

Abstract
Intrinsically disordered (ID) proteins which lack stable secondary and tertiary structure in substantial regions (or throughout) are prevalent in eukaryotes. They exist as ensembles of rapidly fluctuating structures and many undergo coupled folding and binding reactions. Because ID proteins are overrepresented in major disease pathways they are desirable targets for inhibition, however, the feasibility of targeting proteins without defined structures was unclear. Recently, small molecules have been found which bind to the disordered regions of c-Myc, Aβ, EWS-Fli1, and various peptides. As with structured targets, initial hits were further optimized to increase specificity and affinity. Given the number and biological importance of ID proteins, the ability to inhibit their interactions opens tremendous potential in chemical biology and drug discovery.
Introduction
The large majority of drugs target enzymes or cell-surface receptors where small molecules can mimic many of the contacts and interactions of the natural substrate and modulate the protein function [1]. In the past several years, there has been much progress in understanding and inhibiting protein-protein interactions [2,3]. A strong rationale for the expansion of efforts to target protein-protein interactions came from the understanding that the energy of protein-protein interactions was not distributed over the often large contact area in these interactions but rather may be concentrated in regions whose area better matched the surface that may be contacted by a small molecule [4]. These interactions have emerged as viable systems for inhibition by small molecules and as potential drug targets.
In this same period there has been a large expansion of the study and understanding of intrinsically disordered proteins (also known as intrinsically unstructured or natively disordered) [5-7]. Intrinsically disordered (ID) proteins do not have stable secondary or tertiary structure in substantial regions or throughout their sequence. Instead, they exist as an ensemble of rapidly interconverting structures. The prevalence of ID regions increases with increasing complexity in organisms and ID proteins have a wide range of functions that involve various degrees of disorder. Some functions involve coupled folding and binding interactions with partners while other interactions involve remaining essentially disordered, and still others require an intermediate, dynamic state [8,9]. While binding of a small molecule to a disordered protein may seem counterintuitive, the range of intermolecular interactions demonstrated with ID regions makes their ability to generate an adaptable, specific interface for small molecule binding less unexpected. This review will highlight the desirability of targeting ID regions and will discuss several recent examples of small molecules binding to ID proteins.
Why ID proteins make good targets
The foremost reason to target ID proteins is their importance and over-representation in signaling and major disease pathways. Regions of disorder are found to be abundant in proteins associated with signaling, cancer, cardiovascular disease, neurodegenerative disease, and diabetes (Figure 1). The term “disorder in disorders” has been used to emphasize this prevalence of disordered regions within proteins associated with a wide ranges diseases [10]. A feature important to ID protein function is coupled binding and folding [11]. Many ID proteins become structured upon binding to a partner with the energy from specific interactions compensating for the entropic penalty from ordering [12]. These interactions are characterized by high specificity with low affinity which can be desirable in responsive interaction networks were reversible binding is critical.
Figure 1.
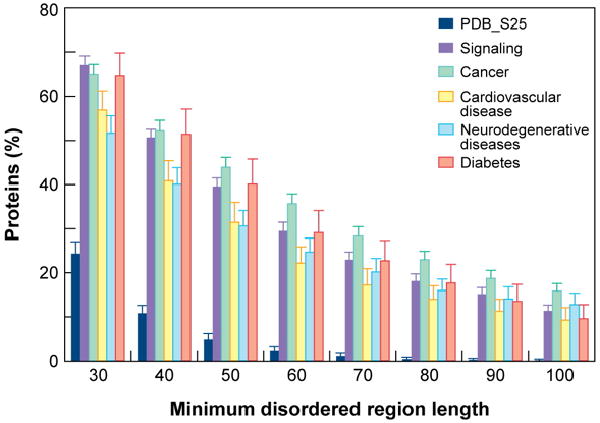
Prevalence of disordered regions in disease-associated proteins. Proteins associated with signaling and the indicated diseases show a much greater abundance of extended regions of disorder than eukaryotic proteins from Swiss-Prot and PDB (PDB_S25). Figure reproduced from reference [10].
Approximately 80% of yeast genes are not harmful if overexpressed, however those which are detrimental tend to be enriched in ID regions. The ID proteins are particularly prone to modulation by their concentration as many bind to multiple partners in dynamic interactions with modest affinities. Since the interactions are controlled by protein concentration, at high concentrations the force of mass-action can overcome the specificity of the interactions (the difference in energy between desired and undesired binding interactions) leading to additional binding [13], thus providing an explanation for the toxicity. In line with this, the concentrations of ID proteins on a proteome scale are tightly regulated by transcript clearance, translation rate, and protein degradation [14]. The interactions of ID proteins with their partners, and the final output of that interaction, will likely be sensitive to modulation by binding of small molecules that can affect their affinity for binding partners.
