation. (A) Bal31 exonuclease protection assay. An overall radioactively labeled DNA substrate with 3′ protruding ends was incubated either in the presence (lanes 3–5) or in the absence of DNA-PK (lanes 1 and 2). During this incubation, ATP (lane 4) or AMP-PNP (lane 5) were present as indicated. After a period known to be sufficient for binding and autophosphorylation of DNA-PK, Bal31 was added. Subsequently, the DNA termini were separated from the DNA substrate by digestion with Eco47III. Denaturing gel electrophoresis separated both strands of the terminal fragment, which are visible as two products with different mobility. The difference in intensity between these bands can be explained by a lower content of radioactive adenosine in the upper band. (B) DNase footprint analysis. An 874 bp DNA substrate with one 5′ radioactively labeled strand was incubated either in the absence (lane 1) or in the presence (lanes 2–4) of DNA-PK. ATP (lane 3) and AMP-PNP (lane 4) were present as indicated.
Upon binding of DNA-PK to DNA in the absence of ATP, both strands of the DNA end were protected against digestion by Bal31 (lane 3). However, in the presence of ATP, the DNA ends became more susceptible to exonuclease digestion, as indicated by a 3-fold reduction in the amount of undigested DNA ends (lane 4). Substitution of ATP by AMP-PNP, an ATP analog that cannot be cleaved at the γ-phosphate, did not result in an increased susceptibility of DNA ends for Bal31 degradation (lane 5). We conclude that ATP hydrolysis between the β- and γ-phosphate is required for remodeling of the DNA end-bound protein complexes.
DNA-PKCS autophosphorylation has been shown to disrupt the DNA-PK complex (23). We therefore analyzed the end-bound DNA-PK complexes in more detail. However, footprint analysis did not reveal dissociation of the bulk of end-bound proteins upon addition of ATP (Fig. 2B). However, the footprint appears to be somewhat weakened after ATP addition, indicating that dissociation of a fraction of bound DNA-PK may occur (see also Discussion).
Autophosphorylation of DNA-PKCS requires synapsis of DNA ends
The previous experiments suggested that the kinase activity of DNA-PKCS, which catalyzes hydrolysis of the phosphodiester bond between the β- and the γ-phosphate of ATP, might be involved in remodeling or disruption of DNA-PK complexes at DNA termini. In order to get a better understanding of the events leading to the alterations in these complexes, we investigated DNA-PKCS autophosphorylation under conditions that favor or disfavor synapsis of DNA ends.
First, we quantified the autophosphorylation rate after incubation of increasing amounts of DNA-PK with a fixed amount of linear DNA. This type of assay yielded a linear dose–response curve (Fig. 3A). Subsequently, we set out to determine whether synapsis of DNA ends is necessary for efficient DNA-PKCS autophosphorylation. If this would be the case, one would expect that this reaction takes place at a rate that depends on the ability of the DNA termini to reach each other. We therefore set up an assay to measure the efficiency of DNA-PKCS autophosphorylation using differently sized DNA substrates (Fig. 3B). When a short (250 bp) DNA substrate is used, which is too short to circularize efficiently, synapsis will mainly depend on inter-molecular end-to-end interactions, whereas the use of a longer (1000 bp) substrate allows efficient circularization, which supports synapsis upon both inter- and intra-molecular interactions. Since intra-molecular DNA end interactions will predominate at low DNA concentrations, autophosphorylation rates should be significantly higher when using the 1000 bp substrate, provided that DNA-PK is present in quantities that allow occupation of both ends of the same DNA molecule. On the other hand, if synapsis is not important, autophosphorylation should only depend on the number of DNA ends present in the reaction mixture, irrespective of the possibility to circularize the activating DNA molecule.
Figure 3.

