

Many pathological changes in the epileptic brain increase the propensity for seizures; however, certain biological mechanisms and molecules appear to keep seizures in check in epileptic animals. Inhibitory neurosteroids, molecules generated in glia from circulating steroid hormones and de novo from cholesterol, are candidate molecules for checking seizures because they can enhance inhibitory transmission mediated by GABAA receptors, and have anticonvulsant action (1). Neurosteroids modulate both synaptic and tonic inhibition mediated by GABAA receptors (2,3).
Anticonvulsant action of naturally-occurring neurosteroids was demonstrated soon after their GABAA receptor action was found (4). The potency of anticonvulsant action of neurosteroids is similar to their potency at GABAA receptors (5). Neurosteroids suppress pentylene-tetrazol, kainate and pilocarpine-induced seizures (6). The anticonvulsant action of the steroid hormone progesterone is mediated by conversion to neurosteroids (7-9). Finally, direct infusion of neurosteroids into the brain blocks ictal activity (10).
These studies, however did not explore the role of endogenous neurosteroids in epileptic animals and seizures were imposed externally on non epileptic animals. In some of these studies effects of externally administered neurosteroids was studied. Finally, there is growing evidence that in epileptic animals, GABAA receptors mediating tonic and synaptic inhibition become less sensitive to neurosteroid modulation than controls (2,11-13).
In order to explore the role of endogenous neurosteroids on seizures in epileptic animals, their synthesis was inhibited in female epileptic animals, and seizures were measured using EEG and video monitoring. Animals were studied in three different hormonal states and in each state inhibition of neurosteroid synthesis caused seizure exacerbation.
Finasteride, an inhibitor of the 5α-reductase enzyme was used to block the natural synthesis of neurosteroids (14) in epileptic animals. Previous studies have demonstrated that finasteride does not lower seizure threshold or cause seizures in naïve animals(6,15). We confirmed these findings. Four naïve animals were implanted with EEG electrodes, finasteride (100 mg/kg) was administered after recording EEG for one day and monitoring was continued for 24 hours. No behavioral or electrographic seizure occurred in the 24 hrs following finasteride administration in any animal.
In order to induce epilepsy, female rats underwent status epilepticus induced by lithium injection followed by pilocarpine injection as previously described for male rats(16). A pair of hippocampal and cortical electrodes was implanted 8-12 weeks after status epilepticus induction for EEG recording(16). EEG and video recording were continuously monitored for occurrence of seizures. A cohort of 22 epileptic animals was identified.
Previous studies in non epileptic animals have suggested that withdrawal from high serum progesterone and neurosteroids lowered seizure threshold (15). This was tested in epileptic animals. The effect of inhibition of neurosteroid synthesis in epileptic animals with persistently high progesterone levels was compared to that in animals where progesterone levels were not manipulated. A high serum progesterone state was induced in epileptic animals using a protocol adapted from Reddy(15). The cohort of 22 epileptic animals were randomly divided into two groups of 11 animals each: Each group was monitored with video-EEG without any treatment for the first 3 days and then the first group received Pregnant Mare Serum Gonadotrophin (PMSG) followed two days later by β Human Chorionic Gonadotrophin (β HCG). The second group (control) received saline injections instead.
The mean serum progesterone levels in five epileptic animals obtained randomly in the 10 days subsequent to HCG administration was 40.1 ± 5.6 ng/ml (n=5), which was more than that in control epileptic animals 15.8 ± 3.7 ng/ml (n=4, p < 0.05). The mean seizure frequency of the PMSG, HCG treated group was 6.3 ± 1.2 seizures per day, and that in the control group was 6.05 ± 0.70 seizures per day, (p > 0.05, grouped t-test, figure).
Figure.
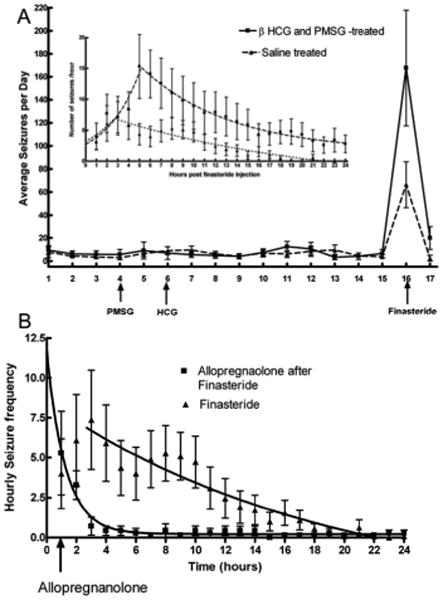
Figure A: Average daily seizure frequency in epileptic animals monitored with video and EEG. Animals treated with PMSG and βHCG (n = 11) were studied in parallel with those treated with saline (n=11). Finasteride caused a marked exacerbation of seizure frequency in both PMSG and HCG-treated and in saline-treated epileptic animals. Inset: The seizure frequency following finasteride administration was plotted on an hourly basis to display the rate of rise and decay of seizure frequency. The lines represent fit to the equation for an exponential curve (Y = start * e(kt)) in both groups of animals. Note gradual increase in seizure frequency and then return to baseline.
