

Abstract
A series of compounds based on the N-amino-4-imidazolidinone scaffold was synthesized and screened against human neutrophil elastase (HNE). These studies lead to the identification of a selective, low micromolar reversible competitive inhibitor of HNE.
Introduction
Chronic obstructive pulmonary disease (COPD) is a major health problem that affects more than 16 million people in the U.S. [1]. The disorder is associated with airflow limitation that is not easily reversible [2–3]. Currently, there are no drugs on the market for the specific treatment of COPD.
Although the pathogenesis of COPD is poorly understood, the disorder is characterized by increased numbers of alveolar macrophages, neutrophils and cytotoxic T lymphocytes, increased elastolysis by an array of proteases [4], oxidative stress [5], apoptosis [6], and the release of multiple inflammatory mediators, including cytokines and chemokines [7–10].
Several lines of evidence suggest that macrophage- and neutrophil-derived oxidative and proteolytic mediators released at sites of inflammation play an important role in the pathophysiology of COPD [11]. Most prominent among these mediators are human neutrophil elastase (HNE), proteinase 3 (Pr 3), and MMP-12. Elucidation of the biochemical and molecular mechanisms related to the pathophysiology of COPD may lead to the emergence of effective therapeutics [11].
HNE and Pr 3 show significant differences in their S2 and S1’-S3’ subsites [12]. For instance, the S1’-S3’ pockets in Pr 3 are smaller and more polar than those of HNE because of the presence of Asp61 and Arg143 [13]. These differences can be exploited by utilizing a scaffold that docks to the active site in a predictable fashion and orients appended recognition elements toward the S’ subsites. Thus, based on previous studies involving the use of functionalized 4-imidazolidinone derivatives as reversible competitive inhibitors of HNE [14], we reasoned that inhibitors based on the N-amino-4-imidazolidinone scaffold (I) (Figure 1) may exhibit high selectivity toward HNE over Pr 3 (and vice versa) through exploitation of differences in the S’ subsites of the two enzymes. We describe herein the results of exploratory studies related to the selective inhibition of HNE by derivatives based on scaffold (I) (Figure 1).
Fig. 1.

General structure of inhibitor (I).
Chemistry
The desired compounds 7–13, 14–18 and 19–26 were synthesized as shown in Scheme 1. Cbz-DL-phenylalanine (or Cbz-DL-leucine) was coupled to t-butyl carbazate using isobutyl chloroformate in the presence of N-methylmorpholine. Removal of the protective group followed by sodium triacetoxyborohydride-mediated reductive amination [15] yielded intermediate 3 which was cyclized with aqueous formaldehyde. The formation of the 4-imidazolidinone ring was also accompanied by N-hydroxymethylation of the product to afford compound 4. Stirring compound 4 with aqueous sodium hydroxide resulted in clean removal of the hydroxymethyl moiety to yield the desired intermediate 5. Initial attempts at removing the Boc group using trifluoroacetic acid or dry HCl lead to intractable mixtures, however, microwave irradiation of 5 in the presence of silica gel [16] yielded the desired N-amino-4-imidazolidinone which could be functionalized via reaction with an array of structurally-diverse isocyanates, carboxylic acids, acid anhydrides, sulfonyl chlorides or N-protected amino acids. The resulting products could be readily debenzylated using catalytic hydrogenation.
Scheme 1.

General synthesis of inhibitor I. Reagents and conditions: a) IBCF/NMM THF then NH2NHBoc; b) H2 /10%Pd-C/CH3OH; c) RCHO/NaBH(OAc)3 / HOAc; d) HCHO or R3CHO; e) NaOH / aq dioxane; f) silica gel/microwave; g) RSO2Cl /base or RN=C=O or (RCO)2O or RCOOH/EDCI/DMF or protected amino acid/EDCI/DMF.
Biochemical Studies
Enzyme assays and inhibition studies using neutrophil elastase and proteinase 3 were conducted as previously described [17]. The inhibition constant (KI) of compound 24 was determined using a Dixon plot [18].
Results and Discussion
Inhibitor Design Rationale
The design of inhibitor (I) was based on the following considerations: (a) recent studies in our laboratory have demonstrated that inhibitor (II) (Figure 2) is a highly efficient mechanism-based irreversible inhibitor of serine proteases that docks to the active site of HNE with the R1 and R2 groups occupying the S1 and S2 subsites [13]; (b) replacement of the sulfonyl group with a methylene group lowers the reactivity of the amide carbonyl in the resulting 4-imidazolidinone ring making possible the transformation of irreversible inhibitor (II) into reversible competitive inhibitor (III)(Figure 2) [14]; (c) groups R4 and R5 in (I) are oriented toward the S’ subsites; (d) HNE is a basic, 218 amino acid single polypeptide glycoprotein whose primary structure shows considerable homology (54%) [13] with Pr 3. Furthermore, the two enzymes have extended binding sites and show a preference for hydrophobic substrates/inhibitors. However, although HNE and Pr 3 exhibit high sequence identity, their S’ subsites show significant differences which can be exploited [13,19]. As mentioned earlier, the S1’-S3’ pockets in Pr 3 are smaller and more polar than those of HNE because of the presence of Asp61 and Arg143 while the presence of Val190, Phe192, Ala213, Val216, Phe228 and the disulfide bridge Cys191–Cys220 makes the active site of HNE hemispherical and hydrophobic.