Autophosphorylation of DNA-PKCS takes place at DNA synapses. (A) The incorporation of radiolabeled phosphate into DNA-PKCS under conditions that allow for DNA-PK autophosphorylation, increases in a linear fashion with the amount of added DNA-PK. DNA-PK was added at amounts ranging from 50 ng (lane 1) to 200 ng (lane 4). These amounts were also used in the assay described in (B) and (C). (B and C) Two differently sized DNA substrates, a 250 and a 1000 bp fragment, were incubated at low concentrations with DNA-PK in the presence of radioactively labeled ATP. Reaction mixtures were separated by denaturing polyacrylamide gel electrophoresis and the efficiency of DNA-PKCS autophosphorylation was determined by measuring the incorporation of radioactive phosphate into DNA-PKCS. The y-axis of the figure represents phosphate incorporation in arbitrary units, the x-axis represents the total amount of DNA-PKCS in the reaction mixtures. (D) Autophosphorylation of DNA-PKCS as a function of DNA concentration. DNA-PKCS concentration was kept constant at 100 ng/reaction. A 1000 bp DNA fragment was added in amounts ranging from 10 to 400 ng/reaction (DNA:DNA-PKCS molar ratio ranging from 1:10 to 3:1). The autophosphorylation level reaches a maximum, estimated at a DNA-PKCS:DNA molar ratio of 2:1 (= 70 ng DNA per reaction).
The level of autophosphorylation was determined by measuring the incorporation of radiolabeled phosphate into DNA-PKCS in reaction mixtures containing a short (250 bp) and a long (1000 bp) DNA substrate in the presence of radiolabeled ATP (Fig. 3B). A much higher level of DNA-PKCS autophosphorylation was measured when the 1000 bp DNA substrate was used, compared with the levels that were determined upon incubation with the shorter 250 bp substrate (Fig. 3C). This finding supports the proposed model that DNA-PKCS autophosphorylation takes place upon synapsis of DNA ends.
To verify that the observed difference in autophosphorylation level is indeed caused by the predicted difference in circularization efficiency of both DNA substrates, Hmg1 protein was added. This protein facilitates DNA bending and thus increases the efficiency of intra-molecular DNA end synapsis for the 250 bp substrate. Indeed, the autophosphorylation level in reactions containing the 250 bp substrate in the presence of Hmg1 is similar to the activity observed in reactions containing the 1000 bp substrate (Fig. 3C), whereas Hmg1 addition to the 1000 bp substrate did not significantly change the autophosphorylation level.
In addition, we verified that autophosphorylation requires DNA end-to-end synapsis and not any other event in which DNA is bent back upon itself. To this end, the autophosphorylation assay was performed with increasing concentrations of DNA (1000 bp fragment) and a constant amount of DNA-PK (Fig. 3D). DNA-PKCS:DNA ratios ranged from 3:1 to 1:10. We observed that such a titration curve has a clear optimum, which finding is consistent with a model in which both DNA ends with bound DNA-PK have to be brought together before autophosphorylation can proceed. Any other event in which DNA bends back upon itself would result in autophosphorylation levels that increase with DNA concentration up to a maximum when all DNA-PK is bound to a DNA end, which should remain constant upon further increase of DNA concentration. We observed that the maximum level of DNA-PKCS autophosphorylation was reached when the ratio of DNA-PKCS:DNA was estimated to be 2:1 (Fig. 3D), suggesting that both ends of the same DNA molecule should be occupied by DNA-PK for optimal activity. Therefore, we conclude that synapsis of two DNA-PK bound DNA ends is required for autophosphorylation.
The kinase activity of DNA-PKCS is required for ligation of DNA ends
The results presented above demonstrate that autophosphorylation of DNA-PKCS takes place at the synapsis of two DNA ends. Furthermore, footprinting and exonuclease digestion assays showed that the protein kinase activity of DNA-PKCS is required to modify protein–DNA complexes. We further investigated how this ATP dependent change in the end-bound DNA-PK complex influences accessibility of DNA ends to DNA ligases under various reaction conditions.
Ligation of DNA ends was studied in vitro, using a 965 bp radiolabeled DNA substrate with 5′ protruding ends, under the same reaction conditions that were used to demonstrate DNA-PK autophosphorylation. Ligation reactions were performed at low DNA concentrations, strongly favoring intra-molecular ligation. Using either T4 DNA ligase (Fig. 4A, lanes 2 and 3) or E.coli DNA ligase (Fig. 4B, lanes 2–4), intra-molecular ligation of the substrate was achieved.
Figure 4.