Because of the absence of a stable three-dimensional structure, the primary sequence is truly the determinant of the ID binding sites. Disordered regions can be predicted with approximately 90% accuracy [15]. Within these regions the sequences that undergo binding interactions, molecular recognition features (MoRFs), have been analyzed based on the type of conformation adopted in their complex [16], and predictors of MoRFs have been developed [17]. MoRFs have been proposed in drug design where they would be used to identify structured binding partners followed by determination of the complex structure. The MoRF would then be substituted by a small molecule [17]. This strategy is complementary to one considered here. Predictions of binding regions in disordered proteins have been carried out on multiple proteomes and their abundance is found to increase with increasing organism complexity [18]. The likely strong overlap in protein interaction sites and small molecule binding sites in ID proteins means there are already tools to predict and compare potential small-molecule binding sites on a proteome scale.
Targeting disordered c-Myc monomers
The c-Myc oncoprotein (hereafter referred to as Myc) is a transcription factor which regulates large numbers of genes important in key cellular processes such as growth, differentiation, metabolism, and apoptosis [19-21]. Myc's function requires association with its partner Max through a basic-helix-loop-helix-leucine zipper (bHLHZip) domain in each protein [22,23]. The individual monomers are disordered and undergo coupled folding and binding upon dimerization (Figure 2a). Myc overexpression occurs in most human cancers [21,24] and the feasibility of targeting Myc was recently demonstrated in a mouse model [25]. Inhibition of its function has been sought by a variety of biochemical strategies (dominant negatives, antisense, and siRNA among others) [26-28]. Despite the difficulties of targeting protein-protein interactions, the potential of targeting Myc in cancer therapy led multiple groups to search for small molecules that could inhibit the Myc-Max interaction. After many of the inhibitors were discovered, several small molecules were demonstrated to be unfolding the Myc-Max dimer and specifically binding to the monomeric, disordered HLHZip region [29,30]. The number of small molecules able to target Myc, the demonstration that the extensive (3206 Å2) [23] Myc-Max protein interface could be disrupted by a small molecule binding to one disordered protein partner, and several other recent examples of small molecules binding to short unstructured peptide segments, have clearly established the feasibility of using small molecules to target ID proteins. Unique to the Myc binding molecules is their ability to force their target protein from a highly structured heterodimer into a monomeric, disordered form in order to bind (Figure 2). Examining the range of strategies, techniques, and outcomes used by multiple groups in finding and analyzing inhibitors of Myc provides useful insight into small molecule-ID protein interactions. The presumption is that all molecules that disrupt the Myc-Max dimer also bind to disordered monomers (either Myc, Max, or both) but that has not been demonstrated in all cases.
Figure 2.
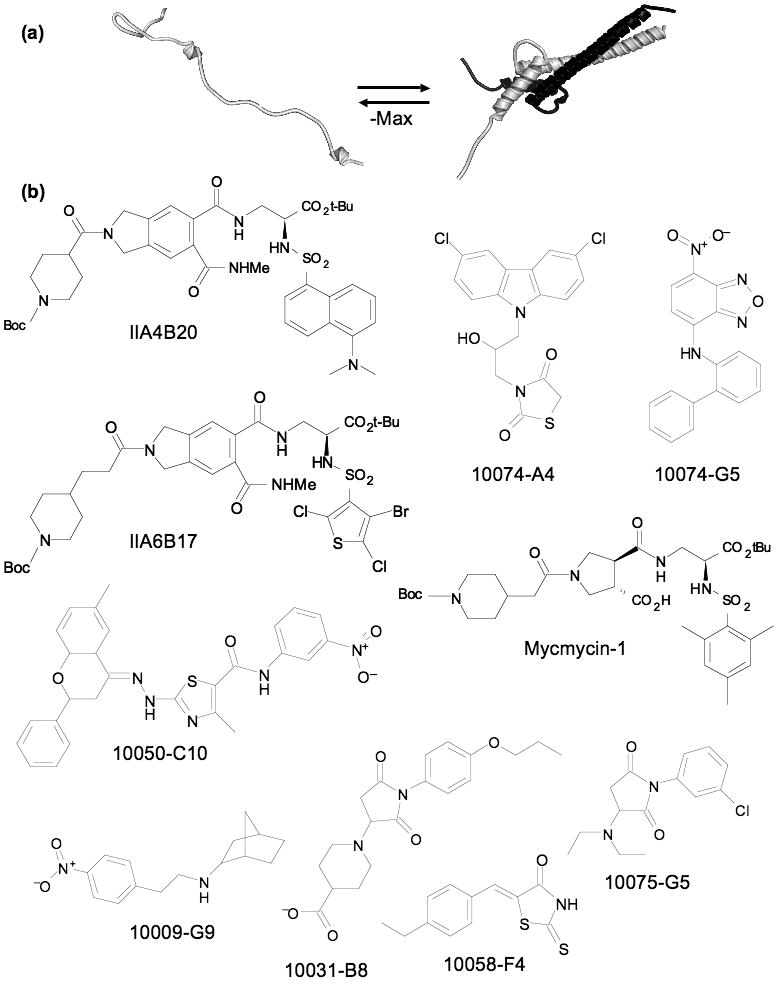
Targeting Myc-Max dimerization: (a) cartoon of Myc-Max dimerization, binding of small molecules shifts the equilibrium to the left. (b) Small molecule inhibitors of Myc-Max dimerization from combinatorial and diversity libraries.