Fig. 2.

Design rationale of reversible competitive inhibitors of human neutrophil elastase and proteinase 3.
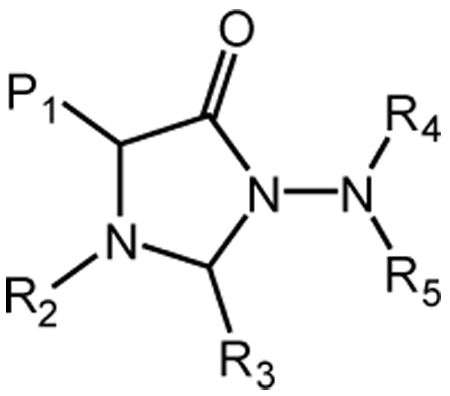
Based on the aforementioned considerations, it was envisaged that derivatives based on scaffold (I) might make possible the attainment of optimal potency and selectivity toward HNE (or Pr 3) through favorable binding interactions with the S’ subsites of the two enzymes. Thus, initial exploratory studies focused on generating a series of focused libraries by varying diversity sites R4 and R5. All compounds were derived from (DL) Phe (P1 = benzyl) and (DL) Leu (P1 = isobutyl) and listed in Table 1. The inhibitory activity against HNE and Pr 3 is summarized in Figure 3. Compound 24 was found to be a highly selective, low micromolar (KI 7.34 µM) reversible competitive inhibitor of HNE, as evidenced by the construction of a Dixon plot [18] (Figure 4). Compound 24 was devoid of any inhibitory activity toward Pr 3 while the rest of the compounds showed minimal inhibition (0–20%) toward both enzymes (Figure 3). Inhibition of HNE and Pr 3 appears to be insensitive to variations in the structural diversity of R4 and R5, suggesting that R4 and R5 are oriented toward the S’ subsites in a non-optimal manner. The results also suggest that it may be possible to design non-covalent inhibitors of HNE and Pr 3 that exhibit optimized potency and selectivity through the utilization of appropriate recognition elements. These inhibitors are unlike most reported inhibitors of mammalian and viral serine proteases which incorporate in their structures a warhead that leads to covalent inhibition or, in the case of transition state inhibitors, the formation of a reversible covalent adduct [20].
Table 1.
N-Amino-4-imidazolidinone derivatives (I)a
 | ||||
|---|---|---|---|---|
| Compound | P1 | R2 | R4 | R5 |
| 7 | benzyl | benzyl | 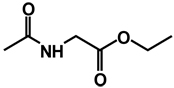 |
H |
| 8 | benzyl | benzyl | 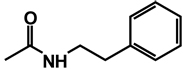 |
H |
| 9 | benzyl | benzyl | 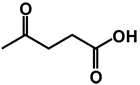 |
H |
| 10b | benzyl | benzyl | 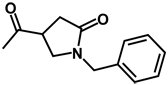 |
H |
| 11b | benzyl | benzyl | 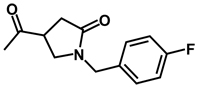 |
H |
| 12b | benzyl | benzyl | 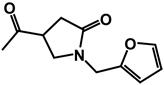 |
H |
| 13 | benzyl | benzyl | 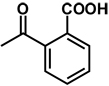 |
H |
| 14 | benzyl | H | 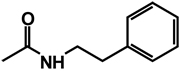 |
H |
| 15 | benzyl | H | 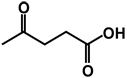 |
H |
| 16b | benzyl | H | 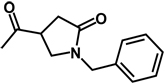 |
H |
| 17b | benzyl | H | 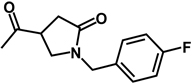 |
H |
| 18 | benzyl | H | 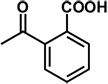 |
H |
| 19 | isobutyl | benzyl | 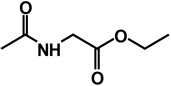 |
H |
| 20 | isobutyl | benzyl | 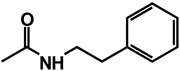 |
H |
| 21 | isobutyl | benzyl | 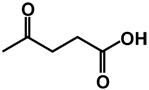 |
H |
| 22 | isobutyl | benzyl | 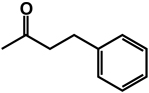 |
H |
| 23 | isobutyl | benzyl | 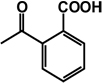 |
H |
| 24b | isobutyl | benzyl | 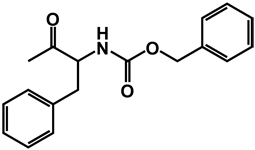 |
H |
| 25 | isobutyl | benzyl | ||
| 26 | isobutyl | benzyl | 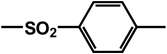 |
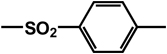 |
R3 = H for all.
