


Abstract
Thermophilic viruses represent a novel source of genetic material and enzymes with great potential for use in biotechnology. We have isolated a number of thermophilic viruses from geothermal areas in Iceland, and by combining high throughput genome sequencing and state of the art bioinformatics we have identified a number of genes with potential use in biotechnology. We have also demonstrated the existence of thermostable counterparts of previously known bacteriophage enzymes. Here we describe a thermostable RNA ligase 1 from the thermophilic bacteriophage RM378 that infects the thermophilic eubacterium Rhodothermus marinus. The RM378 RNA ligase 1 has a temperature optimum of 60–64°C and it ligates both RNA and single-stranded DNA. Its thermostability and ability to work under conditions of high temperature where nucleic acid secondary structures are removed makes it an ideal enzyme for RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE), and other RNA and DNA ligation applications.
INTRODUCTION
As a novel source for nucleic acid modifying enzymes, thermophilic viruses have remained relatively unexplored. Most studies on thermophilic viruses have focused on phages infecting thermophilic Archaea (1,2), but little effort has been put into isolating and characterizing bacteriophages infecting thermophilic eubacteria. We have systematically isolated and characterized thermophilic viruses in recent years for the purpose of isolating genes and gene products for uses in biotechnology and molecular biology applications. One of these studies resulted in isolation of a novel thermophilic bacteriophage that infects the thermophilic eubacterium Rhodothermus marinus (3,4).
The bacteriophage, designated RM378, is a large head and tail phage and a member of the Myoviridae family with an A2 morphology, which includes the well studied bacteriophage T4 that infects Escherichia coli. The bacteriophage RM378 consists of a moderately elongated head, 95 nm long and 85 nm in diameter, and a tail of 150 nm with connector to the head (4). It was completely stable and infectious up to 65°C, but lost viability above that temperature. The genome was fully sequenced and contained a 130 kb, double-stranded DNA helix, with a G+C content of 42.0% (4).
More than 200 open reading frames (ORFs) were found, but the overall sequence similarity to known gene products in public databases was low when using BLAST software (5). The predicted product of one of these ORFs showed homology to the T4 RNA ligase 1.
T4 RNA ligase 1 (EC 6.5.1.3) has the ability to ligate single-stranded (ss) nucleic acids by catalyzing the ATP-dependent formation of phosphodiester bonds between 5′-phosphate and 3′-hydroxyl termini of ssRNA or ssDNA (6,7). The apparent biological role of T4 RNA ligase 1, together with the phage-encoded polynucleotide kinase, is to repair cleaved tRNA molecules, thereby counteracting the defense mechanism of the bacterial host. The tRNALys molecules are cleaved by T4-induced anticodon nuclease (ACNase) 5′ to the wobble position, yielding 2′:3′-cyclic phosphate and 5′ hydroxyl termini (8–10). The anticodon nuclease (PrrC or ACNase) is coded by the prrABC operon, which encodes subunits of a type IC DNA restriction and a modification system called EcoprrI. When E.coli is infected by T4 phage, the phage inactivates the EcoprrI system by means of the T4 Stp polypeptide, causing conformational changes resulting in the activation of the ACNase (9–11). The anticodon nuclease system can be found in some other eubacteria, and examples of other tRNA degrading systems are the bacterial toxins colicins D and E5 (9,12).
The T4 RNA ligase 1 is a workhorse of molecular biology and is used in numerous protocols in biotechnology. Applications include RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) (13,14), ligation of oligonucleotide adaptors to cDNA or single-stranded primer extension products for PCR (15,16), oligonucleotide synthesis (17) and various 5′ nucleotide modifications of nucleic acids (18). Here we describe the isolation and characterization of an RNA ligase, a thermostable homolog of T4 RNA ligase 1.
MATERIALS AND METHODS
Cloning of the RnlA gene, and expression and purification of the gene product
All standard molecular biology methods were performed as described in Sambrook et al. (19). The full-length RNA ligase gene was amplified by standard PCR from RM378 viral DNA (4) using primers Rnl3 [d(GGGAATTCTTATGAACGTAAAATACCCG)] and Rnl4 [d(GGAGATCTTATTTAAATAACCCCTTTTC)] with Dynazyme™ DNA polymerase (Finnzymes Oy), as recommended by the manufacturer. The RnlA gene was cloned into pBTAC1 expression vector and verified by DNA sequencing. A clone (RNAlig6.2.1) containing the gene for RM378 RNA ligase was selected for expression and transformed into BL21 E.coli cells. The strain was cultivated at 37°C and induced with 1 mM IPTG. The cells were harvested and disrupted by sonication. The crude cell extract was centrifuged in an SA-600 rotor (Sorvall Inc.) at 10 400 g for 1 h. The supernatant was collected and ammonium sulfate added to 30%. The solution was stirred at 4°C for 40 min and centrifuged at 10 000 r.p.m. for 1 h. The protein pellet was dissolved in 20 mM Tris pH 8 and centrifuged at 20 000 r.p.m. for 1 h. The supernatant was run through a HiPrep 26/10 (Amersham Biotech Inc.) desalting column in 20 mM Tris pH 8. The protein was then run on a ResQ column and eluted with a KCl solution. The majority of the RNA ligase 1 eluted as a single peak, which was collected. The RM378 RNA ligase was concentrated with ammonium sulfate precipitation and dialysed against 20 mM Tris pH 8. The protein was stored at –20°C in 10 mM Tris pH 8, 50 mM KCl, 1 mM DTT, 0.1 mM EDTA and 50% glycerol.