Ligation in the presence of DNA-PK requires ATP hydrolysis and an active DNA-PKCS kinase. (A) An overall labeled DNA substrate with cohesive ends was incubated with T4 DNA ligase, either in the absence (lanes 1–3) or the presence (lanes 4 and 5) of DNA-PK. ATP or AMP-PNP was present as indicated. Ligation products were separated by agarose gel electrophoresis. The nature of the ligation products, identified as intra- or inter-molecular ligation products, was confirmed by exonuclease V digestion. Note that intra-molecular ligation products can be either ligated on one strand (open circular form) or on both strands (covalently closed circular form). (B) An overall labeled DNA substrate with cohesive ends was incubated with E.coli DNA ligase, either in the absence (lanes 1–4) or the presence (lanes 5 and 6) of DNA-PK. ATP and/or NAD+ were present as indicated. (C) An overall labeled DNA substrate with cohesive ends was incubated with T4 DNA ligase, either in the absence (lanes 1 and 2) or the presence (lanes 3 and 4) of DNA-PK. All reaction mixtures contained ATP. The DNA-PKCS kinase inhibitor wortmannin was added in lane 4. Total levels of ligation products in all lanes were decreased in comparison with (A), due to the presence of DMSO in the reaction mixtures. (D) Wortmannin inhibits autophosphorylation of DNA-PKCS. Incorporation of radiolabeled phosphate into DNA-PKCS was determined in the absence and presence of 1 or 10 µM wortmannin. Even 1 µM wortmannin completely inhibits DNA-PKCS autophosphorylation.
Subsequently, ligation was studied in reaction mixtures that contained the DNA substrate and DNA-PK, either in the presence or the absence of ATP. When incubation took place without ATP, we used several strategies to ensure that ligation could still proceed at the same rate as would be observed in the presence of ATP. First, we used AMP-PNP, an ATP analog that supports activity of T4 DNA ligase, but cannot function as a cofactor for DNA-PKCS (Fig. 4A). In the absence of DNA-PK, the substrate was clearly ligated intra-molecularly (lane 3). Strikingly, a dramatically reduced level of ligation (5-fold) was observed in reaction mixtures to which DNA-PK was added (lane 5). A similar result was obtained when E.coli DNA ligase was used. This ligase requires NAD+ as a cofactor instead of ATP, which allows omission of ATP in these reactions, without loss of ligation activity. Consistent with the previous experiment, addition of DNA-PK inhibited ligation of the DNA substrate in the absence of ATP (Fig. 4B, lane 5). In conclusion, these data indicate that the presence of DNA-PK under conditions that do not allow for phosphorylation renders the DNA ends unavailable for ligation.
In contrast, efficient ligation of the substrate was restored in reaction mixtures containing DNA-PK if ATP was present (Fig. 4A, lane 4 and B, lane 6), indicating that ATP is required for ligation to proceed, even when the ligase itself does not need ATP for its activity.
Interestingly, ligation products were different from the intra-molecular joining products that were formed in reactions without DNA-PK. These products had a lower mobility, suggesting that they were formed via an inter-molecular joining route, even though the intra-molecular event should be highly favored at the low DNA concentrations used in this assay. Addition of the Ku70/80 heterodimer to the reaction mixture did not result in a shift in the nature of ligation products, indicating that this activity requires DNA-PKCS (data not shown). The DNA-PK dependent shift from intra-molecular to inter-molecular ligation was also observed when E.coli DNA ligase was used instead of T4 DNA ligase (Fig. 4B, lane 6). We confirmed that the lower mobility DNA species were linear multimers of the substrate by exonuclease V digestion; intra-molecular ligation products could not be degraded by an exonuclease, whereas non-circular inter-molecular products were exonuclease sensitive (data not shown).
The need for ATP hydrolysis suggested that DNA-PKCS protein kinase activity was required for ligation. We therefore included the DNA-PKCS inhibitor wortmannin in ligation reactions with DNA-PK and ATP and observed that inhibition of DNA-PKCS kinase activity indeed reduced ligation efficiency of the DNA substrate 2-fold (Fig. 4C). As expected, wortmannin did not influence T4 DNA ligase activity (data not shown). Wortmannin was included in the reactions at a final concentration of 10 µM, which effectively inhibits autophosphorylation of DNA-PKCS (Fig. 4D).
Both the increased accessibility of DNA ends upon ATP addition and the shift from intra- to inter-molecular joining products in the presence of DNA-PK have been observed with two differently purified DNA-PK preparations, obtained from human placenta (see Materials and Methods for details of the purification procedure). In addition, similar results were obtained by using a commercially available DNA-PK preparation from HeLa cells (Promega) and a highly pure DNA-PK preparation from HeLa cells. In all cases we observed an identical shift of ligation products and a similar requirement for the presence of ATP for efficient ligation to proceed. These data are reproducible and strongly argue against a possible contaminating factor that might contribute to the observed effects.
