

Introduction
Malaria is the best known protozoal disease. It is one of the most common infectious diseases in at least 100 tropical and subtropical countries in Africa, Southeast Asia, and South America.1 Worldwide, malaria infects 300–600 million people and kills about three million in a year. While the most powerful general weapon against malaria would be a long-lasting vaccine, the failure of some vaccine development indicates that a highly active vaccine is a long way from reality.2–4 The increasing prevalence of multiple drug resistant strains in most malaria endemic areas has significantly reduced the efficacy of current anti-malarial drugs for prophylaxis and treatment of this disease.5 Therefore, medicinal agents based on novel mode of action are required to overcome the emergence of resistance and to control an ever-increasing number of epidemics caused by the malaria parasite.
Febrifugine (1) was isolated as the active components against malaria in the Chinese herb Chang Shan (Dichroa febrifuga Lour),6, 7 which has been employed by the local people as medicine against fevers caused by malaria parasites for a long time. Febrifugine acts by impairing haemazoin formation required for maturation of the parasite at the trophozoite stage.8 The use of febrifugine as anti-malarial agent is initially appealing not only because of its rapid effect and no drug resistance, but also because of its availability. Subsequent pre-clinical researches have found that febrifugine possesses adverse side effects. Strong liver toxicity has precluded febrifugine as a clinical drug.9–11
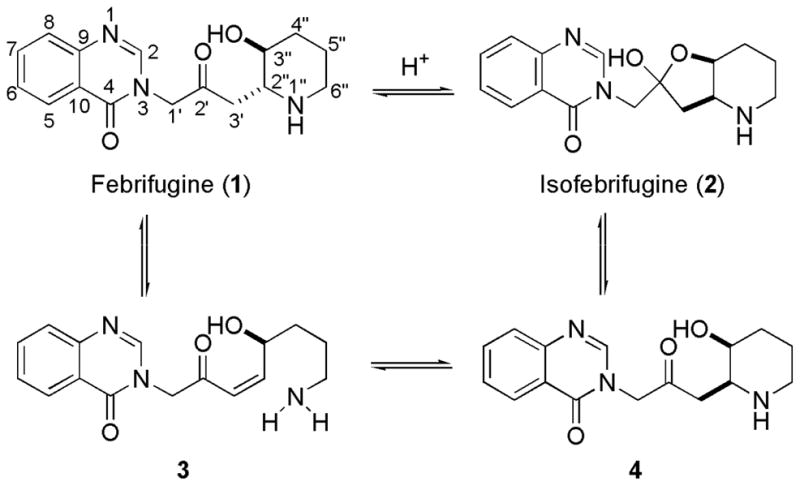
Total synthesis and structural modifications of febrifugine have been made.12–18 It has been elucidated the essential role played by the 4-quinazolinone ring and 1″-amino group in the appearance of activity. It is known that febrifugine (1) and isofebrifugine (2) are in equilibrium when they are in solution (Scheme 1). This equilibrium most likely follows a three-step sequence: (i) retro-Michael reaction (1 → 3); (ii) Michael addition (3 → 4); (iii) hemi-ketal cyclization (4 → 2). Intermediate 3, as a Michael acceptor, is highly electrophilic. If half-life of 3 is sufficiently long, then it is very possible that this electrophile will non-selectively alkylate biomolecules such as proteins, peptides, DNA, or RNA inside the host cell, resulting in the observed toxicity.
Scheme 1.
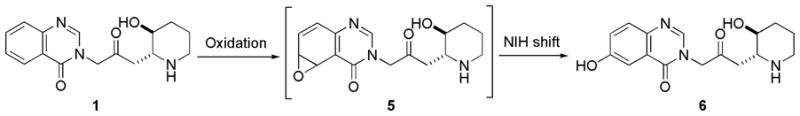
Febrifugine could be metabolized to the corresponding arene oxide 5 by cytochrome P-450 enzymes (Scheme 2). When arene oxide 5 escapes deactivation process by certain enzymes such as epoxide hydrase or glutathione S-transferase, toxicity can result because this reactive electrophile will form covalent adducts with DNA, RNA, proteins, or other biomolecules of the host. Such binding can cause mutations and result in cell damage. In a recent metabolic study of febrifugine, Oshima and coworkers isolated metabolite 6.16 This study indicates that arene oxide 5 is most probably a short-lived reactive metabolic intermediate because metabolite 6 would have been derived from intermediate 5 through the rearrangement known as NIH shift.19
Scheme 2.
As part of an ongoing malaria chemotherapy project in our laboratory, we undertook the synthesis and antimalarial activity evaluation of some febrifugine analogues (compounds 7–18, shown in Figure 1). Lower toxicity of these newly designed compounds will be achieved by reducing or eliminating the tendency to form chemically reactive and toxic intermediates. Two strategies are employed: (I) Blocking the retro-Michael process; (II) Increasing the oxidation potential of the molecule. In compounds 7–18, the 1″-amino group is now attached to the alpha position of the carbonyl group, eliminating the possibility of retro-Michael reaction. For compounds 8–12, an extra nitrogen atom or a substitution group was also introduced on the aromatic ring to block the C-5 or C-6 position of the quinazolinone ring or to increase the oxidation potential of the molecule. Compounds 13–16 each bear an alkoxyl group on C-3″ position of the piperidine ring. These compounds possess increased lipophilicity. Overall, these compounds closely resemble febrifugine itself by possessing a planar aromatic ring, a 1″-amino group and C-2′, C-3″ O-functionality and are therefore expected to possess same or similar mode of action. Meanwhile, they would be much less likely to produce toxic intermediates because these compounds are designed to block the aforementioned unwarranted metabolic pathway.
Figure 1.

As a comparative study, compounds 17 and 18 were also designed. These compounds each bear an electron-donating methoxy group on the C-7 or C-8 position of the aromatic ring. These compounds have comparable or even greater tendency to undergo oxidation. Biological data from these compounds should further validate, or nullify, the hypothesis that some oxidized febrifugine metabolites have contributed to the observed toxicity.