Activity assay and characterization
The standard T4 RNA ligase 1 activity assay, developed by Silber et al. (6), was modified for characterization of the RM378 RNA ligase. The reaction mixture was as follows, in 10-µl aliquots: 50 mM MOPS pH 7, 1 mM DTT, 0.1–1 mM ATP, 5 mM MgCl2, 10 mM KCl, 25 µg/ml BSA, 10 µM 5′-32P-labeled DNA or RNA substrate, and 0.01–0.2 mg/ml protein (final concentration). Reactions times varied from 5 min to a few hours based on the assay or the application. The reactions were terminated by incubation at 90°C for 5 min, followed by cooling on ice, followed by the addition of 30 µl of solution of shrimp alkaline phosphatase (0.2 U/µl) in its standard buffer (USB Corp.) and incubation for 4 h at 37°C. The alkaline phosphatase-resistant 5′-phosphoryl termini were captured on DE81 filters, washed twice in 500 mM phosphate buffer and dried. Radioactivity on the filters was then counted using a scintillation counter (Packard Inc.) after the addition of 5 ml scintillation fluid. All assays were performed in triplicate and the mean calculated.
One unit of RNA ligase activity was determined as the amount of enzyme needed to convert 1 nmol of 5′-phosphoryl termini in 32P-r(A20) to a phosphatase-resistant form in 30 min at 60°C for the RM378 RNA ligase 1 (6). This assay was used for studies of the RM378 RNA ligase, using either DNA or RNA 20-nucleotide oligomers 32P-d(A20), 32P-r(A20) as substrates. For Km measurements of the substrate adenylation, 0.02 mg/ml of enzyme was incubated with 20 µM 32P-r(A10) 3′ dideoxy-blocked at 60°C for 60 min with different amounts of ATP (1, 10, 25, 50, 100, 250, 500 and 1000 µM), and the protection assay employed.
Exonuclease I protection assays
An exonuclease protection assay (EPA) was developed to measure the accumulation of the final product in intra-molecular ligations (circularization), excluding the adenylated intermediate. The DNA intra-molecular ligation reactions were done by assaying the RM378 RNA ligase, using 0.1 mg/ml enzyme, 50 µM ATP and P-d(A20) as a substrate for 8 h at 60°C. After the incubation, the samples were heated to 90°C for 5 min and half the sample was subjected to exonuclease I (New England Biolabs Inc.) digestion (0.5 U/µl for 8 h), while the other half of the sample was incubated with water. The samples were then mixed in a 1:1 ratio with 2× Oligreen Dye® (Molecular Probes Inc.) and measured for fluoresence intensity in an LJL Analyst™ fluoresence meter (LJL Biosystems), according to the manufacturers’ instructions. The background was then subtracted from the samples and the ExoI digested, ligated samples compared with the undigested samples, and the percentage of protected DNA (circular) calculated.
RACE assay
Vector-derived RNA (1 ng) (Takara Inc.) was used as a substrate for the RLM-RACE protocol using the components of the Generacer kit (Invitrogen Inc.) as described by the manufacturer, except that the phosphatase step was excluded and RM378 RNA ligase and buffer were used for the RNA ligation, and incubation was performed at 60°C for 1 h. The cDNA was made using the AMV First Strand Synthesis Kit (Invitrogen Inc.) with random hexamers as primers, according to the manufacturer’s instructions. The template was then amplified using specific primer and an adapter primer from the Generacer kit, using standard PCR with AmpliTaq Gold (Applied Biosystems Inc.), as recommended by the manufacturer. The products were cloned into TOPO TA vectors and sequenced for verification using the BigDye 2.0 DNA sequencing kit, and analyzed on a ABI Prism 3700 genetic analyzer (Applied Biosystems Inc.).