Figure B: Hourly seizure frequency in epileptic animals treated with finasteride alone (control animals in panel A) or finasteride followed 1 hour later by 30 mg/Kg allopregnanolone. Note both groups had similar seizure frequency before administration of allopregnanolone, but seizures slowed down and stopped rapidly in the allopregnanolone-treated animals. The lines represent best fit to the equation for an exponential decay and half life of decay was much shorter in allopregnanolone-treated group.
Epileptic animals received 100 mg/kg finasteride on the morning of the 11th day after receiving β HCG or saline. Following finasteride administration in βHCG and PMSG treated animals, the mean serum progesterone level increased to 146.6 ± 46 ng/ml, confirming inhibition of progesterone metabolism. Each animal in the group had more seizures during the 24-hr period following finasteride administration than the average seizure frequency for the previous 14 days and 10 out of 11 animals had more seizures during that period than on any previous day when seizures were recorded. Mean seizure frequency in this group of animals was 167.7 ± 50.3 seizures in the 24-hr period following finasteride administration, a 26-fold increase over the mean seizure frequency for the previous 14 days (Figure, panel A). The mean time interval between finasteride injection and the first seizure was 70 ± 6 minutes. The seizure frequency increased for a period of time after finasteride administration and then diminished down to baseline (Figure, panel A inset).
Surprisingly, finasteride also caused an exacerbation of seizures in saline-injected control animals. There were 66.5 ± 19.9 seizures in the 24-hr period following the administration of finasteride, an 11-fold increase in seizure frequency over baseline (Figure panel A). The mean seizure frequency doubling time after finasteride injection was 147 minutes (Figure, panel A inset). Mean latency to the first seizure was 51 ± 5 minutes, which was shorter than that observed in HCG-treated animals (p < 0.05, n = 11). In 5 control animals, mean seizure duration for the 7 days prior to injection was 45.4 ± 8.9 sec. Duration of all recorded seizures for the day of the finasteride injection in these animals was 38.09 ± 6.7sec (p = 0.521). Thus duration of seizures after finasteride injection was similar to that in the baseline period.
This experiment suggested that inhibition of neurosteroid synthesis caused seizure exacerbation even when progesterone levels were not elevated to a high level, and basal levels of neurosteroid synthesis in the brain may be keeping seizures in check. If finasteride causes seizures by depriving brain of freshly synthesized neurosteroids, then replenishing should treat seizure exacerbation caused by finasteride. This prediction was tested directly.
Seven female rats were made epileptic and monitored for epilepsy. After baseline recording period 100 mg/Kg finasteride was administered. In the hour following finasteride administration seizure frequency increased (5.3 ± 2.6, n =7) and it was similar to untreated control group, described above (4 ± 2.2 n=11, p = 0.37). Animals (n= 7) were then given allopregnanolone 30 mg/kg intraperitoneally and seizure frequency declined very rapidly. In the next two hours seizures frequency in allopregnanolone treated animals was 3.29 ± 0.89 and 0.71 ± 0.56 whereas in untreated control animals (described above) it was 6.09 ± 2.88 and 7.36 ± 3.1 (p < 0.05 for both comparisons). Seizure frequency decay data obtained from allopregnanolone treated and untreated control animals with their best fit to an equation for exponential decay are presented in Figure panel B. Allopregnanolone rapidly terminated finasteride-induced seizures with a half life of 0.87 hr compared to 13.04 hrs in untreated controls.
We tested dose response relationship of finasteride in 5 animals. A lower dose of finasteride 30 mg/Kg, which is 0.5 Log units lower than the original dose was administered to 5 epileptic animals after 10 days of baseline monitoring. In five epileptic animals the baseline daily seizure frequency was 16.82 ± 3.54 and following administration of 30 mg/Kg finasteride it was 46.20 ± 33.85 (p = 0.33). Lower dose of finasteride did not cause a significant seizure exacerbation.