In conclusion, our data show that DNA-PK renders the DNA termini unavailable for ligation, until an ATP dependent event takes place that requires the kinase activity of DNA-PKCS, most likely DNA-PKCS autophosphorylation.
DISCUSSION
Autophosphorylation of DNA-PKCS requires synapsis of DNA ends
The discovery that DNA-PK is involved in NHEJ was a major breakthrough in this area of research (8). However, the role of this protein kinase in bringing together DNA ends and stimulating the subsequent ligation step is not yet understood in much detail. In this study we have investigated the role of DNA-PK in the early steps of NHEJ. As reported before (30), we found that DNA-PK forms specific complexes at DNA termini, covering approximately three turns of the helix. Upon addition of ATP, DNA-PKCS can undergo autophosphorylation. We demonstrated that this reaction only occurs efficiently when both DNA termini of the same substrate molecule can reach each other and are occupied by DNA-PK, showing that synapsis of two DNA-PK bound termini is required for efficient autophosphorylation. Our data are consistent with either DNA-PKCS autophosphorylation in trans or in cis and with the observation that DNA-PK activity on a peptide substrate was most efficient under conditions that allowed for synapsis (21).
DNA-PKCS kinase activity is required for accessibility of DNA ends
As seen before (31), the DNA-PK complexes blocked access to the DNA ends; both exonucleases and ligases were not able to exert their activity in the presence of DNA-PK. However, the presence of ATP caused a change; the ends became more accessible for Bal31 exonuclease and ligase. AMP-PNP could not substitute for ATP, showing that all these changes required ATP hydrolysis, probably of the γ-phosphodiester bond. The most obvious activity that could be involved in these changes is the protein kinase activity of DNA-PK, which hydrolyzes this phosphodiester bond. Indeed, we found that the PI-3 kinase inhibitor wortmannin reduced the efficiency of DNA end ligation in the presence of DNA-PK, suggesting that DNA-PK autophosphorylation may be required for the remodeling of end-bound protein complexes. We found that four different DNA-PK preparations (two from human placenta and two from HeLa cells), including a highly pure preparation (>90%), had a similar effect in the ligation assay, rendering a contribution of incidental contaminants unlikely.
Although both DNA-PKCS and Ku can be autophosphorylated, only the protein kinase activity of the catalytic subunit has been shown to be regulated by phosphorylation (23). Therefore, we consider the most obvious explanation for our results that the regulation of DNA end accessibility is executed, at least in part, through DNA-PKCS autophosphorylation. This would provide a biochemical explanation for the observation that DNA-PKCS autophosphorylation is required for efficient DSB rejoining in vivo (25,26,28).
The increased accessibility of the DNA ends to Bal31 exonuclease and ligase shows that a change in the protein–DNA complex takes place upon ATP addition. This provides further evidence for the suggestion of Ding et al. (28), that DNA-PKCS autophosphorylation mediates a conformational change that is required for end processing. In agreement with their findings, our footprint analysis did not provide evidence for complete dissociation of the protein complex. This observation is also consistent with the results of Yoo and Dynan (32), but contradicts several other studies (23,24) that presented evidence for decreased affinity of DNA-PKCS for the DNA-Ku complex after autophosphorylation, resulting in disassembly of DNA-PKCS from the DNA and loss of DNA-PK activity. The apparent discrepancy between these findings and our observations can possibly be explained by incomplete phosphorylation of our DNA-PKCS. However, our findings may also be consistent with the observation that DNA-PKCS dissociates from DNA upon autophosphorylation. The molar excess of DNA-PK used in our autophosphorylation assay may allow dissociation of phosphorylated DNA-PKCS and subsequent reloading of a still unphosphorylated DNA-PKCS molecule. This would be consistent with our observation of a slightly less pronounced footprint upon ATP addition. The window of time between dissociation of the phosphorylated DNA-PKCS molecule and re-association of a new DNA-PKCS molecule would allow binding of ligases or nucleases to the DNA termini.