RESULTS
Cloning, overexpression and purification of the RM378 RNA ligase
Over 200 putative ORFs were discovered after complete sequencing of the RM378 genome (4). The RM378 bacteriophage genome has been submitted to the DDBJ/EMBL/GenBank database (accession no. NC_004735). Standard BLAST analysis (5) of these ORFs showed ∼40% similarity to the RnlA gene amino acid sequence which encodes the RNA ligase 1 in the T4 phage (20), and also the RNA ligase part of the pnk/pnl gene (ORF 86) from the plant virus Autographa californica polyhedrovirus (ACNV) genome (21). This putative RNA ligase 1 gene, designated RnlA, from the RM378 genome was 1314 bp in length and coded for a 438-amino-acid polypeptide with a calculated mass of 51.3 kDa. The alignments of RNA ligase 1 from T4, ACNV and RM378 are shown in Figure 1. The RnlA gene was amplified from the viral genome, cloned into pBTAC1 expression vector and sequenced. The gene product was overexpressed in E.coli and purified to near homogeneity. The yield of purified recombinant RM378 RNA ligase was 1 mg/g of cells (wet weight).
Figure 1.

Amino acids alignment of RM378, T4 and ACNV RNA ligase 1 using the Clustal_X program (33). The ATP binding motif (KxDG) is boxed.
Aliquots from the purification steps were collected and run on a 12% SDS polyacylamide gel and stained with Coomassie Brilliant Blue R-250 (22). The purification was estimated to be >95% by SDS–PAGE analysis (Fig. 2).
Figure 2.
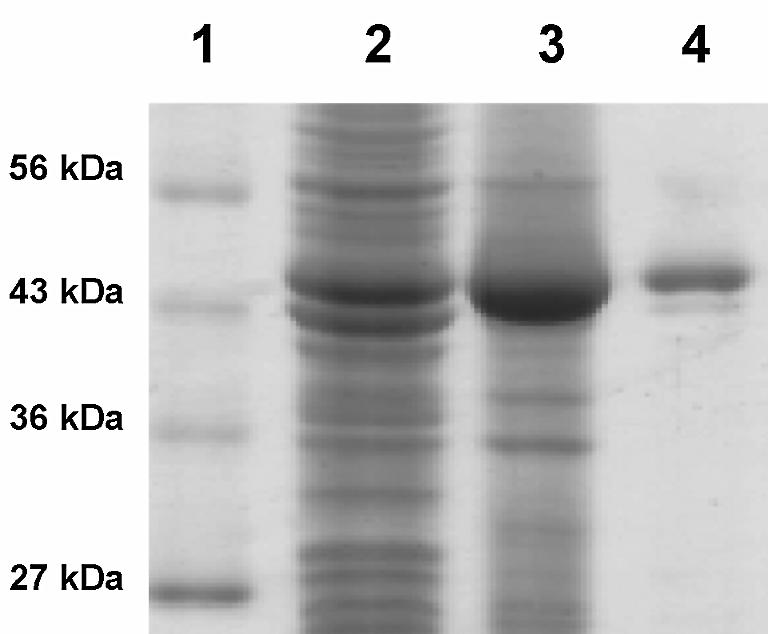
Protein purification of the RM378 RNA ligase on a 12% SDS polyacrylamide gel. Protein marker (lane 1), crude extract (lane 2), crude extract after 30% ammonium sulfate precipitation (lane 3) and RM378 RNA ligase peak from ResQ column (lane 4). Purification was estimated at >95% by SDS–PAGE analysis.
Characterization of the RM378 RNA ligase
Various properties of the purified RNA ligase protein were examined. The first step was to determine the pH optimum of the enzyme. A phosphatase resistance assay (6) was carried out with the enzyme in the reaction buffer (1 mM DTT, 1 mM ATP, 10 mM MgCl2, 25 µg/ml BSA and 10 mM KCl) at 55°C with either 50 mM Tris–HCl or 50 mM MOPS buffers over a broad pH range. The concentration of RNA ligase for the following experiments was 0.02 mg/ml, and the concentration of polynucleotide substrate was 10 µM using 5′-32P-labeled r(A20). As seen in Figure 3A, the optimum pH is 6 and 7 for Tris and MOPS, respectively. Since MOPS was within its useful pH range (6.6–7.8) (19), it was used in the following experiments.
Figure 3.

Characterization of the effects of pH and temperature on the RM378 RNA ligase using the phosphatase-resistant assay. (A) pH profile using both Tris–HCl (filled diamonds) and MOPS buffers (filled squares). The pH optima were 6 and 7, respectively. (B) Temperature optimum. The apparent optimum temperature of the enzyme activity was 64°C. (C) Thermostability measured by heating the enzyme in buffer for 1 h and then adding substrate and assaying for 1 h. Heating at 60°C increases the activity, which suggests that the enzyme is thermoactivated, but after heating at 70°C the activity was ∼50% and all activity was lost after 1 h at 80°C. (D) Thermostability at 60°C (filled squares) and 64°C (filled diamonds) for an extended period of time. The enzyme loses activity rapidly at 64°C, but is relatively stable at 60°C.