Compounds were screened as diastereomeric mixtures.
Fig. 3.

Inhibition of human neutrophil elastase and proteinase 3 by compounds 7–26. Values were expressed as percent inhibition using an inhibitor/enzyme ratio of 500, and an incubation time of 30 minutes for human neutrophil elastase and no incubation for proteinase 3.
Fig. 4.

Dixon plot for the interaction of compound 24 with human neutrophil elastase.
In conclusion, we have demonstrated that the multiple points of diversity afforded by scaffold (I) render it well-suited to the design of non-covalent inhibitors of serine proteases with similar active sites.
Experimental section
General methods
The 1H spectra were recorded on a Varian XL-300 or XL-400 NMR spectrometer. A Hewlett-Packard diode array UV/Vis spectrophotometer was used in the in vitro evaluation of the inhibitors. Human neutrophil elastase, proteinase 3 and Boc-Ala-Ala-Nva thiobenzyl ester were purchased from Elastin Products Company, Owensville, MO. Methoxysuccinyl Ala-Ala-Pro-Val p-nitroanilide and 5,5’-dithio-bis (2-nitrobenzoic acid) were purchased from Sigma Chemicals, St. Louis, MO. Melting points were determined on a Mel-Temp apparatus and are uncorrected. Reagents and solvents were purchased from various chemical suppliers (Aldrich, Acros Organics, TCI American, and Bachem). Silica gel (230–450 mesh) used for flash chromatography was purchased from Sorbent Technologies (Atlanta, GA). Thin layer chromatography was performed using Analtech silica gel plates and the TLC plates were visualized using iodine and/or UV light. All compounds synthesized as diastereomeric mixtures were screened against human neutrophil elastase and proteinase 3 as such without separating the individual diastereomers.
Representative Syntheses
(RS)-Tert-butyl 2-(2-(benzyloxycarbonylamino)-3-phenylpropanoyl)hydrazine-carboxylate 1
A solution of Z-DL-Phe-OH (71.84 g, 240 mmol) in dry THF (200 mL) cooled in an ice-bath was treated with isobutyl chloroformate (32.78 g; 240 mmol), followed with N-methylmorpholine (24.28 g; 240 mmol). The resulting mixture was stirred for 5 minutes, and t-butyl carbazate (31.60 g; 240 mmol) was added. The ice-bath was removed and the reaction mixture was stirred for 7 h at room temperature. The solvent was removed on the rotary evaporator and the residue was taken up in methylene chloride (200 mL). The resulting solution was washed sequentially with 5% aqueous HCl (70 mL), 5% aqueous NaHCO3 (3 × 70 mL) and brine (3 × 70 mL). The organic phase was dried over anhydrous Na2SO4 and the solvent was removed to yield the product (97.25 g; 98% yield) as a white solid, mp 120.0-121.5 °C. 1H NMR (CDCl3): δ 1.45(s, 9H), 3.00-3.20(m, 2H), 4.41-4.52(m, 1H), 5.05(s, 2H), 5.22-5.30(s, 1H), 6.38(s, 1H), 7.08-7.38(m, 10H).
(RS)-Tert-butyl 2-(2-amino-3-phenylpropanoyl)hydrazinecarboxylate 2
A solution of compound 1 (47.79 g; 116 mmol) in methanol (250 mL) was treated with 10% palladium on carbon (7.89 g). The reaction mixture was stirred at room temperature under hydrogen (low pressure) for 3 h. The catalyst was filtered off, rinsed with methanol and the solvent was evaporated, leaving a crude product which was purified by recrystallization (ethyl acetate/hexanes) to yield the product as a white solid (26.53 g, 82% yield), mp 115.0-116.0 °C. 1H NMR (CDCl3): δ 1.48 (s, 9H), 2.68-2.76 (dd, 1H, J = 15, 9 Hz), 3.28-3.36 (dd, 1H, J = 15, 6 Hz), 3.70-3.75 (dd, 1H, J = 9, 6 Hz), 6.45 (s, 1H), 7.20-7.36 (m, 5H).
(RS)-Tert-butyl 2-(2-(benzylamino)-3-phenylpropanoyl)hydrazinecarboxylate 3
A solution of compound 2 (23.40 g; 83.8 mmol) and benzaldehyde (10.68 g; 100.6 mmol) in dry 1, 2-dichloroethane (150 mL) was treated with glacial acetic acid (7.05 g; 117.3 mmol), followed by sodium triacetoxyborohydride (25.56 g; 117.3 mmol). The reaction mixture was stirred for 4 h at room temperature and then neutralized via the dropwise addition of cold 10% aqueous NaOH solution to pH 9–10 while stirring. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (3 × 150 mL), the combined organic extracts were dried over anhydrous Na2SO4 and the solvent removed. The crude product was purified by flash chromatography (silica gel/hexane/ethyl acetate) to yield a pure product as a colorless oil (14.04 g, 45% yield). 1H NMR (CDCl3): δ 1.45 (s, 9H), 2.74-2.82 (m, 1H), 3.19-3.24 (dd, 1H, J = 12, 6 Hz), 3.46-3.54 (dd, 1H, J = 9, 6 Hz), 3.54-3.58 (d, 1H, J = 12 Hz), 3.78-3.52 (d, 1H, J = 12 Hz), 6.82 (s, 1H), 7.08-7.32 (m, 10H).