The nature of the synaptic complexes
As discussed above, the specific DNase footprint indicates the presence of a specific protein–DNA complex under our assay conditions. Interestingly, the presence of DNA-PK severely affected the nature of the joining products after ligation. Inter-molecular ligation events were favored over intra-molecular ones in the presence of DNA-PK, whereas intra-molecular joining products were mainly detected in the absence of DNA-PK. Similar observations have been made previously in ligation reactions in crude nuclear extracts or with purified components. Addition of crude fractions containing DNA-PK to DNA substrates resulted in formation of oligomeric ligation products (31,33). This reaction could be inhibited by addition of wortmannin, suggesting that the kinase activity of DNA-PKCS was involved. Multimeric ligation products were also observed in reactions containing purified DNA-PK and the XRCC4/ligase IV complex, but not in reactions containing only XRCC4/ligase IV (18). We found that the presence of DNA-PK stimulated inter-molecular ligation by the two tested ligases: T4 DNA ligase and E.coli ligase. Therefore, the reported shift from an intra- to an inter-molecular ligation pathway is likely to be independent of the ligase and can be attributed exclusively to the action of DNA-PK.
Chen et al. (18) proposed that DNA-PK might stimulate association of DNA molecules, which would increase the likelihood of inter-molecular joining. However, this cannot explain some of our results; we found that autophosphorylation depended on synapsis of the two ends of the same DNA molecule under the conditions used in the ligation reaction, indicating that association with the other end of the same DNA molecule predominates at these low DNA concentrations. We assume that the two synapsed DNA termini do not dissociate after DNA-PKCS autophosphorylation. However, we found that the two ends of the same DNA molecule are not joined efficiently. These two findings may be reconciled by the following model (see Fig. 5). Upon binding of DNA-PK to DNA termini, the DNA ends can form a synapse, held together by protein–protein contacts. Synapsis activates autophosphorylation activity, which results in remodeling of the complex, allowing access to processing enzymes as well as DNA ligase. However, the ends are held in such a way that ligation of these ends is sterically highly unfavorable. The only way to perform a ligation reaction under these conditions is inter-molecular ligation.
Figure 5.
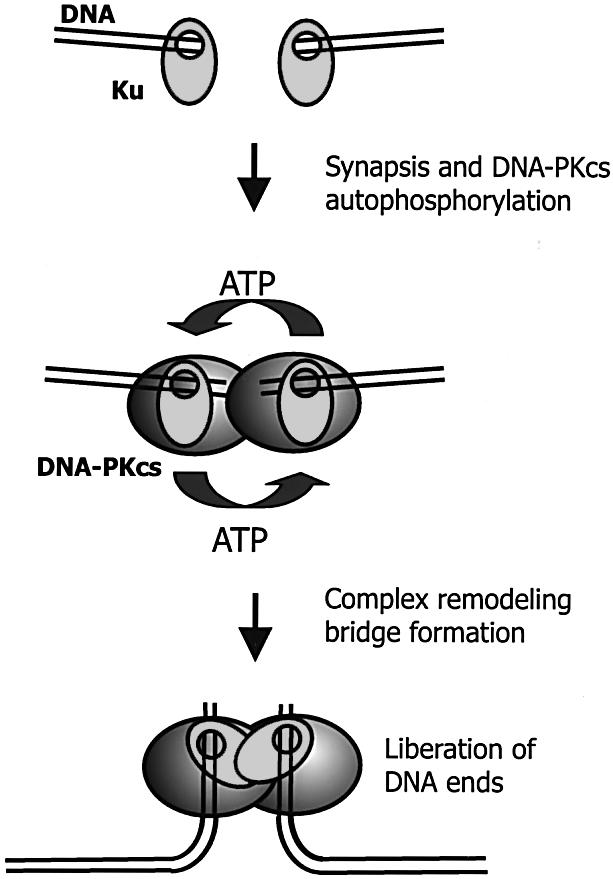
Model for DNA-PKCS autophosphorylation and its possible significance for in vivo end-joining. After the occurrence of a DSB, DNA-PK binds to the termini of both DNA fragments, thereby sterically protecting the termini against degradation by nucleases or (premature) ligation. Upon synapsis, DNA-PKCS autophosphorylation induces a (subtle) remodeling of the protein–DNA complex. This alteration of the end-bound complex makes the extreme ends accessible for ligases (or processing enzymes). Upon DNA-PKCS autophosphorylation, the termini are held together by the protein complexes, which function as a molecular bridge between the broken DNA fragments. Such a bridge would ensure stable association of both ends without blocking access to the extreme termini.
Implications for end-joining in vivo
The model of Figure 5 can explain the results from our in vitro assays, and has several implications for end-joining in vivo. It explains how DNA ends are protected from degradation until they encounter the other end. DNA-PKCS autophosphorylation would enable end processing while the ends are held together. This configuration ensures synapsis of the ends in a tight complex without blocking the extreme termini. This is especially important for the more complex types of breaks generated by ionizing radiation, which may require extensive processing before ligation can take place.