The apparent optimum temperatures for enzyme activity were measured by performing the reaction at numerous temperatures between 4 and 90°C, using the standard circularization assay. As seen in Figure 3B, the optimum temperature is 64°C, which is close to the R.marinus optimal growth conditions (3).
The thermostability at different temperature was studied in two ways: (i) by heating the enzyme for 1 h at a given temperature in its reaction buffer (without the substrate) and measuring activity using the phosphatase resistance assay; and (ii) by heating the enzyme as before at 90°C, and then taking samples at 5 min intervals, for activity measurements at 60°C for 1 h. As seen in Figure 3C, the enzyme was relatively stable at 60°C and showed clear signs of thermoactivation, i.e. increased activity after incubation at 60°C. At higher temperatures the enzyme started to lose activity rapidly, and demonstrated no activity at all after 1 h at 80°C. The enzyme had lost all of its activity after 5 min at 90°C (data not shown). We therefore incubated the protein for an extended time at 60 and 64°C and assayed its activity for 1 h at 60°C afterwards. As seen in Figure 3D, this experiment clearly showed that the protein lost activity quite rapidly at 64°C but was relatively stable at 60°C, with a half life of ∼10 h. This shows that even though apparent optimum activity is at 64°C, the enzyme is not stable at temperatures >60°C.
The effect of ATP concentration was studied by adding different amounts of ATP to the reaction. Since the conversion of ATP to AMP and PPi along with the adenylation of the donor was essential for the reaction, it was clear that no activity should be present without the ATP. As seen in Figure 4A, the optimum ATP concentration is reached at 0.1–0.25 mM ATP, but at higher concentrations the ATP clearly inhibits the reaction and at 10 mM the inhibition is complete. This inhibition could not be rescued by adding Mg2+ (up to 20 mM) to the reaction, which ruled out the possibility that complete chelation of the Mg2+ was causing it.
Figure 4.

Characterization of the RM378 RNA ligase using the phosphatase-resistant assay. (A) The effect of ATP concentration on the activity. Optimum activity was obtained in 100–250 µM ATP and total inhibition was reached at 10 mM ATP. (B) Effect of Mn2+ (filled squares) and Mg2+ (filled diamonds) cations. The enzyme is dependent on divalent cations. The optimum concentrations were 1 and 5 mM for Mn2+ and Mg2+, respectively, with Mn2+ showing 90% of the Mg2+ activity. (C) Effect of NaCl (filled squares) and KCl (filled diamonds) salts; NaCl showed steady inhibition, but low level KCl increased the activity (0.1–10 mM), whereas at higher concentrations it too resulted in steady inhibition. (D) Effect of protein concentration on activity. The enzyme reached maximum accumulation of phosphatase-resistant products at 0.1 mg/ml protein concentration, resulting in 70% phosphatase resistance.
The effects of divalent cations were studied with both Mn2+ and Mg2+ at different concentrations. The results showed that the enzyme was inactive at cation concentrations <100 µM and that optimum activity was reached at 1 mM and 5 mM for Mn2+ and Mg2+, respectively (Fig. 4B), with Mn2+ maintaining ∼90% activity compared with Mg2+. The same metal ion assay (Mn2+ and Mg2) was performed using DNA template and no detectable difference was observed between the cations (data not shown).
The effects of common salts like NaCl and KCl were studied by titration of these salts in the buffers. As seen in Figure 4C, the NaCl steadily inhibited the reaction, completely inhibiting enzyme activity at a concentration of 500 mM. A low concentration of KCl (0.1–10 mM) increased the activity, but showed similar inhibition at higher concentrations.
The effect of protein concentration was studied on the standard 10 µM 32P-r(A20) template concentration and the reactions were incubated for 1 h (Fig. 4D). The graph was linear up to 0.025 mg/ml protein, but at 0.1 mg/ml concentration 70% of the template was phosphatase resistant. The specific activity of the RM378 RNA ligase was calculated 150 and 300 U/mg in its standard buffer, at 60 and 64°C, respectively, containing 0.1 mM ATP, 10 µM 32P-r(A20) substrate, and following incubation for 30 min, using the standard unit definition by Silber et al. (6). The turnover numbers were estimated to be 0.25 (at 60°C) and 0.5 (at 64°C) pmol of ligated RNA ends per pmol of enzyme per minute.