The first small molecule inhibitors of Myc-Max dimerization were found by screening 7000 compounds from combinatorial libraries. The initial screen yielded four related molecules from a peptidomemetic library [31]. Two of the molecules, IIA4B20 and IIA6B17 (Figure 2), showed inhibition of Myc dependent cell growth. Both of these compounds also showed activity against cells transformed with Jun. Recently another combinatorial library was assembled using the same substituents as the original but built from a racemic, trans-3, 4 dicarboxylic acid core. Two molecules, mycmycin-1 and 2, were found that effectively inhibited Myc dependent cell growth; mycmycin-1 (Figure 2) also showed strong inhibition of Myc-Max dimerization [32]. Neither mycmycin inhibited Jun dependent cell growth. This gain in specificity demonstrated an important aspect of ID proteins as targets: although they may interact with a variety of molecules, a subset of these molecules can display high specificity for the protein target.
The ability of ID proteins to bind diverse molecules was made evident by the discovery of a variety of inhibitors from a 10,000 member diversity library screened using a yeast two-hybrid system to detect disruption of Myc-Max dimers [33]. The compounds were simultaneously tested for disruption of Id2-E47 HLH protein dimers. A set of structurally varied inhibitors were found: seven specific for Myc-Max, ten specific for Id2-E47, and 28 termed dual specific which inhibited both. Compounds were further tested against 32 additional HLH, HLHZip, or bZip interaction pairs. Compounds that disrupted only one of the original targets averaged less than one additional strong interaction, whereas the dual specific inhibitors tested averaged over three. The study showed that small molecules can readily be found that interact with ID proteins, but a majority of these molecules are likely to interact with additional, related proteins. Synthesis and screening of analogs of one of the parent compounds (10058-F4) led to modest increases in potency with several sites tolerating a wide range of groups, a potential general feature of these flexible complexes [34]. A set of these molecules was used to generate a pharmacophore model for 10058-F4 that was able to successfully predict additional inhibitory molecules with diverse structures [35]. The addition of 10058-F4 to Myc-Max dimers caused reversion to dissociated, disordered monomers with circular dichroism (CD) spectra that matched separated monomers [29]. Inhibitors that increase the flexibility of their binding sites are receiving increased attention as some molecules, such BCR-ABL inhibitors, appear to work in part by increasing backbone dynamics in some regions of their binding site [36,37]. In this context, Myc inhibitors can be seen as extreme allosteric effectors [38].
Screening of additional combinatorial libraries, diversity libraries, and natural products all yielded Myc-Max dimerization inhibitors. A 285 compound “credit card” library specifically designed to target protein-protein interactions was tested [39]. The molecules were screened for their ability to disrupt Myc-Max dimerization, inhibit specific DNA binding, and inhibit oncogenic transformation. Two compounds, NY2267 and NY2280 (Figure 3), were active in each assay. In transcriptional assays the selected compounds reduced Myc dependent transcription but also showed similar inhibition of a c-Jun dependent transcript. The screening of diversity libraries identified Mycro1 and Mycro2 (Figure 3) as inhibitors of Myc-Max DNA binding and transcription from a Myc reporter gene [40]. However, inhibition of Max-Max DNA binding and repression of transcription from an AP-1 dependent reporter was also seen. Screening >1400 Mycro derivatives identified Mycro3 which showed strong suppression of Myc based transcription with no inhibition against AP-1 [41]. Dihydroxycapnellene (Figure 3), a sesquiterpene isolated from soft coral, exhibited antiproliferative activity against cancer cells and disrupted Myc-Max interaction in a yeast two-hybrid assay [42]. Given the range of structures demonstrated to bind to Myc, it would have been surprising if natural products with similar activity were not found. Several groups have also isolated molecules that interfered with Myc activity but did not disrupt dimerization [43-45].
Figure 3.