One major problem has not been solved by our experiments: why are the synapsed ends not ligated to each other? If a similar block would exist in vivo, it would cause a very high level of chromosomal aberrations. Obviously, there should be factors that modulate the structure of this complex in vivo in such a way that the ends that are brought together can be ligated. As the inter-molecular ligation does not depend on the DNA ligase, we assume that other factor(s) may be required for this switch. We are currently investigating whether DNA bending proteins such as Hmg1 or Hmg2 might be involved in the modulation of this process.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are grateful to Dr Susan P. Lees-Miller (Departments of Biochemistry and Molecular Biology and Biological Sciences, University of Calgary, Calgary, Alberta, Canada) for kindly providing preparations of highly pure DNA-PK. We would also like to thank Dr Ellen Smid-Koopman for help with obtaining human placentas and Drs Jaspers, Florea and Wyman for helpful discussions. This work was supported by grants from the Netherlands Scientific Organization (NWO) to E.W. and H.T.B., and the Netherlands Cancer Foundation (KWF) to N.S.V.
REFERENCES
- 1.Kanaar R., Hoeijmakers,J.H. and van Gent,D.C. (1998) Molecular mechanisms of DNA double strand break repair. Trends Cell. Biol., 8, 483–489. [DOI] [PubMed] [Google Scholar]
- 2.van Gent D.C., Hoeijmakers,J.H. and Kanaar,R. (2001) Chromosomal stability and the DNA double-stranded break connection. Nature Rev. Genet., 2, 196–206. [DOI] [PubMed] [Google Scholar]
- 3.Critchlow S.E. and Jackson,S.P. (1998) DNA end-joining: from yeast to man. Trends Biochem. Sci., 23, 394–398. [DOI] [PubMed] [Google Scholar]
- 4.Chappell C., Hanakahi,L.A., Karimi-Busheri,F., Weinfeld,M. and West,S.C. (2002) Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J., 21, 2827–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma Y., Pannicke,U., Schwarz,K. and Lieber,M.R. (2002) Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell, 108, 781–794. [DOI] [PubMed] [Google Scholar]
- 6.Dai Y., Kysela,B., Hanakahi,L.A., Manolis,K., Riballo,E., Stumm,M., Harville,T.O., West,S.C., Oettinger,M.A. and Jeggo,P.A. (2003) Nonhomologous end joining and V(D)J recombination require an additional factor. Proc. Natl Acad. Sci. USA, 100, 2462–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Udayakumar D., Bladen,C.L., Hudson,F.Z. and Dynan,W.S. (2003) Distinct pathways of nonhomologous end joining that are differentially regulated by DNA-dependent protein kinase mediated phosphorylation. J. Biol. Chem., 278, 41631–41635. [DOI] [PubMed] [Google Scholar]
- 8.Smith G.C. and Jackson,S.P. (1999) The DNA-dependent protein kinase. Genes Dev., 13, 916–934. [DOI] [PubMed] [Google Scholar]
- 9.Walker J.R., Corpina,R.A. and Goldberg,J. (2001) Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature, 412, 607–614. [DOI] [PubMed] [Google Scholar]
- 10.Bliss T.M. and Lane,D.P. (1997) Ku selectively transfers between DNA molecules with homologous ends. J. Biol. Chem., 272, 5765–5773. [DOI] [PubMed] [Google Scholar]
- 11.Chiu C.F., Lin,T.Y. and Chou,W.G. (2001) Direct transfer of Ku between DNA molecules with nonhomologous ends. Mutat. Res., 486, 185–194. [DOI] [PubMed] [Google Scholar]
- 12.Cary R.B., Peterson,S.R., Wang,J., Bear,D.G., Bradbury,E.M. and Chen,D.J. (1997) DNA looping by Ku and the DNA-dependent protein kinase. Proc. Natl Acad. Sci. USA, 94, 4267–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pang D., Yoo,S., Dynan,W.S., Jung,M. and Dritschilo,A. (1997) Ku proteins join DNA fragments as shown by atomic force microscopy. Cancer Res., 57, 1412–1415. [PubMed] [Google Scholar]