Chemistry
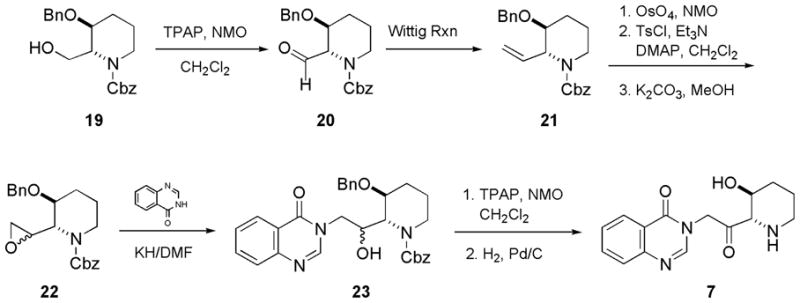
Scheme 3 illustrated the synthesis of compound 7. TPAP/NMO oxidation of the readily available alcohol 1920 gave aldehyde 20. The yield of direct conversion ofaldehyde 20 to oxirane 22 by the Corey-Chaykowsky ylide21 method was poor. So aldehyde 20 was first converted to alkene 21 under Wittig condition. Although initial oxidation of alkene 21 by mCPBA to give oxirane 22 was not successful, the transformation was smooth and high-yielding via a three-step sequence: (i) dihydroxylation; (ii) tosylation of the primary alcohol; and (iii) epoxide ring formation facilitated by potassium carbonate in methanol. 3H-Quinazolin-4-one condensed with oxirane 22, to furnish corresponding alcohol 23, as a pair of diastereomers, this follows the methods of Ogasawara, et al.12 Compound 23 then underwent TPAP oxidation22 and hydrogenolysis, to afford compound 7. The synthesis of compounds 8–18 follows similar route. The respective aromatic moieties are available from previously published procedures.23–25
Scheme 3.
Results and Discussion
Synthesized compounds (7–18) were tested against Plasmodium falciparum clones W-2, a chloroquine resistant cell line for in vitro efficacy. All compounds were also evaluated for antimalarial efficacy and toxicity in mouse models. The ED50s (effective dose leading to 50% reduction in parasitemia), MCDs (minimum clearance dose), and MTDs (maximum tolerated dose) were determined. The therapeutic indices, which were obtained by using the MCDs as the effective parameters, were calculated. The data are summarized in Table 1. All new compounds (7–18) have exhibited comparable or superior in vitro and in vivo antimalarial activity, as compared to parent natural product febrifugine. This confirms earlier observation that it is crucial to possess a planar aromatic ring, a 1″-amino group and C-2′, C-3″ O-functionality in order to preserve the excellent antimalarial activity of febrifugine.
Table 1.
Antimalarial Efficacy and Toxicity Evaluationa
| Test Compounds | In Vitro W2 Strain (IC50, nM) | In Vivo P. berghei (ED50, mg/kg) | MCD (mg/kg) | MTD (mg/kg) | TIb |
|---|---|---|---|---|---|
| 7 | 1.8 | 2.2 | 9.4 | 185 | 20 |
| 8 | 0.43 | 0.61 | 3.3 | >200 | >61 |
| 9 | 1.2 | 1.3 | 6.5 | >200 | >31 |
| 10 | 0.98 | 1.7 | 9.2 | >200 | >22 |
| 11 | 0.49 | 0.54 | 2.7 | >200 | >74 |
| 12 | 0.94 | 1.4 | 7.0 | >200 | >29 |
| 13 | 1.9 | 1.7 | 6.5 | 137 | 21 |
| 14 | 0.55 | 0.39 | 2.1 | 160 | 76 |
| 15 | 0.39 | 0.41 | 1.9 | 124 | 65 |
| 16 | 1.0 | 1.2 | 4.8 | 170 | 35 |
| 17 | 2.1 | 3.4 | 13 | 66 | 5 |
| 18 | 1.6 | 1.9 | 8.8 | 54 | 6 |
| Febrifugine | 2.5 | 2.3 | 12 | 35 | 3 |
| Chloroquine | 670 | 4.5 | 40.0 | 240 | 6 |
Antimalarial activities were determined after oral administration of the compounds once daily for 4 days to infected mice.
Therapeutic indices were calculated as MTD divided by MCD.
Compound 7, where the 1″-amino group is now attached to the alpha position of the carbonyl group, becomes 5 times less toxic as compared to febrifugine itself. Compounds 8–12 possess an extra nitrogen atom or an electron-withdrawing group on the aromatic ring. Those compounds have shown drastically reduced toxicity. Compounds 13–16 each bears an ethyloxy or benzyloxy group on C-3″ position of the piperidine ring. They also possess excellent in vitro and in vivo antimalarial activity. The efficacy is comparable to that of compounds 7, 8, 11 and 12, bearing a free hydroxyl group on C-3″ position of the piperidine ring. It is interesting to note that the more lipophilic compounds 13–16 have shown slightly elevated level of toxicity as compared to compounds 7, 8, 11 and 12.
Although the antimalarial efficacy of compounds 17 and 18 is about the same as that of compound 7, these compounds are considerably more toxic. Compounds 17 and 18 each possess an electron-donating methoxy group attached to the C-7 or C-8 of the aromatic ring and possess even greater tendency to undergo oxidation than that of compound 7. These biological data further validate the hypothesis that some oxidized febrifugine metabolites have contributed to the observed toxicity.
In conclusion, novel febrifugine analogues were designed and synthesized. New compounds possess excellent in vivo antimalarial activity and most of them become less toxic than the natural product febrifugine. Some of the compounds (14 and 15) possess a therapeutic index over ten times superior to that of febrifugine and the commonly used antimalarial drug chloroquine. These compounds, as well as the underlying design rationale, may find usefulness in the discovery and development of new antimalarial drugs.