(RS)-Tert-butyl 3,4-dibenzyl-5-oxoimidazolidin-1-yl(hydroxymethyl)carbamate 4
A mixture of compound 3 (11.20 g; 30.31 mmol) and 37% aqueous formaldehyde (50 mL) was refluxed for 4 h. The reaction mixture was cooled to room temperature, water (50 mL) was added, and the solution was extracted with CH2Cl2 (3 × 70 mL). The organic phase was dried over anhydrous Na2SO4 and the solvent was removed in vacuo. The crude product was purified by flash chromatography (silica gel/hexane/ethyl acetate) to yield a pure product as a colorless oil (6.48 g; 52% yield). 1H NMR (CDCl3): δ 1.42 (s, 9H), 2.98-3.02 (m, 1H), 3.08-3.15 (m, 1H), 3.45-3.60 (m, 2H), 3.80-3.85 (s, 1H), 4.05 (s, 2H), 4.70-4.82 (m, 2H), 7.05-7.50 (m, 10H).
(RS)-Tert-butyl 3,4-dibenzyl-5-oxoimidazolidin-1-ylcarbamate 5
Compound 4 (12.47 g; 30.31 mmol) in 1, 4-dioxane (50 mL) was treated with a solution of KOH (29.68 g; 530 mmol) in H2O (53 mL) and the reaction mixture was stirred at room temperature for 1h. The solvent was removed on a rotary evaporator and the resulting residue was extracted with ethyl acetate (3 × 70 mL). The organic phase was dried over anhydrous Na2SO4 and the solvent was removed. The crude product was purified by flash chromatography (silica gel/hexane/ethyl acetate) to yield a pure product as a colorless oil (7.51g; 65% yield). 1H NMR (CDCl3): δ 1.42 (s, 9H), 2.95-3.08 (m, 1H), 3.12-3.22 (dd, 1H, J = 12, 6 Hz), 3.44-3.50 (d, 1H, J = 12 Hz), 3.54-3.58 (s, 1H), 3.78-3.86 (d, 1H J = 12 Hz), 3.96 (s, 1H), 4.02-4.12 (m, 1H), 6.30-6.42 (s, 1H), 7.10-7.22 (m, 10H).
(RS)-3-Amino-1,5-dibenzylimidazolidin-4-one 6
Compound 5 (10.53 g; 27.60 mmol) was dissolved in CH2Cl2 (300 mL) and silica gel (Sorbent Technologies, 60 Å, 32–63 µm) (140 g) was added. The solvent was removed on a rotary evaporator and the dry powdered solid was irradiated in a microwave oven in an open crystallizing dish for 10 minutes at 860 watts. The solid was thoroughly washed with methanol and the solvent was removed. The crude product was purified by flash chromatography (silica gel/hexane/ethyl acetate) to yield a pure product as a colorless oil (2.33 g; 30% yield). 1H NMR (CDCl3): δ 2.85-3.02 (m, 1H), 3.09-3.17 (dd, 1H, J = 12, 6 Hz), 3.38-3.46 (d, 1H, J = 12 Hz), 3.46-3.56 (m, 1H), 3.72-3.84 (m, 2H), 3.95-4.04 (d, 1H, J = 12 Hz), 7.08-7.18 (m, 10H).
(RS)-Ethyl 2-(3-(3,4-dibenzyl-5-oxoimidazolidin-1-yl)ureido)acetate 7
Compound 6 (0.56 g; 2 mmol) in dry CH2Cl2 (3 mL) was cooled to 0 °C and then treated with triethylamine (0.33 mL, 2.4 mmol) and ethyl isocyanatoacetate (0.26 g; 2 mmol). The reaction mixture was stirred overnight at room temperature. The mixture was treated with ethyl acetate (40 mL) and then washed successively with 5% aqueous HCl (10 mL), 5% aqueous NaHCO3 (10 mL) and brine (10 mL). The organic phase was dried over anhydrous Na2SO4 and the solvent was removed. The crude product was purified by flash chromatography (silica gel/hexane/ethyl acetate) to yield a pure product as a colorless oil (0.15 g; 18%). 1H NMR (CDCl3): δ 1.20-1.28 (t, 3H, J = 6 Hz), 2.92-3.15 (m, 2H), 3.48-3.55 (d, 1H, J = 9 Hz), 3.55-3.61 (m, 1H), 3.75-3.92 (m, 3H), 4.00-4.19 (m, 4H), 5.80 (s, 1H), 7.15-7.28 (m, 10H); 13C NMR (CDCl3): δ 173.37, 170.86, 156.88, 137.83, 137.22, 130.06, 129.78, 128.85, 128.64, 128.55, 128.52, 128.41, 127.71, 126.68, 69.65, 65.44, 61.51, 59.03, 41.94, 37.11, 14.27.