We also studied the ssDNA ligation using 32P-d(A20) substrate, similar to the RNA phosphatase protection assay (Fig. 5A). The results showed that up to 40% of the oligomer was phosphatase resistant, but after a much longer incubation time. The specific activity was estimated to be only ∼20 U/mg. The phosphatase resistance assay measures both the adenylated intermediate and the final ligation. To see the actual circular ligation we developed the EPA, which is based on the resistance of circular ssDNA to degradation by exonuclease I. The resistant products from the DNA assay was measured using OliGreen® single-stranded quantification reagent (Molecular Probes Inc.). We assayed 5′P-d(A20) oligomer using this assay, after assaying the mixure for 8 h at 60°C, and subjected it to exonuclease I digestion. As seen in Figure 5B, the protection varies according to the addition of PEG 6000 and hexamine cobalt chloride (HCC), which are known to facilitate ssDNA ligation using T4 RNA ligase 1 and DNA ligase (16). Specific activity of the RM378 RNA ligase using this assay was only ∼1.5 U/mg.
Figure 5.

(A) Single-stranded DNA ligation using the phosphatase-resistant assay, using 10 µM 32P-d(A20) as template, showing the accumulation of products over extended periods of time. (B) Circularization of P-d(A20) using 0.1 and 0.2 mg/ml enzyme concentration with 1 µM (white bar) and 2.5 µM (black bar) template with and without 1 mM HCC and 25% PEG6000. The additives increase the activity 3–4-fold, reaching the maximum of 20% after 8 h at 60°C. (C) A Lineweaver–Burk plot, derived from adenylation of 32P-r(A10) 3′-dideoxy-blocked oligomer, was measured using ATP titration. ATP inhibited adenylation at >100 µM ATP concentration. (D) RACE experiment using vector-derived RNA template and components from the Generacer™ kit (Invitrogen Inc.) followed by cDNA synthesis and PCR. RM378 RNA ligase 1 and buffer were used instead of T4 RNA ligase 1, and were incubated at 60°C for 1 h. Results show that 300-bp PCR products were obtained from the sample at different dilutions.
We measured the Km of the adenylation step on 32P-r(A10) donor with 3′ dideoxy blocker, and found that the Km for the adenylation step was 7 µM and the Vmax was 0.2 µmol mg–1 h–1. As in the titration of the phosphatase resistance assay, elevated ATP concentration inhibited the adenylation. ATP concentration started to inhibit the RNA adenylation reaction at a concentration of 100 µM. The results are shown in Figure 5C as a Lineweaver–Burk plot.
We also looked at the binding of ATP to the protein, to see the efficiency of the binding and subsequent conversion, and to see if the enzyme needed to bind RNA before the hydrolysis. The results showed that the protein had reached maximum adenylation in 20 µM ATP (data not shown).
RACE
RNA ligations were carried out on vector-derived RNA transcript and the sample was taken using the Generacer™ standard RLM-RACE protocol, except that phosphatase digestion was omitted and RM378 RNA ligase was used instead of the conventional T4 RNA ligase 1, and the sample was incubated for 60 min at 60°C. Standard PCR was then employed after the cDNA synthesis. As shown in Figure 5D, the PCR yielded 300 bp products of the expected size. The PCR products were then verified by DNA sequencing.
DISCUSSION
Sequence analysis, expression and purification of the RM378 RNA ligase
After analyzing the putative ORFs of the bacteriophage RM378 genome, it became apparent that one of the ORFs was a putative RNA ligase gene. This suggests that the RM378 bacteriphage most likely needs to counter similar host defense systems analogous to the T4 phage with the E.coli anticodon nuclease system (9,10). Sequence analysis showed that the RM378 RNA ligase had limited identity to both T4 RNA ligase 1 (20) and A.californica nuclear polyhedrovirus (ACNV) polynucleotide kinase/ligase (21). The sequence identities were 17% and 19%, respectively, counted overall (i.e. total number of identities divided by average length of the two sequences; see Fig. 1).
RNA ligases 1 belong to a superfamily of covalent nucleotidyl transferases, which also includes RNA capping enzymes and DNA ligases, both ATP- and NAD+-dependent (23). These enzymes share a number of sequence motifs, which are mostly confined to the central active site region bordering a presumed nucleic acid-binding cleft, as seen in the available structures of DNA ligases and mRNA capping enzyme (24,25). However, the presence of all these six sequence motifs in the very few available RNA ligase 1 sequences is not obvious for all different motifs. The most conserved motif, characterized by the consensus KXDG, corresponds to residues 124KLDG127 in the RM378 RNA ligase 1 (Fig. 1). This motif includes an active site Lys residue, which becomes covalently bound to AMP, and an acidic Asp residue that is known to play a role in adenylation as well as the ligation reaction. The present sequence of RM378 RNA ligase 1 is a valuable addition to the very small number of known sequences in this family of RNA ligases 1 (Fig. 1) and as such gives important clues to the identity of conserved residues of functional importance and helps to establish the location of sequence motifs characteristic of the subfamily, as well as the superfamily in general.