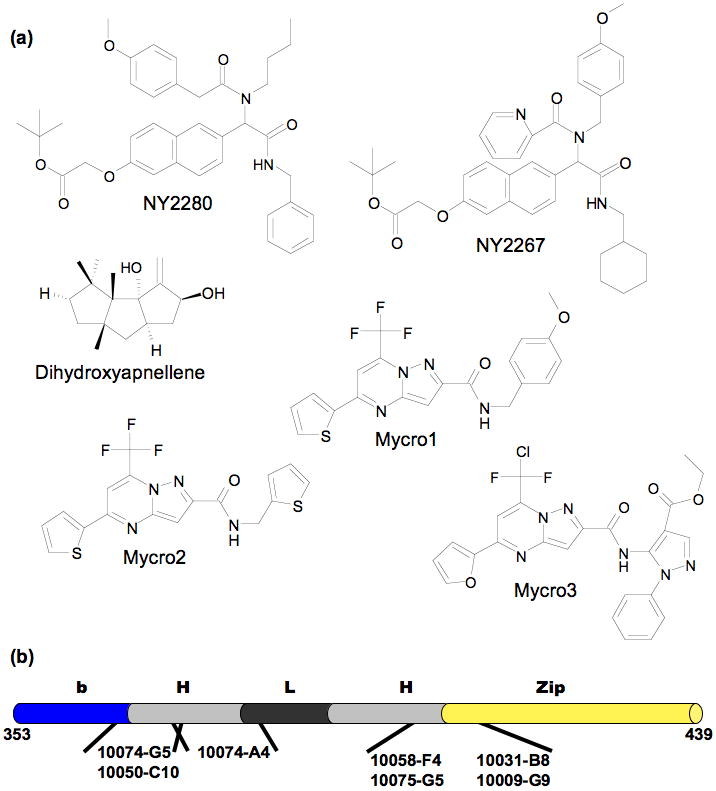
Additional Myc-Max dimerization inhibitors: (a) “credit-card” molecules, natural product, and Mycro series. (b) Known binding sites localized on the Myc bHLHZip; each binding site can be occupied independently and simultaneously.
Since there are no ‘normal’ binding sites on ID proteins, the whole region whose binding is inhibited serves as the potential binding site. For Myc-Max dimer disruption, the binding site could be located anywhere along the HLHZip region of either partner in the dimer. The Myc-Max inhibitors identified by Prochownik and coworkers have all been shown to bind to monomeric Myc [30]. Initially, the intrinsic fluorescence of 10058-F4 and another parent compound 10074-G5 were used to measure directly their binding to Myc. Using point mutants and deletions of Myc, two distinct binding sites were located [29]. These were confirmed by CD and NMR analysis using short, synthetic peptides comprising just the individual binding regions. Each of the remaining original seven inhibitors was tested for competition with 10074-G5 or 10058-F4 (Figure 3). All except 10074-A4 bound to one of the above sites. CD changes in Myc fragments and changes in backbone chemical shifts were used to identify 10074-A4's site as immediately adjacent to the 10074-G5 site. Inhibitors can simultaneously occupy each of the three sites with no detected changes in their affinity [30]. The identification of three separate binding sites within only 85 residues on Myc is a potential indicator of the prevalence in recognition regions of ID sequences that can support small molecule binding. Although the NMR data indicated that the complexes are rigidified in the binding region relative to the uncomplexed form, there is still substantial flexibility. The development and use of computational methods that explicitly deal with flexibility in ligand binding is increasing dramatically [46]. How well cases of very high flexibility, including backbone motion, can be dealt with is uncertain.
Substrate targeting and unbiased screening
Results with small-molecule γ-secretase modulators (GSMs) provide an example of the inversion of the typical paradigm: these inhibitors bind to a disordered substrate rather than an enzyme active site. The γ-secretase complex mediates cleavage of the amyloid-B peptide (Aβ) from the amyloid precursor protein (APP). When Kukar, Golde, and coworkers probed binding with biotin tagged photoactivated GSMs (Fen-B and Fluri-BpB, Figure 4), they found binding occurred on the APP substrate rather than the enzyme target [47]. Using various truncations, the binding sites of both molecules was isolated to residues 29-36 of the soluble, presumably disordered [48,49], Aβ peptide. Labeling of the APP was competed by a minimal peptide Aβ28-36 confirming that this short sequence was capable of specific binding to the molecules. These molecules represent substrate targeted inhibitors (also called molecular clamps) which regulate enzyme activity by binding to and blocking a specific substrate leaving an enzyme free to act on other substrates [50,51]. Sites of post-translational modification, such as proteolysis, are prevalent in ID regions [52], making these a large class of targets for small molecules that bind ID regions.