Experimental
Chemistry
Melting points were determined on a Mettler FP62 melting point apparatus and are uncorrected. Unless otherwise noted, all nonaqueous reactions were performed under an oxygen-free atmosphere of nitrogen with rigid exclusion of moisture from reagents and glassware. Analytical thin layer chromatography (TLC) was performed using EM Reagents 0.25 mm silica gel 60-F plates. Visualization of the developed chromatogram was performed by UV absorbance, aqueous potassium permanganate, or ethanolic anisaldehyde. Liquid chromatography was performed using a force flow (flash chromatography) of the indicated solvent system on EM Reagents Silica Gel 60 (70–230 mesh). Preparative TLC was performed using Whatman Silica Gel C8 TLC plates (PLK5F). 1H NMR spectra were recorded in deuteriochloroform, unless otherwise noted, on a Bruker Avance 600 spectrometer at the frequency of 600.1 MHz. Chemical shifts are reported in parts per million on the δ scale from an internal standard of tetramethylsilane. Data are reported as follows: chemical shifts, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, m = multiplet, and br = broad), coupling constant in Hertz, integration, and assignment. 13C NMR spectra were recorded in deuteriochloroform, unless otherwise noted, on a Bruker Avance 600 spectrometer at the frequency of 150.9 MHz. Chemical shifts are reported in parts per million on the δ scale from an internal standard of tetramethylsilane. Combustion analyses were performed by Atlantic Microlab, Inc. (Norcross, Georgia). All purified compounds possess a purity of at least 95%. When necessary, solvents and reagents were dried as follows: ether, tetrahydrofuran, benzene, and toluene were stored and distilled from sodium benzophenone ketyl; dichloromethane, triethylamine, pyridine, and hexane were distilled over calcium hydride. Unless otherwise stated, the reagents were purchased from Fisher Scientific, Aldrich Chemical Company, Lancaster, or Fluka, and used as received.
3-Benzyloxy-2-formylpiperidine-1-carboxylic acid benzyl ester (20)
A solution of alcohol 19 (5.5 g) in 25 mL of CH2Cl2 was added into a stirred slurry of tetrapropylammonium perruthenate (TPAP, 100 mg), N-methylmorpholine N-oxide (NMO, 5.4 g), and grounded molecular sieve (4.5 g) in 30 mL of CH2Cl2 at room temperature. The reaction completed in 2 hours. Solid molecular sieves were filtered off. The dark liquid was concentrated and loaded directly into a short column of silica gel and eluted with 25% ethyl acetate in hexanes. Concentration of the eluant under reduced pressure afforded the corresponding aldehyde 20 as colorless oil. Yield: 92%. [α]25D +14.2 (c 0.25, CH2Cl2). 1H NMR: 9.82 (s, 1H), 7.30-7.19 (m, 10H), 5.67 (s, 2H), 4.81 (d, J = 13.5 Hz, 1H), 4.75 (d, J = 13.5 Hz, 1H), 4.61 (d, J = 6.8 Hz, 1H), 3.44 (m, 2H), 3.29 (m, 1H), 1.69 (m, 2H), 1.55 (m, 2H). 13C NMR: 200.1, 162.4, 140.9, 138.1, 129.1, 128.7, 128.4, 127.6, 127.3, 127.1, 74.1, 73.2, 71.9, 66.7, 49.1, 28.9, 22.4. HRESIMS m/z 376.1532 [M+Na]+ (calcd for C21H23NNaO4+, 376.1525) (100%). Anal. Calcd for C21H23NO4: C, 71.37; H, 6.56; N, 3.96. Found: C, 71.43; H, 6.54; N, 3.90.
3-Benzyloxy-2-vinylpiperidine-1-carboxylic acid benzyl ester (21)
The salt Ph3PCH3+I− (7.1 g) was suspended in anhydrous THF (50 mL). It was cooled to −20°C and n-BuLi (11.5 mL, 1.6M in hexanes) was slowly added in while the solution was stirred vigorously. 20 minutes later, a solution of compound 20 (4.9 g) in 10 mL THF was added in. The resulting solution was allowed to warm up to ambient temperature and stirred for additional 2 hours. The reaction mixture was quenched by the addition of aqueous NaHCO3 (5 mL), and then partitioned between ethyl acetate (120 mL) and water (120 mL). The separated organic layer was washed with brine (50 mL), dried over anhydrous sodium sulfate, and concentrated in a rotary evaporator under reduced pressure. Silica gel flash chromatography (15% ethyl acetate in hexanes) furnished 21 as colorless oil. Yield: 82%. [α]25D −19.1 (c 0.21, CH2Cl2). 1H NMR: 7.28-7.17 (m, 10H), 5.88 (m, 1H), 5.71 (s, 2H), 5.22 (m, 1H), 5.14 (m, 1H), 5.04 (d, J = 6.6 Hz, 1H), 4.84 (d, J = 13.8 Hz, 1H), 4.76 (d, J = 13.8 Hz, 1H), 3.48 (m, 2H), 3.02 (m, 1H), 1.66 (m, 2H), 1.52 (m, 2H). 13C NMR: 160.8, 141.9, 140.5, 137.9, 129.2, 128.9, 128.5, 127.8, 127.4, 127.0, 115.1, 73.9, 72.2, 71.3, 59.7, 50.1, 29.3, 21.8. HRESIMS m/z 374.1736 [M+Na]+ (calcd for C22H25NNaO3+, 374.1732) (100%). Anal. Calcd for C22H25NO3: C, 75.19; H, 7.17; N, 3.99. Found: C, 75.23; H, 7.14; N, 3.92.