A number of residues in the homologous enzyme from T4 have been mutated followed by functional characterization of the corresponding mutated proteins (26). Of the residues found to be essential for T4 RNA ligase 1 activity, most are conserved in RM378 RNA ligase 1 also, which further supports the notion of conservation of the general structure and catalytic mechanism in the superfamily (26). This includes residues in motif I (Lys124 and Gly127), motif IV (Asp284 and Gly285) and motif V containing residues Lys208 and Lys210, which are possibly involved in contacts with the α-phosphate of ATP. However, the proposal of a new essential motif in the superfamily [(S/T/D)-(R/K), called motif 1a (26)] is difficult to reconcile with the present results, since the location of the allegedly essential basic residue is occupied by a hydrophobic Leu residue in RM378 RNA ligase 1 (Fig. 1).
We note the conservation of specific residues (Arg60, Lys80 and Phe82) N-terminal to motif I that have been found to be essential in the T4 enzyme and proposed to be a signature feature of this specific family of RNA ligase 1 (Fig. 1). From the sequence alignment, we further note the absence or less obvious conservation of motifs III and IIIa found in DNA ligases and RNA capping enzymes (26). It is thus difficult with any certainty to identify residues of particular function such as the one involved in binding ATP through stacking against the adenine base of ATP in analogy with a Tyr residue seen in the co-crystal structure of bacteriophage T7 DNA ligase and ATP (27). Although the speculation may hold true that the relative order of fuctionally equivalent motifs is different in different subfamilies of the superfamily, such that residue Phe80 (Phe77 in T4; 26), which is N-terminal to motif I, would stack against the adenine base, we would like to point to alternative possible residues for this function, i.e. Phe194 or Tyr207 (Phe158 and Tyr171 in T4), located between motifs I and IV, in favor of an unchanged relative order of analogous motifs.
Futhermore, since covalent nucleotidyl transferases may utilize two divalent metal ion mechanisms for catalysis similar to DNA polymerases (28), conserved acidic residues are expected to be found as ligands to the metal ions. Residues Asp126 and Asp284 seem possible candidates for this function after examination of available crystal structures and sequence alignments of the available RNA ligase I sequences (Fig. 1) (26) and other members of the superfamily (23–25).
Characterization of the RM378 RNA ligase
When comparing the properties of RM378 RNA ligase 1 to T4 RNA ligase 1, the difference in thermostability and optimum temperature for activity is most striking (60 and 37°C, respectively). This observation is not surprising since the habitats of the T4 and RM378 hosts differ greatly with respect to temperature. However, the RM378 RNA ligase 1 must only be considered a moderately thermostable enzyme (stable at 60°C). The RM378 RNA ligase showed clear signs of thermoactivation, and can therefore be pre-incubated at 60°C for some time to obtain better ligation. As seen in Figure 5A, the enzyme shows no decrease in activity after 6–8 h of incubation at 60°C, but at higher temperatures it loses activity rapidly, as seen in Figure 3D. The specific activity of RM378 RNA ligase was determined to be ∼300 U/mg protein at 64°C, compared with 77 U/mg for T4 RNA ligase as shown by Silber et al. (6). The specific activity measurements differ greatly for T4 RNA ligase 1 preparations from commercial sources (1000–12 000 U/mg). We noted that when we assayed T4 RNA ligase that was commercially obtained we observed only one-tenth of the claimed activity. This may be due to different setup and substrates of the assays. By titrating the ATP in the circularization assay, the Km for ATP concentration was determined as 1 µM compared with 0.2 µM for T4 RNA ligase 1 (6). When measuring just the ATP optimum concentration for the substrate adenylation step, the Km was found to be 7 µM for P-r(A10dd). This difference can be explained by the difference in the assays, i.e. circularization versus adenylation. The ATP optimum experiments also revealed inhibition of activity at high ATP concentration. This is in contrast with T4 RNA ligase 1, which is known to increase both yield and accumulation of products (29). The reason for this is unknown, but it can clearly be seen in both the adenylation assay and the circularization assay. A possible explanation would be that at higher ATP concentrations, ATP would either (i) block binding of the nucleic acid donor to the enzyme or (ii) compete with the covalently bound AMP (to K124) for the binding site, thereby blocking the formation of the adenylated donor intermediate and inhibiting the reaction.