(RS)-1-(3,4-Dibenzyl-5-oxoimidazolidin-1-yl)-3-phenethylurea 8
Prepared from compound 6 and 2-phenethyl isocyanate using a procedure similar to that used in the synthesis of compound 7 to give a pure product as a colorless oil (0.61 g; 72% yield). 1H NMR (CDCl3): δ 2.56-2.79 (m, 2H), 2.91-3.12 (m, 2H), 3.14-3.44 (m, 2H), 3.44-3.54 (d, 1H, J = 12 Hz), 3.51-3.63 (m, 1H), 3.80-4.00 (m, 3H), 4.64-4.78 (m, 1H), 6.64-6.76 (s, 1H), 7.00-7.44 (m, 15H); 13C NMR (CDCl3): δ 173.05, 156.87, 139.07, 137.68, 137.11, 130.15, 128.99, 128.85, 128.72, 128.65, 128.39, 127.82, 126.79, 126.49, 69.72, 65.33, 58.87, 41.54, 37.01, 36.22.
(RS)-4-(3,4-Dibenzyl-5-oxoimidazolidin-1-ylamino)-4-oxobutanoic acid 9
To compound 6 (0.56 g; 2 mmol) in dry CH2Cl2 (10 mL) was added succinic anhydride (0.20 g; 2 mmol) and the reaction mixture was stirred overnight at room temperature. The solvent was removed on a rotary evaporator and the crude product was washed with 20 mL diethyl ether to give a pure product as a white solid (0.54 g; 71% yield), mp 131.5-133.0 °C. 1H NMR (CDCl3): δ 2.39-2.59 (m, 2H), 2.64-2.74 (m, 2H), 2.91-3.19 (m, 2H), 3.49-3.58 (d, 1H, J = 12 Hz), 3.56-3.64 (m, 1H), 3.73-3.82 (d, 1H, J = 12 Hz), 4.04-4.23 (m, 2H), 7.12-7.36 (m, 10H), 8.33-8.46 (s, 1H); 13C NMR (CD3OD): δ 174.69, 172.41, 171.46, 137.59, 137.03, 129.67, 128.57, 128.36, 128.17, 127.44, 126.43, 68.61, 65.32, 58.95, 36.85, 28.63, 28.15.
(RS)-1-Benzyl-N-[(RS)-3,4-dibenzyl-5-oxoimidazolidin-1-yl]-5-oxopyrrolidine-3-carboxamide 10
A solution of 1-benzyl-5-oxopyrrolidine-3-carboxylic acid (0.44 g; 2 mmol) dissolved in 20 mL DMF was treated with 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.46 g; 2.4 mmol) and the solution was stirred for 20 minutes. Compound 6 (0.56 g; 2 mmol) was added and the reaction mixture was stirred at room temperature overnight. The solvent was removed in vacuo and the residue was dissolved in CH2Cl2 (70 mL) and then was washed sequentially with 5% aqueous HCl (3 × 20 mL), 5% aqueous NaHCO3 (3 × 20 mL) and brine (3 × 20 mL). The organic phase was dried over anhydrous Na2SO4 and the solvent was removed. The crude product was purified by flash chromatography (silica gel/hexane/ethyl acetate) to yield a pure product as a colorless oil (0.28g; 29% yield). 1H NMR (CDCl3): δ 2.50-2.80 (m, 2H), 2.86-2.98 (m, 1H), 3.00-3.17 (m, 2H), 3.28-3.49 (m, 3H), 3.48-3.56 (m, 1H), 3.71-3.86 (m, 1H), 3.93-4.02 (t, 1H, J = 6 Hz), 4.02-4.11 (m, 1H), 4.30-4.42 (t, 2H, J = 6 Hz), 7.05-7.40 (m, 15H), 9.16-9.22 (d, 1H, J = 15 Hz); 13C NMR (CDCl3): δ 172.75, 172.47, 171.77, 137.74, 137.09, 135.73, 129.79, 128.91, 128.75, 128.62, 128.43, 128.09, 127.89, 127.71, 126.69, 68.73, 65.29, 59.09, 49.08, 46.63, 37.09, 34.88, 34.56, 34.39.