3-Benzyloxy-2-oxiranylpiperidine-1-carboxylic acid benzyl ester (22)
Compound 21 (3.8 g), osmium tetraoxide (OsO4, 4% in water, 0.5 mL), and N-methyl morpholine N-oxide (NMO, 4.0 g) were dissolved in 25 mL of water and 25 mL of THF. The resulting heterogeneous solution is stirred vigorously for 3 days at room temperature. After partition between ethyl acetate (80 mL) and water (80 mL), the separated organic layer was washed with brine (30 mL), dried over anhydrous sodium sulfate, and evaporated in a rotary evaporator under reduced pressure to furnish the essentially pure diol. Tosyl chloride (TsCl, 2.0 g) was added into a stirred solution of this diol (3.9 g), triethylamine (Et3N, 2.0 mL) and 4-dimethylaminopyridine (DMAP, 80 mg) in 30 mL of methylene chloride (CH2Cl2) in an ice-water bath. The reaction mixture was then warmed to room temperature and stirred for additional 2 h. Solvent was evaporated. The resulting slurry was dissolved in ethyl acetate (40 mL), washed with water (30 mL), then aqueous sodium bicarbonate (20 mL), and brine (10 mL), dried over anhydrous sodium sulfate, and evaporated in a rotary evaporator under reduced pressure to furnish the essentially pure mono-tosylate. Potassium carbonate (300 mg) was added into a stirred solution of this mono-tosylate (5.1 g) in 15 mL of methanol at room temperature. After 4 h, reaction mixture was partitioned between ethyl acetate (30 mL) and water (30 mL), the separated organic layer was then washed with brine (10 mL), dried over anhydrous sodium sulfate, and evaporated in a rotary evaporator under reduced pressure to furnish the crude product. Silica gel flash chromatography (10% ethyl acetate in hexanes) furnishes 22 as colorless oil. Yield: 78% (3 steps). 1H NMR: 7.28-7.20 (m, 10H), 5.47 (m, 2H), 4.75 (m, 2H), 4.63 (m, 1H), 3.57 (m, 2H), 3.34 (m, 1H), 3.05 (m, 1H), 2.69 (m, 2H), 1.74 (m, 2H), 1.61 (m, 2H). 13C NMR: 162.9, 141.1, 138.2, 128.9, 128.1, 127.7, 127.4, 127.2, 126.9, 73.1, 70.9, 68.2, 55.1, 50.5, 47.1, 43.4, 28.9, 21.6. HRESIMS m/z 390.1676 [M+Na]+ (calcd for C22H25NNaO4+, 390.1681) (100%). Anal. Calcd for C22H25NO4: C, 71.91; H, 6.86; N, 3.81. Found: C, 71.84; H, 6.91; N, 3.85.
3-Benzyloxy-2-[1-hydroxy-2-(4-oxo-4H-quinazolin-3-yl)-ethyl]-piperidine-1-carboxylic acid benzyl ester (23)
Potassium hydride (30% in mineral oil, 1.9 g) was suspended in 25 mL of DMF. It was cooled in an ice-water bath, and solid 3H-quinazolin-4-one (2.0 g) was added in. After 20 min, a solution of compound 22 (2.5 g) in 8 mL of DMF was added in. The reaction mixture was then heated at 80°C for 12 h under nitrogen atmosphere. It was partitioned between ethyl acetate (50 mL) and water (50 mL), separated organic layer was washed with water (3 × 30 mL), then brine (25 mL), dried over anhydrous sodium sulfate, and evaporated in a rotary evaporator under reduced pressure to furnish the crude product. Silica gel flash chromatography (60% ethyl acetate in hexanes) furnished 23 as off white solid. Yield: 82%. MP: 178°C. 1H NMR: 8.32 (d, J = 7.2 Hz, 1H), 8.26 (s, 1H), 7.75 (m, 2H), 7.44 (m, 1H), 7.30-7.26 (m, 10H), 5.47 (m, 2H), 5.09 (br, 1H), 4.80 (m, 2H), 4.29 (m, 1H), 4.12 (m, 1H), 3.87 (m, 2H), 3.61 (m, 2H), 3.19 (m, 1H), 1.86 (m, 2H), 1.59 (m, 2H). 13C NMR: 168.3, 163.2, 158.9, 147.3, 141.1, 138.5, 133.8, 129.1, 128.9, 128.5, 127.8, 127.6, 127.4, 127.1, 127.0, 126.8, 124.8, 76.4, 71.6, 68.5, 66.4, 55.1, 51.7, 47.2, 33.4, 25.8. HRESIMS m/z 536.2156 [M+Na]+ (calcd for C30H31N3NaO5+, 536.2161) (100%). Anal. Calcd for C30H31N3O5.HCl: C, 65.51; H, 5.86; Cl, 6.45; N, 7.64. Found: C, 65.58; H, 5.81; Cl, 6.41; N, 7.68.
3-[2-(3-Hydroxypiperidin-2-yl)-2-oxoethyl]-3H-quinazolin-4-one (7)
A solution of compound 23 (2.2 g) in 5 mL of CH2Cl2 was added into a stirred slurry of TPAP (60 mg), NMO (950 mg), and grounded molecular sieve (600 mg) in 10 mL of CH2Cl2 at room temperature. After 1 h, the reaction mixture was loaded directly into a short column of silica gel and eluted with 5% MeOH/EtOAc. Concentration of the eluant under reduced pressure afforded the corresponding ketone. This ketone (2.0 g) was dissolved in 15 mL of 95% EtOH/H2O. 200 mg of 10% Pd on carbon was added in. It was then treated with hydrogen (60 psi) in a Parr apparatus for 16 h. Solid was filtered off and the solution was evaporated under vacuum to dryness. Recrystallization from ethanol-water (with addition of dilute aqueous HCl solution, 4–5 equiv. of HCl) furnishes compound 7 (HCl salt) as off-white crystals. Yield: 74% (two steps). MP: 187–188°C. [α]25D +27.1 (c 0.49, EtOH). 1H NMR (CD3OD): 8.32 (s, 1H), 8.26 (d, J = 7.2 Hz 1H), 7.77 (dd, J = 7.2, 6.7 Hz, 1H), 7.65 (d, J = 6.7 Hz, 1H), 7.51 (t, J = 7.2 Hz, 1H), 5.06 (d, J = 16.6 Hz, 1H), 4.92 (d, J = 16.6 Hz, 1H), 3.88 (d, J = 6.6 Hz, 1H), 3.49 (m, 1H), 2.89 (m, 2H), 1.88 (m, 2H), 1.61 (m, 2H). 13C NMR (CD3OD): 205.2, 168.4, 164.1, 148.3, 132.9, 128.5, 127.6, 126.1, 122.3, 72.2, 66.9, 52.9, 46.2, 32.6, 23.5. HRESIMS m/z 310.1162 [M+Na]+ (calcd for C15H17N3NaO3+, 310.1168) (100%). Anal. Calcd for C15H17N3O3.HCl: C, 55.64; H, 5.60; Cl, 10.95; N, 12.98. Found: C, 55.60; H, 5.63; Cl, 10.99; N, 12.89.