Figure 4.
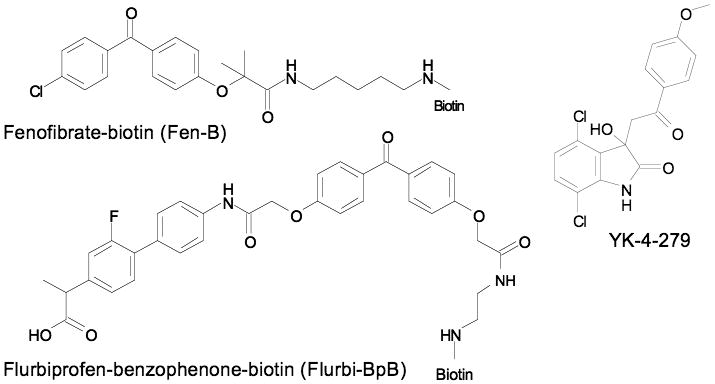
Substrate targeted γ-secretase inhibitors and EWS-FLI1 binding molecule (YK-4-279.
The oncogenic fusion protein EWS-FLI1 contains a structured Ets domain but is predominantly disordered outside that region [53]. Using SPR, a library of 3000 molecules was screened for binding to immobilized EWS-FLI1 [54]. An initial hit was identified (NSC635437) and optimized to yield YK-4-279 (Figure 4). The optimized molecule's affinity for its target (10 μM) was similar to those of many Myc inhibitors. The interaction of RNA helicase A with EWS-FLI1 was inhibited by YK-4-279 and was the presumed mechanism by which the small molecule inhibited EWS-FLI1 function in vitro and in vivo. The location of the YK-4-279 binding site and whether it is fully disordered is not known. However, the results show that small molecule binding to a largely disordered, full-length, protein can be directly detected via SPR and that binding of the small molecule occurs at a protein-protein interaction site despite the initial screen not being biased in any way.
A variety of unbiased screens have also been conducted in the opposite sense where short random (presumably disordered) peptides are displayed on phage and screened for binding to a particular small molecule [55]. In two separate experiments, immobilized paclitaxel captured consensus sequences on phage. In one case the peptide resembled a sequence in the disordered loop region of Bcl-2 and paclitaxel was subsequently shown to bind to recombinant Bcl-2 and increase the amount of structure in the protein [56]. The other consensus sequence mapped to NFX1 and paclitaxel binding was confirmed by pull-down assays and SPR [57]. Using phage display, in conjunction with SPR and quartz crystal microbalance (QCM) measurements, peptides capable of binding camptothecin, anti-tumor molecule NK109, and a trimannoside were also found [58-60].
There are likely other cases of small molecules binding to disordered regions but the specific binding or the protein state is unconfirmed. Among these, there are many involving amyloidogenic proteins [61]. Congo red has recently been shown to act in a detergent-like manner [62], and other small molecule amyloid inhibitors were found to act via colloidal aggregates [63]. Improvements in methods to monitor protein-protein and protein-small molecule interactions along with the knowledge that ID proteins can be the target for specific small-molecule binding will likely lead to increased examples of small molecule-ID protein interactions through confirmation of known interactions and discovery of novel ones.
Summary and Opinion
A question that arises in considering small molecules directed against ID proteins is whether the binding can truly be specific. There are two perspectives on this question. First, how many small molecules can a particular ID recognition sequence bind? Second, to how many other proteins can a small molecule that binds an ID sequence also bind? The malleability that ID proteins tend to display in binding to multiple, specific protein targets [64] is recapitulated in their interaction with small molecules. Presumably, the conformational flexibility of the peptide recognition element allows the plasticity to bind molecules with divergent structures with similar affinity. Some of the molecules selected for Myc binding also bound other transcription factors, however further optimization allowed the Myc binding to be selected. Thus it appears that ID proteins do bind to a large range of small molecules. This promiscuous binding is a major advantage in that finding an initial binding compound may be relatively simple. The affinity and especially the specificity of the compound can then be enhanced in standard molecular optimization procedures.
On the second part of the question, molecules that bind to ID sequences with very high specificity (based on screening a series of closely related proteins) have been found. However, finding a molecule from a screen with only partial selectivity for a given ID target is more likely. Counter screening at the library level or on selected compounds is a critical aspect of finding specific ID protein binding molecules. A recent study found that for 802 drugs, each one interacted with six different targets on average [65]. Thus even known, targeted drugs may not be as selective as was once believed. Most of the molecules that have been selected for ID protein binding come from standard ‘drug-like’ libraries or conform to similar constraints. The question can thus be reversed: whether one is targeting an ID protein or not, the potential for a given molecule to bind off-target sequences that are ID has to accepted.