(RS)-N-[(RS)-3,4-Dibenzyl-5-oxoimidazolidin-1-yl]-1-(4-fluorobenzyl)-5-oxopyrrolidine-3-carboxamide 11
Prepared from compound 6 and 1-(4-fluorobenzyl)-5-oxopyrrolidine-3-carboxylic acid using a procedure similar to that used in the synthesis of compound 10 to give a pure product as a colorless oil (0.34 g; 34% yield). 1H NMR (CDCl3): δ 2.54-2.83 (m, 2H), 2.91-3.18 (m, 3H), 3.30-3.51 (m, 3H), 3.51-3.62 (m, 1H), 3.78-3.90 (t, 1H, J = 12 Hz), 3.97-4.12 (m, 2H), 4.27-4.51 (m, 2H), 6.94-7.08 (m, 2H), 7.12-7.40 (m, 12H), 7.66-7.80 (d, 1H, J = 24 Hz); 13C NMR (CDCl3): δ 172.65, 172.61, 172.49, 172.43, 171.74, 164.04, 160.78, 137.68, 137.07, 137.04, 131.63, 131.59, 129.93, 129.82, 128.75, 128.66, 128.44, 127.78, 126.73, 115.96, 115.67, 68.74, 65.25, 58.99, 48.88, 45.93, 37.02, 34.83, 34.54.
(RS)-N-[(RS)-3,4-Dibenzyl-5-oxoimidazolidin-1-yl]-1-(furan-2-ylmethyl)-5-oxopyrrolidine-3-carboxamide 12
Prepared from compound 6 and 1-(furan-2-ylmethyl)-5-oxopyrrolidine-3-carboxylic acid using a procedure similar to that used in the synthesis of compound 10 to give a pure product as a colorless oil (0.51g; 40% yield). 1H NMR (CDCl3): δ 2.50-2.75 (m, 2H), 2.90-3.01 (m, 1H), 3.01-3.17 (m, 2H), 3.37-3.63 (m, 4H), 3.76-3.90 (m, 1H), 4.00-4.15 (m, 2H), 4.28-4.53 (m, 2H), 6.20-6.30 (m, 2H), 7.07-7.42 (m, 11H), 8.67-8.82 (d, 1H, J = 27 Hz); 13C NMR (CDCl3): δ 172.63, 172.54, 171.78, 149.32, 142.80, 137.74, 137.11, 129.81, 128.78, 128.63, 128.45, 127.73, 126.71, 110.59, 108.90, 68.77, 65.33, 59.13, 49.16, 39.35, 37.09, 34.92, 34.51.
(RS)-2-(3,4-Dibenzyl-5-oxoimidazolidin-1-ylcarbamoyl)benzoic acid 13
Prepared from compound 6 and phthalic anhydride using a similar procedure as that used in the synthesis of compound 9 to give a pure product as a white solid (0.47 g; 47% yield), mp 132.0-134.0 °C. 1H NMR (CDCl3): δ 2.91-3.17 (m, 2H), 3.64-3.74 (m, 2H), 3.74-3.82 (d, 1H, J = 15 Hz), 4.29-4.40 (dd, 1H, J = 18, 6 HZ), 7.04-7.67 (m, 10H), 7.75-8.08 (m, 4H), 9.22(s, 1H); 13C NMR (CD3OD): δ 172.51, 164.16, 163.66, 137.08, 136.61, 134.89, 134.86, 132.30, 130.68, 129.48, 129.45, 129.38, 128.82, 128.47, 128.33, 128.18, 128.18, 127.34, 126.35, 123.79, 123.77, 67.96, 64.74, 59.04, 36.65.
(RS)-1-(4-Benzyl-5-oxoimidazolidin-1-yl)-3-phenethylurea 14
A solution of compound 8 (0.50 g; 1.2 mmol) in dry methanol (50 mL) was treated with 10% palladium on carbon (0.35 g) under hydrogen gas and the reaction mixture was stirred for 1h at room temperature. The catalyst was filtered off and the solvent was removed on a rotary evaporator leaving a crude product which was washed with diethyl ether (2 × 5 mL) to yield a pure product as a white solid (0.23 g; 59% yield), mp 130.0-131.0 °C. 1H NMR (CDCl3): δ 2.69-2.85(m, 2H), 2.92-3.14 (m, 2H), 3.33-3.43 (m, 2H), 3.70-3.82 (m, 1H), 4.04-4.15 (t, 1H, J = 9 Hz), 4.26-4.37 (dd, 1H, J = 12, 6 Hz), 4.98-5.13 (t, 1H, J = 9 Hz), 6.97-7.11 (s, 1H), 7.11-7.38 (m, 10H); 13C NMR (CDCl3): δ 175.59, 156.93, 139.06, 136.95, 129.63, 129.03, 128.88, 128.75, 127.25, 126.64, 64.73, 59.28, 41.56, 36.82, 36.22.
(RS)-4-(4-Benzyl-5-oxoimidazolidin-1-ylamino)-4-oxobutanoic acid 15
Prepared from compound 9 using a similar procedure as that of compound 14 to give a pure product as a light yellow oil (0.34 g; 89% yield). 1H NMR (CDCl3): δ 2.43-2.73 (m, 4H), 3.09-3.26 (m, 1H), 3.30-3.47 (m, 1H), 4.23-4.38 (m, 1H), 4.51-4.69 (m, 2H), 7.16-7.46 (m, 5H); 13C NMR (CD3OD): δ 174.22, 171.94, 165.82, 133.83, 129.34, 129.03, 128.70, 128.61, 127.33, 57.67, 33.68, 28.45, 28.08, 26.08.