3-[2-(3-Hydroxypiperidin-2-yl)-2-oxoethyl]-3H-pyrido[3,2-d]pyrimidin-4-one (8)
Pale yellow crystal. MP: 213–214°C. [α]25D −29.8 (c 0.45, EtOH). 1H NMR (CD3OD): 8.71 (d, J = 6.9 Hz, 1H), 8.43 (d, J = 6.9 Hz, 1H), 8.29 (s, 1H), 7.72 (t, J = 6.9 Hz, 1H), 5.17 (d, J = 17.1 Hz, 1H), 4.99 (d, J = 17.1 Hz, 1H), 3.95 (d, J = 6.8 Hz, 1H), 3.61 (m, 1H), 2.95 (m, 2H), 1.91 (m, 2H), 1.57 (m, 2H). 13C NMR: 205.8, 168.9, 162.7, 151.1, 149.4, 142.9, 137.1, 133.7, 71.8, 66.1, 51.5, 46.6, 33.2, 22.8. HRESIMS m/z 311.1115 [M+Na]+ (calcd for C14H16N4NaO3+, 311.1120) (100%). Anal. Calcd for C14H16N4O3.HCl: C, 51.78; H, 5.28; Cl, 10.92; N, 17.25. Found: C, 51.82; H, 5.26; Cl, 10.88; N, 17.28.
3-[2-(3-Hydroxypiperidin-2-yl)-2-oxoethyl]-3H-pyrido[4,3-d]pyrimidin-4-one (9)
Pale yellow crystal. MP: 218–219°C. [α]25D +14.3 (c 0.51, EtOH). 1H NMR (CD3OD): 8.74 (s, 1H), 8.45 (d, J = 7.3 Hz, 1H), 8.29 (s, 1H), 7.89 (d, J = 7.3 Hz, 1H), 5.21 (d, J = 17.4 Hz, 1H), 5.08 (d, J = 17.4 Hz, 1H), 3.91 (d, J = 6.7 Hz, 1H), 3.66 (m, 1H), 2.91 (m, 2H), 1.88 (m, 2H), 1.55 (m, 2H). 13C NMR: 204.6, 168.1, 162.9, 158.4, 153.7, 148.6, 132.5, 129.9, 72.2, 66.4, 50.9, 47.9, 35.1, 23.4. HRESIMS m/z 311.1127 [M+Na]+ (calcd for C14H16N4NaO3+, 311.1120) (100%). Anal. Calcd for C14H16N4O3.HCl: C, 51.78; H, 5.28; Cl, 10.92; N, 17.25. Found: C, 51.72; H, 5.32; Cl, 10.87; N, 17.30.
3-[2-(3-Hydroxypiperidin-2-yl)-2-oxoethyl]-3H-pyrido[2,3-d]pyrimidin-4-one (10)
Pale yellow needles. MP: 215–216°C. [α]25D −18.6 (c 0.42, EtOH). 1H NMR (CD3OD): 8.83 (d, J = 6.8 Hz, 1H), 8.71 (d, J = 6.8 Hz, 1H), 8.35 (s, 1H), 8.15 (t, J = 6.8 Hz, 1H), 5.08 (d, J = 17.1 Hz, 1H), 4.95 (d, J = 17.1 Hz, 1H), 3.96 (d, J = 6.8 Hz, 1H), 3.61 (m, 1H), 2.95 (m, 2H), 1.85 (m, 2H), 1.63 (m, 2H). 13C NMR: 205.8, 170.1, 166.2, 163.9, 151.8, 143.5, 134.0, 130.5, 71.9, 67.0, 51.4, 48.2, 34.7, 22.7. HRESIMS m/z 311.1117 [M+Na]+ (calcd for C14H16N4NaO3+, 311.1120) (100%). Anal. Calcd for C14H16N4O3.HCl: C, 51.78; H, 5.28; Cl, 10.92; N, 17.25. Found: C, 51.83; H, 5.25; Cl, 10.98; N, 17.19.
5-Fluoro-3-[2-(3-hydroxypiperidin-2-yl)-2-oxoethyl]-3H-quinazolin-4-one (11)
White solid. MP: 172–173°C. [α]25D +24.1 (c 0.5, EtOH). 1H NMR (CD3OD): 8.24 (s, 1H), 8.18 (d, J = 7.2 Hz, 1H), 7.69 (d, J = 7.2 Hz, 1H), 7.51 (t, J = 7.2 Hz, 1H), 5.11 (d, J = 16.9 Hz, 1H), 4.92 (d, J = 16.9 Hz, 1H), 3.89 (d, J = 6.7 Hz, 1H), 3.68 (m, 1H), 2.90 (m, 2H), 1.89 (m, 2H), 1.58 (m, 2H). 13C NMR: 205.8, 167.1, 164.9, 146.1, 141.9, 133.6, 131.4, 128.9, 127.7, 71.3, 67.6 51.8, 49.1, 32.8, 23.4. HRESIMS m/z 328.1068 [M+Na]+ (calcd for C15H16FN3NaO3+, 328.1073) (100%). Anal. Calcd for C15H16FN3O3.HCl: C, 52.71; H, 5.01; Cl, 10.37; F, 5.56; N, 12.30. Found: C, 52.67; H, 5.05; Cl, 10.44; F, 5.50; N, 12.28.
3-[2-(3-Hydroxypiperidin-2-yl)-2-oxoethyl]-5-trifluoromethyl-3H-quinazolin-4-one (12)
White solid. MP: 180–181°C. [α]25D −14.5 (c 0.45, EtOH). 1H NMR (CD3OD): 8.19 (s, 1H), 7.87 (d, J = 6.9 Hz, 1H), 7.69 (d, J = 6.9 Hz, 1H), 7.51 (t, J = 6.9 Hz, 1H), 5.06 (d, J = 16.8 Hz, 1H), 4.84 (d, J = 16.8 Hz, 1H), 3.93 (d, J = 6.8 Hz, 1H), 3.65 (m, 1H), 2.96 (m, 2H), 1.86 (m, 2H), 1.63 (m, 2H). 13C NMR: 204.4, 165.7, 162.9, 149.6, 135.1, 133.8, 128.6, 128.1, 126.5, 116.2, 71.9, 68.1 51.4, 48.8, 33.6, 23.1. HRESIMS m/z 378.1038 [M+Na]+ (calcd for C16H16F3N3NaO3+, 378.1041) (100%). Anal. Calcd for C16H16F3N3O3.HCl: C, 49.05; H, 4.37; Cl, 9.05; F, 14.55; N, 10.73. Found: C, 49.11; H, 4.35; Cl, 9.01; F, 14.61; N, 10.75.