When binding to small molecules, ID proteins seem to behave as they do when binding to partner proteins: they interact using specific segments of sequence (MoRFs or similar), they form various complexes with the same sequence binding to multiple targets, and they bind with high specificity but low affinity. Generating large increases in affinity will be a challenge in the further development of small molecule binders of ID proteins. While the interactions the molecules are competing with may also be low-affinity, tighter binding is desirable. In the case of Myc, the presence of multiple binding sites within even the compact bHLHZip region may provide substantial gains in affinity through the use of bivalent molecules.
Acknowledgments
The author would like to thank E. V. Prochownik for comments on the manuscript. This work was supported by a grant from NIH (1R01CA140624).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5:821–834. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- 2.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 3.Yin H, Hamilton AD. Strategies for targeting protein-protein interactions with synthetic agents. Angew Chem-Int Edit. 2005;44:4130–4163. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]
- 4.Clackson T, Wells JA. A Hot-Spot of Binding-Energy in a Hormone-Receptor Interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 5.Dunker AK, Silman I, Uversky VN, Sussman JL. Function and structure of inherently disordered proteins. Curr Opin Struct Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Eliezer D. Biophysical characterization of intrinsically disordered proteins. Curr Opin Struct Biol. 2009;19:23–30. doi: 10.1016/j.sbi.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This review provides an up to date and broad synopsis of developments in the field of intrinsically disordered proteins.
- 8.Mittag T, Kay LE, Forman-Kay JD. Protein dynamics and conformational disorder in molecular recognition. J Mol Recognit. 2010;23:105–116. doi: 10.1002/jmr.961. [DOI] [PubMed] [Google Scholar]
- 9.Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 11.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 13.Vavouri T, Semple JI, Garcia-Verdugo R, Lehner B. Intrinsic Protein Disorder and Interaction Promiscuity Are Widely Associated with Dosage Sensitivity. Cell. 2009;138:198–208. doi: 10.1016/j.cell.2009.04.029. [DOI] [PubMed] [Google Scholar]; • The authors use first principles (mass action) and correlations to support their theory that ID proteins are dosage sensitive due to their tendency to engage in promiscuous interactions at high concentrations.
- 14.Gsponer J, Futschik ME, Teichmann SA, Babu MM. Tight Regulation of Unstructured Proteins: From Transcript Synthesis to Protein Degradation. Science. 2008;322:1365–1368. doi: 10.1126/science.1163581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He B, Wang KJ, Liu YL, Xue B, Uversky VN, Dunker AK. Predicting intrinsic disorder in proteins: an overview. Cell Research. 2009;19:929–949. doi: 10.1038/cr.2009.87. [DOI] [PubMed] [Google Scholar]
- 16.Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, Dunker AK. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res. 2007;6:2351–2366. doi: 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng YG, Oldfield CJ, Meng JW, Romero P, Uversky VN, Dunker AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007;46:13468–13477. doi: 10.1021/bi7012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meszaros B, Simon I, Dosztanyi Z. Prediction of Protein Binding Regions in Disordered Proteins. PLos Computational Biology. 2009;5:e1000376. doi: 10.1371/journal.pcbi.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adhikary S, Eilers M. Transcriptional regulation and transformation by MYC proteins. Nat Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 20.Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci USA. 2000;97:3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 23.Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- 24.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 25.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–683. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ponzielli R, Katz S, Barsyte-Lovejoy D, Penn LZ. Cancer therapeutics: targeting the dark side of Myc. Eur J Cancer. 2005;41:2485–2501. doi: 10.1016/j.ejca.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 27.Prochownik EV. c-Myc as a therapeutic target in cancer. Expert Rev Anticancer Ther. 2004;4:289–302. doi: 10.1586/14737140.4.2.289. [DOI] [PubMed] [Google Scholar]
- 28.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Sem Cancer Biol. 2006;16:318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 29.Follis AV, Hammoudeh DI, Wang HB, Prochownik EV, Metallo SJ. Structural Rationale for the Coupled Binding and Unfolding of the c-Myc Oncoprotein by Small Molecules. Chem Biol. 2008;15:1149–1155. doi: 10.1016/j.chembiol.2008.09.011. [DOI] [PubMed] [Google Scholar]; •• This paper was the first to define specific small molecule binding regions on Myc and to provide a model for how small molecules were recognized by the protein. Circular dichroism data indicated that Myc shifts to a globally disordered conformation upon inhibitor binding which is incompatible with dimerization to Max.
- 30.Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc. 2009;131:7390–7401. doi: 10.1021/ja900616b. [DOI] [PubMed] [Google Scholar]; • This work describes the discovery a third binding site along the HLHZip of Myc and the ability of Myc to bind to compounds at all three sites simultaneously and independently.