(RS)-1-Benzyl-N-[(RS)-4-benzyl-5-oxoimidazolidin-1-yl]-5-oxopyrrolidine-3-carboxamide 16
Prepared from compound 10 using a similar procedure as that of compound 14 to give a pure product as a colorless oil (0.40 g; 65% yield). 1H NMR (CDCl3): δ 2.57-2.84 (m, 2H), 2.85-2.98 (m, 1H), 2.98-3.23 (m, 2H), 3.30-3.58 (m, 3H), 3.68-3.78 (m, 1H), 4.20-4.28 (m, 1H), 4.34-4.41 (m, 1H), 4.44-4.56 (m, 1H), 7.05-7.42 (m, 10H), 8.68-8.86 (d, 1H, J = 18 Hz); 13C NMR (CDCl3): δ 174.90, 172.89, 171.69, 137.81, 137.02, 129.64, 129.41, 128.94, 128.75, 128.42, 128.07, 127.03, 126.64, 66.72, 59.11, 49.08, 46.68, 40.93, 36.56, 34.81.
(RS)-N-[(RS)-4-Benzyl-5-oxoimidazolidin-1-yl]-1-(4-fluorobenzyl)-5-oxopyrrolidine-3-carboxamide 17
Prepared from compound 11 using a similar procedure as that of compound 14 to give a pure product as a colorless oil (0.29 g; 71% yield). 1H NMR (CDCl3): δ 2.63-2.82 (m, 2H), 2.90-3.04 (m, 1H), 3.04-3.21 (m, 23H), 3.34-3.57 (m, 2H), 3.73-3.81 (m, 1H), 4.21-4.34 (m, 1H), 4.34-4.53 (m, 2H), 6.93-7.07 (t, 2H, J = 9 Hz), 7.10-7.38 (m, 7H), 8.07-8.25 (d, 1H, J = 21 Hz); 13C NMR (CDCl3): δ 171.79, 171.73, 164.09, 160.83, 136.92, 131.53, 129.96, 129.85, 129.68, 129.44, 128.83, 128.48, 127.13, 116.02, 115.74, 63.78, 59.16, 48.93, 46.02, 36.49, 34.86.
(RS)-2-(4-Benzyl-5-oxoimidazolidin-1-ylcarbamoyl)benzoic acid 18
Prepared from compound 13 using a similar procedure as that of compound 14 to give a pure product as a light yellow solid (0.32 g; 81% yield), mp 123.5-125.0 °C. 1H NMR (DMSO): δ 2.66-2.80 (m, 1H), 2.92-3.06 (m, 1H), 3.15 (s, 1H), 3.51-3.63 (m, 1H), 4.00-4.16 (m, 1H), 4.22-4.34 (m, 1H), 7.12-7.40 (m, 5H), 7.42-7.74 (m, 4H), 7.07 (s, 1H), 10.43 (s, 1H); 13C NMR (CD3OD): δ 173.01, 164.66, 164.16, 135.51, 134.50, 131.82, 131.36, 130.63, 130.19, 129.50, 128.51, 124.23, 65.24, 59.54, 37.15.
The compounds derived from Z-DL-Leu-OH (compounds 19–26) were synthesized using procedures as those used in the synthesis of compounds 1–18.
(RS)-Ethyl 2-(3-(3-benzyl-4-isobutyl-5-oxoimidazolidin-1-yl)ureido)acetate 19
White solid (50% yield), mp 114.0-115.5 °C. 1H NMR (DMSO): δ 0.68-0.90 (m, 6H), 1.10-1.24 (t, 3H, J = 9 Hz), 1.42-1.58 (m, 2H), 1.78-1.93 (m, 1H), 3.17-3.26 (m, 1H), 3.57-3.66 (d, 1H, J = 12 Hz), 3.80-3.95 (m, 3H), 3.97-4.14 (m, 3H), 4.22-4.30 (d, 1H, J = 6 Hz), 6.24-6.36 (t, 1H, J = 6 Hz), 7.12-7.36 (m, 5H), 8.05 (s, 1H); 13C NMR (CDCl3): δ 175.49, 170.92, 156.76, 137.58, 129.14, 128.74, 127.87, 69.68, 62.75, 61.66, 59.45, 42.10, 39.08, 24.88, 23.17, 22.51, 14.33.