We further confirmed that inhibition of neurosteroid synthesis would cause seizure exacerbation even when animals had low progesterone levels. In a separate experiment in epileptic animals, fluctuations in the progesterone and estrogen levels were eliminated by ovariectomy and the hormones were replaced by progesterone and estrogen pellets placed subcutaneously(17). The mean serum progesterone level in ovariectomized implanted epileptic animals was 13.3 ± 0.9 ng/ml and the mean daily seizure frequency during a 10-day observation period was 3.3 ± 0.7. In the 24-hr period following the administration of 100 mg/kg finasteride, animals experienced 81.5 ± 30.5 seizures in 24 hrs, a 25-fold increase in seizure frequency.
These studies demonstrate that inhibition of neurosteroid synthesis in female epileptic rats caused exacerbation of seizures regardless of preceding progesterone levels. Seizure exacerbation occurred in animals with persistently high progesterone levels, progesterone fixed in a low physiological range and in animals with naturally-cycling progesterone levels. The fluctuations in serum progesterone levels during the rat estrous cycle are well described(18). In the current study, finasteride was administered to animals whose progesterone levels were close to those observed in proestrous (45-55 ng/ml) and those observed in diestrous (5-15 ng/ml), and in both cases seizure exacerbation occurred.
Seizure exacerbation observed in the current study is likely due to reduced neurosteroid levels in the brain. Finasteride blocks the synthesis of neurosteroids such as allopregnanolone and THDOC in the brain in a dose-dependent fashion(14). Other studies demonstrated that finasteride causes serum neurosteroid levels to decline and progesterone levels to rise(15). The breakdown of existing neurosteroids and the decline in brain neurosteroid levels is likely to take some time after finasteride administration. Consistent with this explanation, there was a distinct latent period between finasteride administration and the onset of seizures and a further slow rise to peak seizure frequency. Furthermore, the seizure frequency declined to baseline slowly from a peak likely reflecting the time to replenish neurosteroids in the brain. Finally replenishing allopregnanolone after finasteride administration terminated seizures quickly and lower dose of finasteride did not cause significant seizure exacerbation. Our attempt to measure serum neurosteroid levels was unsuccessful but finasteride administration increased serum progesterone levels consistent with the enzyme inhibition effect of finasteride. Seizure exacerbation as a result of finasteride administration has been reported in one patient with epilepsy(19).
These findings suggest that basal neurosteroid synthesis in the brain help keep seizures in check in epileptic animals. Low concentrations of neurosteroids (10-30 nM) enhance GABA-mediated synaptic and tonic inhibition on hippocampal dentate granule cells(2). However, in epileptic animals, the sensitivity of synaptic GABAA receptors is diminished and higher concentrations (50-100 nM) are necessary to enhance synaptic and tonic currents (3,11-13). This reduction in neurosteroid sensitivity occurs in dentate granule cells, which play a key role in keeping seizures in check(20). The current study in combination with previous studies suggests that reduced neurosteroid sensitivity of GABAA receptors on dentate granule cells creates a window of vulnerability to seizures, allowing seizures to occur when neurosteroids synthesis is inhibited by drugs. Further studies are needed to determine whether natural modulation in brain neurosteroid levels relates to occurrence of “spontaneous” seizures in epileptic animals.
Acknowledgments
This study was supported by NIH grants RO1 NS 040337 and RO1 NS 044370.