- 31.Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL, Vogt PK. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc Nat Acad Sci USA. 2002;99:3830–3835. doi: 10.1073/pnas.062036999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi J, Stover JS, Whitby LR, Vogt PK, Boger DL. Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg Med Chem Let. 2009;19:6038–6041. doi: 10.1016/j.bmcl.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; • The authors provide an important proof of principle in that a promiscuous initial structure was converted into a specific binder of Myc demonstrating that specificity for ID proteins does arises from the particular array of interacting groups on the molecule.
- 33.Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22:6151–6159. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, Prochownik EV. Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther. 2007;6:2399–2408. doi: 10.1158/1535-7163.MCT-07-0005. [DOI] [PubMed] [Google Scholar]
- 35.Mustata G, Follis AV, Hammoudeh DI, Metallo SJ, Wang HB, Prochownik EV, Lazo JS, Bahar I. Discovery of Novel Myc-Max Heterodimer Disruptors with a Three-Dimensional Pharmacophore Model. J Med Chem. 2009;52:1247–1250. doi: 10.1021/jm801278g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crespo A, Fernandez A. Induced disorder in protein-ligand complexes as a drug-design strategy. Mol Pharm. 2008;5:430–437. doi: 10.1021/mp700148h. [DOI] [PubMed] [Google Scholar]
- 37.Lee GM, Craik CS. Trapping Moving Targets with Small Molecules. Science. 2009;324:213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci U S A. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Y, Shi J, Yamamoto N, Moss JA, Vogt PK, Janda KD. A credit-card library approach for disrupting protein-protein interactions. Bioorg Med Chem. 2006;14:2660–2673. doi: 10.1016/j.bmc.2005.11.052. [DOI] [PubMed] [Google Scholar]
- 40.Kiessling A, Sperl B, Hollis A, Eick D, Berg T. Selective inhibition of c-Myc/Max dimerization and DNA binding by small molecules. Chem Biol. 2006;13:745–751. doi: 10.1016/j.chembiol.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 41.Kiessling A, Wiesinger R, Sperl B, Berg T. Selective inhibition of c-Myc/Max dimerization by a pyrazolo[1,5-a]-pyrimidine. ChemMedChem. 2007;2:627–630. doi: 10.1002/cmdc.200600294. [DOI] [PubMed] [Google Scholar]
- 42.Grote D, Hanel F, Dahse HM, Seifert K. Capnellenes from the soft coral Dendronephthya rubeola. Chemistry & Biodiversity. 2008;5:1683–1693. doi: 10.1002/cbdv.200890157. [DOI] [PubMed] [Google Scholar]
- 43.Jiang H, Bower KE, Beuscher AE, Zhou B, Bobkov AA, Olson AJ, Vogt PK. Stabilizers of the Max Homodimer Identified in Virtual Ligand Screening Inhibit Myc Function. Mol Pharmacol. 2009;76:491–502. doi: 10.1124/mol.109.054858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mo H, Henriksson M. Identification of small molecules that induce apoptosis in a Myc-dependent manner and inhibit Myc-driven transformation. Proc Natl Acad Sci USA. 2006;103:6344–6349. doi: 10.1073/pnas.0601418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mo H, Vita M, Crespin M, Henriksson M. Myc overexpression enhances apoptosis induced by small molecules. Cell Cycle. 2006;5:2191–2194. doi: 10.4161/cc.5.19.3320. [DOI] [PubMed] [Google Scholar]
- 46.Cozzini P, Kellogg GE, Spyrakis F, Abraham DJ, Costantino G, Emerson A, Fanelli F, Gohlke H, Kuhn LA, Morris GM, et al. Target Flexibility: An Emerging Consideration in Drug Discovery and Design. J Med Chem. 2008;51:6237–6255. doi: 10.1021/jm800562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, et al. Substrate-targeting gamma-secretase modulators. Nature. 2008;453:925–929. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper elegantly demonstrates substrate targeting of the Aβ peptide with small-molecule binding localized to short peptide segment. It shows that molecules may preferentially bind to disordered targets over the more commonly observed enzyme active site. The authors provide evidence that a range of secretase modulators are targeting the substrate.