- 14.Yaneva M., Kowalewski,T. and Lieber,M.R. (1997) Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy studies. EMBO J., 16, 5098–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurimasa A., Kumano,S., Boubnov,N.V., Story,M.D., Tung,C.S., Peterson,S.R. and Chen,D.J. (1999) Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol. Cell. Biol., 19, 3877–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu H.L., Yannone,S.M. and Chen,D.J. (2002) Defining interactions between DNA-PK and ligase IV/Xrcc4. DNA Rep., 1, 225–235. [DOI] [PubMed] [Google Scholar]
- 17.Nick McElhinny S.A., Snowden,C.M., McCarville,J. and Ramsden,D.A. (2000) Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol. Cell. Biol., 20, 2996–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L., Trujillo,K., Sung,P. and Tomkinson,A.E. (2000) Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J. Biol. Chem., 275, 26196–2205. [DOI] [PubMed] [Google Scholar]
- 19.Calsou P., Delteil,C., Frit,P., Drouet,J. and Salles,B. (2003) Coordinated assembly of Ku and p460 subunits of the DNA-dependent protein kinase on DNA ends is necessary for XRCC4-ligase IV recruitment. J. Mol. Biol., 326, 93–103. [DOI] [PubMed] [Google Scholar]
- 20.Leber R., Wise,T.W., Mizuta,R. and Meek,K. (1998) The XRCC4 gene product is a target for and interacts with the DNA-dependent protein kinase. J. Biol. Chem., 273, 1794–1801. [DOI] [PubMed] [Google Scholar]
- 21.DeFazio L.G., Stansel,R.M., Griffith,J.D. and Chu,G. (2002) Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J., 21, 3192–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roth D.B., Menetski,J.P., Nakajima,P.B., Bosma,M.J. and Gellert,M. (1992) V(D)J recombination: broken DNA molecules with covalently sealed (hairpin) coding ends in scid mouse thymocytes. Cell, 70, 983–991. [DOI] [PubMed] [Google Scholar]
- 23.Chan D.W. and Lees-Miller,S.P. (1996) The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J. Biol. Chem., 271, 8936–8941. [DOI] [PubMed] [Google Scholar]
- 24.Merkle D., Douglas,P., Moorhead,G.B., Leonenko,Z., Yu,Y., Cramb,D., Bazett-Jones,D.P. and Lees-Miller,S.P. (2002) The DNA-dependent protein kinase interacts with DNA to form a protein-DNA complex that is disrupted by phosphorylation. Biochemistry, 41, 12706–12714. [DOI] [PubMed] [Google Scholar]
- 25.Chan D.W., Chen,B.P., Prithivirajsingh,S., Kurimasa,A., Story,M.D., Qin,J. and Chen,D.J. (2002) Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev., 16, 2333–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soubeyrand S., Pope,L., Pakuts,B. and Hache,R.J. (2003) Threonines 2638/2647 in DNA-PK are essential for cellular resistance to ionizing radiation. Cancer Res., 63, 1198–1201. [PubMed] [Google Scholar]
- 27.Douglas P., Moorhead,G.B., Ye,R. and Lees-Miller,S.P. (2001) Protein phosphatases regulate DNA-dependent protein kinase activity. J. Biol. Chem., 276, 18992–18998. [DOI] [PubMed] [Google Scholar]
- 28.Ding Q., Reddy,Y.V., Wang,W., Woods,T., Douglas,P., Ramsden,D.A., Lees-Miller,S.P. and Meek,K. (2003) Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell. Biol., 23, 5836–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan D.W., Mody,C.H., Ting,N.S. and Lees-Miller,S.P. (1996) Purification and characterization of the double-stranded DNA-activated protein kinase, DNA-PK, from human placenta. Biochem. Cell Biol., 74, 67–73. [DOI] [PubMed] [Google Scholar]
- 30.Gottlieb T.M. and Jackson,S.P. (1993) The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell, 72, 131–142. [DOI] [PubMed] [Google Scholar]
- 31.Calsou P., Frit,P., Humbert,O., Muller,C., Chen,D.J. and Salles,B. (1999) The DNA-dependent protein kinase catalytic activity regulates DNA end processing by means of Ku entry into DNA. J. Biol. Chem., 274, 7848–7856. [DOI] [PubMed] [Google Scholar]
- 32.Yoo S. and Dynan,W.S. (1999) Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res., 27, 4679–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanakahi L.A., Bartlet-Jones,M., Chappell,C., Pappin,D. and West,S.C. (2000) Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair. Cell, 102, 721–729. [DOI] [PubMed] [Google Scholar]