Reference List
- 1.Reddy DS. The role of neurosteroids in the pathophysiology and treatment of catamenial epilepsy. Epilepsy Res. 2009 doi: 10.1016/j.eplepsyres.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun C, Mtchedlishvili Z, Erisir A, Kapur J. Diminished neurosteroid sensitivity of synaptic inhibition and altered location of the alpha4 subunit of GABA(A) receptors in an animal model of epilepsy. J Neurosci. 2007;27:12641–50. doi: 10.1523/JNEUROSCI.4141-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABAA receptors. Proc Natl Acad Sci U S A. 2003;100:14439–44. doi: 10.1073/pnas.2435457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belelli D, Bolger MB, Gee KW. Anticonvulsant profile of the progesterone metabolite 5 alpha-pregnan-3 alpha-ol-20-one. Eur J Pharmacol. 1989;166:325–9. doi: 10.1016/0014-2999(89)90077-0. [DOI] [PubMed] [Google Scholar]
- 5.Kokate TG, Svensson BE, Rogawski MA. Anticonvulsant activity of neurosteroids: correlation with gamma-aminobutyric acid-evoked chloride current potentiation. J Pharmacol Exp Ther. 1994;270:1223–9. [PubMed] [Google Scholar]
- 6.Kokate TG, Cohen AL, Karp E, Rogawski MA. Neuroactive steroids protect against pilocarpine- and Kainic acid-induced limbic seizures and status epilepticus in mice. Neuropharmacology. 1996;35:1049–56. doi: 10.1016/s0028-3908(96)00021-4. [DOI] [PubMed] [Google Scholar]
- 7.Kokate TG, Banks MK, Magee T, Yamaguchi S, Rogawski MA. Finasteride, a 5alpha-reductase inhibitor, blocks the anticonvulsant activity of progesterone in mice. Journal of Pharmacology & Experimental Therapeutics. 1999;288:679–84. [PubMed] [Google Scholar]
- 8.Frye CA, Scalise TJ, Bayon LE. Finasteride blocks the reduction in ictal activity produced by exogenous estrous cyclicity. Journal of Neuroendocrinology. 1998;10:291–6. doi: 10.1046/j.1365-2826.1998.00202.x. [DOI] [PubMed] [Google Scholar]
- 9.Reddy DS, Castaneda DC, O'Malley BW, Rogawski MA. Anticonvulsant Activity of Progesterone and Neurosteroids in Progesterone Receptor Knockout Mice. J Pharmacol Exptl Therap. 2004;310:230–9. doi: 10.1124/jpet.104.065268. [DOI] [PubMed] [Google Scholar]
- 10.Frye CA, Manjarrez J, Camacho-Arroyo I. Infusion of 3alpha,5alpha-THP to the pontine reticular formation attenuates PTZ-induced seizures. Brain Res. 2000;881:98–102. doi: 10.1016/s0006-8993(00)02897-3. [DOI] [PubMed] [Google Scholar]
- 11.Mtchedlishvili Z, Bertram EH, Kapur J. Diminished allopregnanolone enhancement of GABA(A) receptor currents in a rat model of chronic temporal lobe epilepsy. J Physiol. 2001;537:453–65. doi: 10.1111/j.1469-7793.2001.00453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng Z, Huang CS, Stell BM, Mody I, Houser CR. Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci. 2004;24:8629–39. doi: 10.1523/JNEUROSCI.2877-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang N, Wei W, Mody I, Houser CR. Altered Localization of GABAA Receptor Subunits on Dentate Granule Cell Dendrites Influences Tonic and Phasic Inhibition in a Mouse Model of Epilepsy. J Neurosci. 2007;27:7520–31. doi: 10.1523/JNEUROSCI.1555-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mukai Y, Higashi T, Nagura Y, Shimada K. Studies on neurosteroids XXV. Influence of a 5alpha-reductase inhibitor, finasteride, on rat brain neurosteroid levels and metabolism. Biol Pharm Bull. 2008;31:1646–50. doi: 10.1248/bpb.31.1646. [DOI] [PubMed] [Google Scholar]
- 15.Reddy DS, Kim HY, Rogawski MA. Neurosteroid withdrawal model of perimenstrual catamenial epilepsy. Epilepsia. 2001;42:328–36. doi: 10.1046/j.1528-1157.2001.10100.x. [DOI] [PubMed] [Google Scholar]
- 16.Martin BS, Kapur J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia. 2008;49:248–55. doi: 10.1111/j.1528-1167.2007.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan SD, Moenter SM. GABAergic integration of progesterone and androgen feedback to gonadotropin-releasing hormone neurons. Biol Reprod. 2005;72:33–41. doi: 10.1095/biolreprod.104.033126. [DOI] [PubMed] [Google Scholar]
- 18.Freeman ME. Neuroendocrine control of the ovarian cycle of the rat. In: Neill JD, editor. Knobil and Neil's Physiology of Reproduction. Elsevier; 2006. pp. 2327–88. [Google Scholar]
- 19.Herzog AG, Frye CA. Seizure exacerbation associated with inhibition of progesterone metabolism. Ann Neurol. 2003;53:390–1. doi: 10.1002/ana.10508. [DOI] [PubMed] [Google Scholar]
- 20.Lothman EW, Stringer JL, Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl. 1992;7:301–13. [PubMed] [Google Scholar]