When studying the DNA ligation it was clear that the phosphatase resistance assay, which measures both the adenylated and ligated product, was insufficient, due to the poor reactivity of a DNA acceptor. Therefore we developed the EPA to attain more direct measurements of DNA circularization. The DNA ligation studies showed much less activity on DNA than RNA, similar to the properties of T4 RNA ligase 1. RM378 RNA ligase 1 demonstrates relatively good adenlyation of the donor substrate, but the actual phosphodiester bond formation creates the ‘bottleneck’, as seen for T4 RNA ligase 1 (29,30). The use of gel and a radioactivity-free system for ligase activity offers the potential for high-throughput screening of ligase variants using methods such as in vitro evolution with DNA shuffling (31). A similar assay could be used for high-throughput screening based on RNA circularization with Exonuclease T digestion and Ribogreen® (Molecular Probes Inc.) RNA quantification reagent.
Applications for RM378 RNA ligase
Following its discovery several decades ago, T4 RNA ligase 1 has been used in a variety of molecular biology applications. It is used for RNA and ssDNA ligation methods such as RLM-RACE, and other PCR-based methods for cloning unknown flanking sequences. Since the secondary structure of mRNA is greatly influenced by temperature, we expect that elevated temperatures can increase the likelihood of successful RLM-RACE (15,16). More specifically, using thermostable RNA ligase at elevated temperatures can facilitate ligations of complex synthetic RNA molecules that were almost impossible using its mesophilic counterparts, since secondary structure of the RNA would prevent access to its termini, making it a poor substrate. A good example is the production and manipulation of hammerhead ribozymes, and circularization of complex RNA molecules for use in structure–function relationship studies (32). The ability to ligate ssDNA at elevated temperatures may also lead to a major advancement in specific applications in molecular biology, such as the increasingly important gene synthesis technology. The discovery of the first known thermostable homolog of T4 RNA ligase 1 provides an enzyme with well-known basic functions, yet with novel and useful new properties.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Dr Adam Baker, deCODE Genetics Inc., for proofreading the manuscript and helpful discussions.
DDBJ/EMBL/GenBank accession no. NC_004735
REFERENCES
- 1.Sharp R.J., Ahmad,S.I., Munster,A., Dowsett,B. and Atkinson,T. (1986) The isolation and characterization of bacteriophages infecting obligately thermophilic strains of Bacillus. J. Gen. Microbiol., 132, 1709–1722. [DOI] [PubMed] [Google Scholar]
- 2.Arnold H.P., Ziese,U. and Zillig,W. (2000) SNDV, a novel virus of the extremely thermophilic and acidophilic archaeon Sulfolobus. Virology, 272, 409–416. [DOI] [PubMed] [Google Scholar]
- 3.Alfredsson G.A., Kristjánsson,J.K., Hjörleifsdóttir,S. and Stetter,K.O. (1988) Rhodothermus marinus gen. Nov., sp. Nov., a thermophilic halophilic bacterium from submarine hot springs in Iceland. J. Gen. Microbiol., 134, 299–306. [Google Scholar]
- 4.Hjörleifsdottir S.H., Hreggvidsson,G.O., Fridjonsson,O.H., Aevarsson,A. and Kristjansson,J.K. (2002) US Patent 6,492,161.
- 5.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 6.Silber R., Malathi,V.G. and Hurwitz,J. (1972) Purification and properties of bacteriophage T4-induced RNA ligase. Proc. Natl Acad. Sci. USA, 69, 3009–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugino A., Snoper,T.J. and Cozzarelli,N.R. (1977) Bacteriophage T4 RNA ligase. Reaction intermediates and interaction of substrates. J. Biol. Chem., 252, 1732–1738. [PubMed] [Google Scholar]
- 8.Jabbar M.A. and Snyder,L. (1984) Genetic and physiological studies of an Escherichia coli locus that restricts polynucleotide kinase- and RNA ligase-deficient mutants of bacteriophage T4. J. Virol., 51, 522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaufmann G., David,M., Borasio,G.D., Teichmann,A., Paz,A. and Amitsur,M. (1986) Phage and host genetic determinants of the specific anticodon loop cleavages in bacteriophage T4-infected Escherichia coli CTr5X. J. Mol. Biol., 188, 15–22. [DOI] [PubMed] [Google Scholar]
- 10.Amitsur M., Levitz,R. and Kaufmann,G. (1987) Bacteriophage T4 anticodon nuclease, polynucleotide kinase and RNA ligase reprocess the host lysine tRNA. EMBO J., 6, 2499–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyndall C., Meister,J. and Bickle,T.A. (1994) The Escherichia coli prr region encodes a functional type IC DNA restriction system closely integrated with an anticodon nuclease gene. J. Mol. Biol., 237, 266–274. [DOI] [PubMed] [Google Scholar]