- 48.Tompa P. Structural disorder in amyloid fibrils: its implication in dynamic interactions of proteins. FEBS J. 2009;276:5406–5415. doi: 10.1111/j.1742-4658.2009.07250.x. [DOI] [PubMed] [Google Scholar]
- 49.Uversky VN. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front Biosci. 2009;14:5188–5238. doi: 10.2741/3594. [DOI] [PubMed] [Google Scholar]
- 50.Kodadek T. Inhibition of proteolysis and other posttranslational modifications with substrate-targeted inhibitors. Biopolymers. 2002;66:134–140. doi: 10.1002/bip.10233. [DOI] [PubMed] [Google Scholar]
- 51.Weiss ST, McIntyre NR, McLaughlin ML, Merkler DJ. The development of molecular clamps as drugs. Drug Discovery Today. 2006;11:819–824. doi: 10.1016/j.drudis.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 52.Xie HB, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uren A, Tcherkasskaya O, Toretsky JA. Recombinant EWS-FLI1 oncoprotein activates transcription. Biochemistry. 2004;43:13579–13589. doi: 10.1021/bi048776q. [DOI] [PubMed] [Google Scholar]
- 54.Erkizan HV, Kong YL, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan LS, Abaan OD, Chou TH, Dakshanamurthy S, Brown ML, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma. Nature Medicine. 2009;15:750–758. doi: 10.1038/nm.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper demonstrates that in a completely unbiased screen a small molecule can be found that targets a disordered protein and that the binding site was important for the physiological function of the molecule. Further, targeting of the ID protein was demonstrated in animal model of cancer.
- 55.Rodi DJ, Agoston GE, Manon R, Lapcevich R, Green SJ, Makowski L. Identification of small molecule binding sites within proteins using phage display technology. Comb Chem High Throughput Screen. 2001;4:553–572. doi: 10.2174/1386207013330779. [DOI] [PubMed] [Google Scholar]
- 56.Rodi DJ, Janes RW, Sanganee HJ, Holton RA, Wallace BA, Makowski L. Screening of a library of phage-displayed peptides identifies human bcl-2 as a taxol-binding protein. J Mol Biol. 1999;285:197–203. doi: 10.1006/jmbi.1998.2303. [DOI] [PubMed] [Google Scholar]
- 57.Aoki S, Morohashi K, Sunoki T, Kuramochi K, Kobayashi S, Sugawara F. Screening of paclitaxel-binding molecules from a library of random peptides displayed on t7 phage particles using paclitaxel-photoimmobilized resin. Bioconjugate Chem. 2007;18:1981–1986. doi: 10.1021/bc700287v. [DOI] [PubMed] [Google Scholar]
- 58.Morohashi K, Yoshino A, Yoshimori A, Saito S, Tanuma S, Sakaguchi K, Sugawara F. Identification of a drug target motif: an anti-tumor drug NK109 interacts with a PNxxxxP. Biochem Pharmacol. 2005;70:37–46. doi: 10.1016/j.bcp.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 59.Nishiyama K, Takakusagi Y, Kusayanagi T, Matsumoto Y, Habu S, Kuramochi K, Sugawara F, Sakaguchi K, Takahashi H, Natsugari H, et al. Identification of trimannoside-recognizing peptide sequences from a T7 phage display screen using a QCM device. Bioorg Med Chem. 2009;17:195–202. doi: 10.1016/j.bmc.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 60.Takakusagi Y, Kobayashi S, Sugawara F. Camptothecin binds to a synthetic peptide identified by a T7 phage display screen. Bioorg Med Chem Lett. 2005;15:4850–4853. doi: 10.1016/j.bmcl.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 61.Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, Hisanaga S, Goedert M, Hasegawa M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry. 2006;45:6085–6094. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- 62.Lendel C, Bolognesi B, Wahlstrom A, Dobson CM, Graslund A. Detergent-like Interaction of Congo Red with the Amyloid beta Peptide. Biochemistry. 2010;49:1358–1360. doi: 10.1021/bi902005t. [DOI] [PubMed] [Google Scholar]
- 63.Feng BY, Toyama BH, Wille H, Colby DW, Collins SR, May BCH, Prusiner SB, Weissman J, Shoichet BK. Small-molecule aggregates inhibit amyloid polymerization. Nat Chem Biol. 2008;4:197–199. doi: 10.1038/nchembio.65. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This paper provides an important demonstration that apparent binding of small molecules to disordered regions, especially those prone to aggregation, may not correlate to specific complex formation and wariness is warranted in examining such binding data.
- 64.Fuxreiter M, Tompa P, Simon I, Uversky VN, Hansen JC, Asturias FJ. Malleable machines take shape in eukaryotic transcriptional regulation. Nat Chem Biol. 2008;4:728–737. doi: 10.1038/nchembio.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mestres J, Gregori-Puigjane E, Valverde S, Sole RV. The topology of drug-target interaction networks: implicit dependence on drug properties and target families. Molecular BioSystems. 2009;5:1051–1057. doi: 10.1039/b905821b. [DOI] [PubMed] [Google Scholar]