3-[2-(3-Ethoxypiperidin-2-yl)-2-oxoethyl]-3H-quinazolin-4-one (13)
Off white crystals. MP: 169–170°C. [α]25D +16.8 (c 0.43, EtOH). 1H NMR (CD3OD): 8.34 (s, 1H), 8.29 (d, J = 7.0 Hz 1H), 7.69 (dd, J = 7.0, 6.7 Hz, 1H), 7.61 (d, J = 6.7 Hz, 1H), 7.58 (t, J = 7.2 Hz, 1H), 5.11 (d, J = 16.9 Hz, 1H), 4.95 (d, J = 16.9 Hz, 1H), 3.98 (d, J = 6.8 Hz, 1H), 3.41 (q, J = 7.1 Hz, 2H), 3.19 (m, 1H), 2.87 (m, 2H), 1.85 (m, 2H), 1.59 (m, 2H), 1.17 (t, J = 7.1 Hz, 3H). 13C NMR (CD3OD): 205.9, 167.9, 165.0, 148.7, 133.2, 128.7, 127.4, 126.6, 123.1, 70.3, 68.7, 60.8, 53.4, 47.1, 30.6, 22.9, 15.5. HRESIMS m/z 338.1477 [M+Na]+ (calcd for C17H21N3NaO3+, 338.1481) (100%). Anal. Calcd for C17H21N3O3.HCl: C, 58.03; H, 6.30; Cl, 10.08; N, 11.94. Found: C, 58.10; H, 6.27; Cl, 10.12; N, 11.88.
3-[2-(3-Ethoxypiperidin-2-yl)-2-oxoethyl]-5-fluoro-3H-quinazolin-4-one (14)
White solid. MP: 165–166°C. [α]25D −22.4 (c 0.45, EtOH). 1H NMR (CD3OD): 8.29 (s, 1H), 8.21 (d, J = 7.2 Hz, 1H), 7.62 (d, J = 7.2 Hz, 1H), 7.55 (t, J = 7.2 Hz, 1H), 5.03 (d, J = 17.1 Hz, 1H), 4.91 (d, J = 17.1 Hz, 1H), 3.97 (d, J = 6.8 Hz, 1H), 3.44 (q, J = 6.7 Hz, 2H), 3.31 (m, 1H), 2.83 (m, 2H), 1.91 (m, 2H), 1.60 (m, 2H), 1.16 (t, J = 6.7 Hz, 3H). 13C NMR: 206.2, 167.6, 165.1, 147.3, 140.8, 133.7, 130.8, 128.6, 126.9, 70.1, 68.8, 61.2, 50.9, 49.4, 30.9, 23.6, 15.4. HRESIMS m/z 356.1391 [M+Na]+ (calcd for C17H20FN3NaO3+, 356.1386) (100%). Anal. Calcd for C17H20FN3O3.HCl: C, 55.21; H, 5.72; Cl, 9.59; F, 5.14; N, 11.36. Found: C, 55.27; H, 5.69; Cl, 9.63; F, 5.19; N, 11.33.
3-[2-(3-Benzyloxypiperidin-2-yl)-2-oxoethyl]-3H-pyrido[3,2-d]pyrimidin-4-one (15)
Pale yellow crystal. MP: 195–196°C. [α]25D +22.4 (c 0.47, EtOH). 1H NMR (CD3OD): 8.69 (d, J = 7.0 Hz, 1H), 8.51 (d, J = 7.0 Hz, 1H), 8.27 (s, 1H), 7.63 (t, J = 7.0 Hz, 1H), 7.29–7.19 (m, 5H), 5.25 (s, 2H), 5.12 (d, J = 17.0 Hz, 1H), 4.94 (d, J = 17.0 Hz, 1H), 4.05 (d, J = 6.8 Hz, 1H), 3.27 (m, 1H), 2.89 (m, 2H), 1.87 (m, 2H), 1.62 (m, 2H). 13C NMR: 206.4, 168.4, 163.1, 151.6, 148.9, 142.5, 137.8, 136.9, 132.9, 128.9, 127.8, 127.1, 72.6, 70.8, 69.0, 50.7, 47.2, 31.2, 22.5. HRESIMS m/z 401.1586 [M+Na]+ (calcd for C21H22N4NaO3+, 401.1590) (100%). Anal. Calcd for C21H22N4O3.HCl: C, 60.79; H, 5.59; Cl, 8.55; N, 13.50. Found: C, 60.82; H, 5.55; Cl, 8.58; N, 13.48.
3-[2-(3-Benzyloxypiperidin-2-yl)-2-oxoethyl]-5-trifluoromethyl-3H-quinazolin-4-one (16)
White needles. MP: 182–183°C. [α]25D -22.3 (c 0.42, EtOH). 1H NMR (CD3OD): 8.24 (s, 1H), 7.82 (d, J = 6.9 Hz, 1H), 7.71 (d, J = 6.9 Hz, 1H), 7.47 (t, J = 6.9 Hz, 1H), 7.30-7.20 (m, 5H), 5.17 (s, 2H), 5.09 (d, J = 16.7 Hz, 1H), 4.88 (d, J = 16.7 Hz, 1H), 3.97 (d, J = 6.7 Hz, 1H), 3.25 (m, 1H), 2.91 (m, 2H), 1.83 (m, 2H), 1.57 (m, 2H). 13C NMR: 206.1, 164.9, 161.3, 150.0, 137.3, 135.5, 131.2, 128.9, 128.5, 128.0, 127.6, 127.1, 126.9, 116.5, 73.2, 69.8, 67.8, 51.9, 47.6, 29.7, 23.0. HRESIMS m/z 468.1505 [M+Na]+ (calcd for C23H22F3N3NaO3+, 468.1511) (100%). Anal. Calcd for C23H22F3N3O3.HCl: C, 57.32; H, 4.81; Cl, 7.36; F, 11.83; N, 8.72. Found: C, 57.37; H, 4.77; Cl, 7.41; F, 11.75; N, 8.75.