- 12.Tomita K., Ogawa,T., Uozumi,T., Watanabe,K. and Masaki,H. (2000) A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc. Natl Acad. Sci. USA, 97, 8278–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X. and Gorovsky,M.A. (1993) Mapping the 5′ and 3′ ends of Tetrahymena thermophila mRNAs using RNA ligase mediated amplification of cDNA ends (RLM-RACE). Nucleic Acids Res., 21, 4954–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maruyama K. and Sugano,S. (1994) Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene, 138, 171–174. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X.H. and Chiang,V.L. (1996) Single-stranded DNA ligation by T4 RNA ligase for PCR cloning of 5′-noncoding fragments and coding sequence of a specific gene. Nucleic Acids Res., 24, 990–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tessier D.C., Brousseau,R. and Vernet,T. (1986) Ligation of single-stranded oligodeoxyribonucleotides by T4 RNA ligase. Anal. Biochem., 158, 171–178. [DOI] [PubMed] [Google Scholar]
- 17.Kaluz S., Kaluzova,M. and Flint,A.P. (1995) Enzymatically produced composite primers: an application of T4 RNA ligase-coupled primers to PCR. Biotechniques, 19, 182–186. [PubMed] [Google Scholar]
- 18.Kinoshita Y., Nishigaki,K. and Husimi,Y. (1997) Fluorescence-, isotope- or biotin-labeling of the 5′-end of single-stranded DNA/RNA using T4 RNA ligase. Nucleic Acids Res., 25, 3747–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: a Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 20.Rand K.N. and Gait,M.J. (1984) Sequence and cloning of bacteriophage T4 gene 63 encoding RNA ligase and tail fibre attachment activities. EMBO J., 3, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durantel D., Croizier,L., Ayres,M.D., Croizier,G., Possee,R.D. and Lopez-Ferber,M. (1998) The pnk/pnl gene (ORF 86) of Autographa californica nucleopolyhedrovirus is a non-essential, immediate early gene. J. Gen. Virol., 79, 629–637. [DOI] [PubMed] [Google Scholar]
- 22.Hames B.D. and Rickwood,D. (1990) Gel Electrophoresis of Proteins. IRL Press, Oxford. [Google Scholar]
- 23.Shuman S. and Schwer,B. (1995) RNA capping enzyme and DNA ligase: a superfamily of covalent nucleotidyl transferases. Mol. Microbiol., 17, 405–410. [DOI] [PubMed] [Google Scholar]
- 24.Timson D.J., Singleton,M.R. and Wigley,D.B. (2000) DNA ligases in the repair and replication of DNA. Mutat. Res., 460, 301–318. [DOI] [PubMed] [Google Scholar]
- 25.Doherty A.J. and Suh,S.W. (2000) Structural and mechanistic conservation in DNA ligases. Nucleic Acids Res., 28, 4051–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L.K., Ho,C.K., Pei,Y. and Shuman,S. (2003) Mutation analysis of bacteriophage T4 RNA ligase 1: differential functional groups are required for the nucleotidyl transfer and phosphodiester bond formation steps of the ligation reaction. J. Biol. Chem., 278, 29454–29462. [DOI] [PubMed] [Google Scholar]
- 27.Subramanya H.S., Doherty,A.J., Ashford,S.R. and Wigley,D.B. (1996) Crystal structure of an ATP-dependent DNA ligase from bacteriophage T7. Cell, 85, 607–615. [DOI] [PubMed] [Google Scholar]
- 28.Lee J.Y., Chang,C., Song,H.K., Moon,J., Yang,J.K., Kim,H.K., Kwon,S.T. and Suh,S.W. (2000) Crystal structure of NAD(+)-dependent DNA ligase: modular architecture and functional implications. EMBO J., 19, 1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinton D.M., Baez,J.A. and Gumport,R.I. (1978) T4 RNA Ligase joins 2′-deoxyribonucleoside 3′,5′-bisphosphates to oligodeoxyribonucleotides. Biochemistry, 17, 5091–5097. [DOI] [PubMed] [Google Scholar]
- 30.McCoy M.I. and Gumport,R.I. (1980) T4 ribonucleic acid ligase joins single-strand oligo(deoxyribonucleotides). Biochemistry, 19, 635–642. [DOI] [PubMed] [Google Scholar]
- 31.Stemmer W.P. (1994) Rapid evolution of a protein in vitro by DNA shuffling. Nature, 370, 389–391. [DOI] [PubMed] [Google Scholar]
- 32.Wang L. and Ruffner,D.E. (1998) Oligoribonucleotide circularization by ‘template-mediated’ ligation with T4 RNA ligase: synthesis of circular hammerhead ribozymes. Nucleic Acids Res., 26, 2502–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeanmougin F., Thompson,J.D., Gouy,M., Higgins,D.G. and Gibson,T.J. (1998) Multiple sequence alignment with Clustal X. Trends Biochem. Sci., 23, 403–405. [DOI] [PubMed] [Google Scholar]