3-[2-(3-Hydroxypiperidin-2-yl)-2-oxoethyl]-7-methoxy-3H-quinazolin-4-one (17)
White solid. MP: 191–192°C. [α]25D +18.8 (c 0.51, EtOH). 1H NMR (CD3OD): 8.18 (s, 1H), 7.79 (s, 1H), 7.27 (d, J = 7.1 Hz, 1H), 7.33 (d, J = 7.1 Hz, 1H), 5.08 (d, J = 16.8 Hz, 1H), 4.92 (d, J = 16.8 Hz, 1H), 3.81 (s, 3H), 3.78 (d, J = 6.7 Hz, 1H), 3.46 (m, 1H), 2.82 (m, 2H), 1.91 (m, 2H), 1.58 (m, 2H). 13C NMR: 206.1, 168.4, 166.1, 162.2, 149.1, 129.1, 119.4, 113.7, 108.2, 71.1, 65.7, 57.9, 51.9, 46.1, 31.4, 22.6. HRESIMS m/z 340.1281 [M+Na]+ (calcd for C16H19N3NaO4+, 340.1273) (100%). Anal. Calcd for C16H19N3O4.HCl: C, 54.32; H, 5.70; Cl, 10.02; N, 11.88. Found: C, 54.33; H, 5.68; Cl, 10.11; N, 11.84.
3-[2-(3-Ethoxypiperidin-2-yl)-2-oxoethyl]-8-methoxy-3H-quinazolin-4-one (18)
White needles. MP: 188–183°C. [α]25D −19.4 (c 0.45, EtOH). 1H NMR (CD3OD): 8.17 (s, 1H), 7.61 (t, J = 7.2 Hz, 1H), 7.39 (d, J = 7.2 Hz, 1H), 7.12 (d, J = 7.2 Hz, 1H), 5.05 (d, J = 17.1 Hz, 1H), 4.93 (d, J = 17.1 Hz, 1H), 3.94 (d, J = 6.7 Hz, 1H), 3.79 (s, 3H), 3.41 (q, J = 6.9 Hz, 2H), 3.24 (m, 1H), 2.85 (m, 2H), 1.89 (m, 2H), 1.63 (m, 2H), 1.12 (t, J = 6.9 Hz, 3H). 13C NMR: 205.7, 167.4, 162.9, 155.8, 134.2, 128.9, 128.1, 120.4, 118.9, 69.1, 68.4, 61.2, 56.9, 50.8, 46.7, 28.9, 22.1, 15.6. HRESIMS m/z 368.1589 [M+Na]+ (calcd for C18H23N3NaO4+, 368.1586) (100%). Anal. Calcd for C18H23N3O4.HCl: C, 56.62; H, 6.34; Cl, 9.28; N, 11.00. Found: C, 56.65; H, 6.31; Cl, 9.34; N, 10.94.
Parasite culture
Chloroquine resistant W2 strains of Plasmodium falciparum were cultivated in RPMI 1640 media with 6% human erythrocytes supplemented with 10% of human serum. The parasites were cultured in an atmosphere of 5% CO2, 5% O2 and 90% N2 at 37°C.
In vitro drug susceptibility assay26, 27
Febrifugine and analogs were tested in a cell-based in vitro drug susceptibility assay to determine if they were capable of interrupting Plasmodium metabolism and growth. The semi-automated micro-dilution technique was used to assess the sensitivity of the parasites to the selected compounds. The incorporation of [3H]hypoxanthine into the parasites was measured as a function of compound concentration to determine IC50 values.
In vivo efficacy test28
In conducting the research described in this report, the investigators adhered to the Guide for the Care and Use of Laboratory Animals by the Institute of Laboratory Animal Resources, National Research Council. Typically, 4–5 weeks old IRC mice were housed in plastic cages with free access to water and food. P. berghei parasite-infected erythrocytes were obtained from donor mice. On experiment day 0, the donor mice were anesthetized and exsanguinated via cardiac puncture. The pooled blood from the donor mice was then diluted with normal mouse serum to a concentration of 1 × 106 P. berghei-infected erythrocytes per inoculum (0.1 ml). The groups of experimental and control mice were inoculated with this parasitized blood on day 0. The tested mice were treated with either candidate antimalarial drugs or with vehicle alone to serve as negative controls from day 3 to day 10 orally (PO) or subcutaneously (SC) once a day for eight days. Each experimental group received a different dose level, with up to 5 different dose groups per compound. Blood films and body weights were taken on the third and sixth days post-infection, then at weekly intervals through day 60. Films were Giemsa stained and examined microscopically to determine parasitemia. All mice were observed twice a day to assess their clinical signs that were recorded. All treated mice with negative smear on day 60 were considered cured.
Acknowledgments
This research was partially supported by a grant from the National Institutes of Health (AI81308 to S.Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agency.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WHO. World Malaria Report. 2008. [Google Scholar]
- 2.Bojang KA, Obaro SK, Leach A, D’Alessandro U, Benett S, Metzger W, Ballou WR, Targett GA, Greenwood BM. Follow-up of Gambian children recruited to a pilot safety and immunogenicity study of the malaria vaccine SPf66. Parasite Immunol. 1997;19:579–581. doi: 10.1046/j.1365-3024.1997.d01-171.x. [DOI] [PubMed] [Google Scholar]
- 3.Ambroise-Thomas P. Vaccination against malaria. Disappointments and hopes [Article in French] Bull Acad Natl Med. 1997;181:1637–1648. [PubMed] [Google Scholar]
- 4.Doolan DL, Hedstrom RC, Gardner MJ, Sedegah M, Wang H, Gramzinski RA, Margalith M, Hobart P, Hoffman SL. DNA vaccination as an approach to malaria control: current status and strategies. Curr Top Microbiol Immunol. 1998;226:37–56. doi: 10.1007/978-3-642-80475-5_3. [DOI] [PubMed] [Google Scholar]
- 5.Winstanley PA. Chemotherapy for falciparum malaria: the armoury, the problems and the prospects. Parasitology Today. 2000;16:146–153. doi: 10.1016/s0169-4758(99)01622-1. [DOI] [PubMed] [Google Scholar]
- 6.Koepfly JB, Mead JF, Brockman JA., Jr An Alkaloid with High Antimalarial Activity from Dichroa febrifuga. J Am Chem Soc. 1947;69:1837. doi: 10.1021/ja01199a513. [DOI] [PubMed] [Google Scholar]
- 7.Koepfly JB, Mead JF, Brockman JA., Jr Alkaloids of Dichroa febrifuga. I. Isolation and Degradative Studies. J Am Chem Soc. 1949;71:1048–1054. doi: 10.1021/ja01171a080. [DOI] [PubMed] [Google Scholar]
- 8.WHO Report. Meeting on Antimalarial Drug Development; Shanghai, China. 16–17 November 2001. [Google Scholar]
- 9.Ablondi F, Gordon S, Morton J, II, Williams JH. An Antimalarial Alkaloid from Hydrangea. II. Isolation. J Org Chem. 1952;17:14–18. [Google Scholar]
- 10.Hewitt RI, Wallace WS, Gill ER, Williams JH. An Antimalarial Alkaloid from Hydrangea. Am J Trop Med Hyg. 1952;1:768–772. [PubMed] [Google Scholar]
- 11.Kato M, Inaba H, Itahana H, Ohara E, Nakamura K, Uesato S, Inouye H, Fujita T. Studies on Anticoccidial Constituents of Crude Drugs and Related Plants. (I). Isolation and Biological Activities of cis- and trans-Febrifugine from Hydrangea Macrophylla. Shoyakugaku Zasshi. 1990;44:288–292. [Google Scholar]
- 12.Taniguchi T, Ogasawara K. A Diastereocontrolled Synthesis of (+)-Febrifugine: A Potent Antimalarial Piperidine Alkaloid. Org Lett. 2000;2:3193–3195. doi: 10.1021/ol006384f. [DOI] [PubMed] [Google Scholar]
- 13.Takaya Y, Tasaka H, Chiba T, Uwai K, Tanitsu M, Kim HS, Wataya Y, Miura M, Takeshita M, Oshima Y. New Type of Febrifugine Analogues, Bearing a Quinolizidine Moiety, Show Potent Antimalarial Activity against Plasmodium Malaria Parasite. J Med Chem. 1999;42:3163–3166. doi: 10.1021/jm990131e. [DOI] [PubMed] [Google Scholar]
- 14.Fishman M, Cruickshank PA. Febrifugine Antimalarial Agents. I. Pyridine Analogs of Febrifugine. J Med Chem. 1970;13:155–156. doi: 10.1021/jm00295a050. [DOI] [PubMed] [Google Scholar]
- 15.Chien PL, Cheng CC. Structural Modification of Febrifugine. Some Methylenedioxy analogs. J Med Chem. 1970;13:867–870. doi: 10.1021/jm00299a018. [DOI] [PubMed] [Google Scholar]
- 16.Hirai S, Kikuchi H, Kim HS, Begum K, Wataya Y, Tasaka H, Miyazawa Y, Yamamoto K, Oshima Y. Metabolites of Febrifugine and Its Synthetic Analogue by Mouse Liver S9 and Their Antimalarial Activity against Plasmodium Malaria Parasite. J Med Chem. 2003;46:4351–4359. doi: 10.1021/jm0302086. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi Y, Koike M, Azuma K, Nishioka H, Abe H, Kim HS, Wataya Y, Harayama T. Synthesis and Antimalarial Activity of Febrifugine Derivatives. Chem Pharm Bull. 2001;49:721–725. doi: 10.1248/cpb.49.721. [DOI] [PubMed] [Google Scholar]
- 18.Zhu S, Meng L, Zhang Q, Wei L. Synthesis and Evaluation of Febrifugine Analogues As Potential Antimalarial Agents. Bioorg Med Chem Lett. 2006;16:1854–1858. doi: 10.1016/j.bmcl.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guroff G, Daly JW, Jerina DM, Renson J, Witkop B, Udenfriend S. Hydroxylation-Induced Migration: the NIH shift. Science. 1967;157:1524–1530. doi: 10.1126/science.157.3796.1524. [DOI] [PubMed] [Google Scholar]
- 20.Kim IS, Oh JS, Zee OP, Jung YH. Synthesis and conformational analysis of 3-hydroxypipecolic acid analogs via CSI-mediated stereoselective amination. Tetrahedron. 2007;63:2622–2633. [Google Scholar]
- 21.Corey EJ, Chaykovsky M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J Am Chem Soc. 1965;87:1353–1364. [Google Scholar]
- 22.Ley SV, Norman J, Griffith WP, Marsden SP. Tetrapropylammonium Perruthenate, Pr4N+RuO4−, TPAP: Catalytic Oxidant For Organic Synthesis. Synthesis. 1994:639–666. [Google Scholar]
- 23.Williams EJ, Kenny PW, Kettle JG, Mwashimba PG. Synthesis of a 5-Alkoxypyrido[4,3-d]pyrimidin-4(3H)-one Derivative via a Regioselective Meisenheimer N-oxide Rearrangement. Tetrahedron Lett. 2004;45:3737–3739. [Google Scholar]
- 24.Alexandre FR, Berecibar A, Besson T. Microwave-assisted Niementowski Reaction. Back to the Roots. Tetrahedron Lett. 2002;43:3911–3913. [Google Scholar]
- 25.Armarego WLF, Smith JIC. Quinazolines. Part IX. Covalent Hydration in the Neutral Species of Substituted Quinazolines. J Chem Soc B. 1967:449–454. [Google Scholar]
- 26.Desjardins RE, Canfield CJ, Haynes DE, Chulay JD. Quantitative Assessment of Antimalarial Activity In Vitro by a Semiautomated Microdilution Technique. Antimicrob Agents Chemother. 1979;16:710–718. doi: 10.1128/aac.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chulay JD, Haynes JD, Diggs CL. Plasmodium falciparum: Assessment of In Vitro Growth by [3H]hypoxanthine Incorporation. Exp Parasitol. 1983;55:138–146. doi: 10.1016/0014-4894(83)90007-3. [DOI] [PubMed] [Google Scholar]
- 28.Jiang S, Zeng Q, Gettayacamin M, Tungtaeng A, Wannaying S, Lim A, Hansukjariya P, Okunji CO, Zhu S, Fang D. Antimalarial activities and therapeutic properties of febrifugine analogs. Antimicrob Agents Chemother. 2005;49:1169–1176. doi: 10.1128/AAC.49.3.1169-1176.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]