(RS)-1-(3-Benzyl-4-isobutyl-5-oxoimidazolidin-1-yl)-3-phenethylurea 20
Oil (33% yield). 1H NMR (DMSO): δ 0.75-0.98 (m, 6H), 1.46-1.64 (m, 2H), 1.84-1.98 (m, 1H), 2.67-2.82 (t, 2H, J = 9 Hz), 3.19-3.28 (m, 1H), 3.30-3.45 (m, 2H), 3.57-3.67 (d, 1H, J = 12 Hz), 3.90-4.00 (d, 1H, J = 12 Hz), 4.00-4.07 (d, 1H, J = 6 Hz), 4.23-4.51 (d, 1H, J = 6 Hz), 5.52-5.63 (t, 1H, J = 6 Hz), 7.08-7.42 (m, 10H), 7.72 (s, 1H); 13C NMR (CDCl3): δ 175.15, 156.82, 139.03, 137.34, 129.08, 128.81, 127.97, 126.67, 69.74, 62.57, 59.22, 41.53, 39.17, 36.23, 24.87, 23.10, 22.63.
(RS)-4-(3-Benzyl-4-isobutyl-5-oxoimidazolidin-1-ylamino)-4-oxobutanoic acid 21
Oil (52% yield). 1H NMR (DMSO): δ 0.70-0.82 (m, 3H), 0.82-0.95 (m, 3H), 1.47-1.63 (m, 2H), 1.78-1.95 (m, 1H), 2.40-2.58 (m, 2H), 2.58-2.74 (m, 2H), 3.23-3.33 (m, 1H), 3.64-3.74 (d, 1H, J = 12 Hz), 3.90-3.98 (d, 1H, J = 12 Hz), 4.05-4.13 (d, 1H, J = 6 Hz), 4.28-4.35 (d, 1H, J = 6 Hz), 7.16-7.37 (m, 5H), 9.06 (s, 1H); 13C NMR (CDCl3): δ 173.56, 172.79, 172.29, 137.50, 129.27, 129.19, 128.77, 128.69, 127.96, 67.40, 62.46, 59.89, 38.79, 26.91, 26.77, 24.91, 23.20, 22.21.
(RS)-N-(3-Benzyl-4-isobutyl-5-oxoimidazolidin-1-yl)-3-phenylpropanamide 22
White solid (60% yield), mp 102.0-103.5 °C. 1H NMR (DMSO): δ 0.74-0.97 (m, 6H), 1.47-1.67 (m, 2H), 1.84-1.99 (m, 1H), 2.42-2.58 (t, 2H, J = 9 Hz), 2.84-2.98 (t, 2H, J = 9 Hz), 3.23-3.31 (m, 1H), 3.62-3.70 (d, 1H, J = 12 Hz), 3.89-3.98 (d, 1H, J = 12 Hz), 3.98-4.06 (d, 1H, J = 6 Hz), 4.17-4.24 (d, 1H, J = 6 Hz), 7.04-7.41 (m, 10H), 9.17 (s, 1H); 13C NMR (CDCl3): δ 174.34, 171.11, 140.36, 137.72, 129.17, 128.85, 128.76, 128.57, 127.89, 126.69, 68.63, 62.69, 59.67, 39.01, 36.06, 31.32, 24.93, 23.23, 22.44.
(RS)-2-(3-Benzyl-4-isobutyl-5-oxoimidazolidin-1-ylcarbamoyl)benzoic acid 23
White solid (53% yield), mp 105.5-107.0 °C. 1H NMR (DMSO): δ 0.78-1.07 (m, 6H), 1.60-1.85 (m, 2H), 1.92-2.08 (m, 1H), 3.38-3.47 (m, 1H), 3.86-3.95 (d, 1H, J = 12 Hz), 4.04-4.12 (d, 1H, J = 12 Hz), 4.17-4.23 (d, 1H, J = 6 Hz), 4.52-4.59 (d, 1H, J = 6 Hz), 7.20-7.48 (m, 5H), 7.71-7.96 (m, 4H); 13C NMR (CDCl3): δ 173.95, 164.77, 164.11, 137.82, 136.00, 135.03, 130.10, 130.01, 129.17, 128.66, 128.02, 125.94, 124.12, 68.09, 62.25, 59.87, 38.91, 24.93, 23.10, 22.45.
(RS)-Benzyl 1-[(RS)-3-benzyl-4-isobutyl-5-oxoimidazolidin-1-ylamino)-1-oxo-3-phenylpropan-2-ylcarbamate 24
Oil (48% yield). 1H NMR (DMSO): δ 0.73-0.85 (m, 3H), 0.85-0.98 (m, 3H), 1.47-1.66 (m, 2H), 1.84-1.96 (m, 1H), 2.92-3.17 (m, 2H), 3.20-3.28 (m, 1H), 3.57-3.68 (m, 1H), 3.78-3.86 (m, 1H), 3.86-3.99 (m, 2H), 3.99-4.08 (m, 1H), 4.85-5.06 (m, 2H), 7.05-7.40 (m, 10H), 8.64-8.80 (d, 1H, J = 24 Hz); 13C NMR (CDCl3): δ 174.01, 170.35, 156.25, 137.79, 136.21, 129.81, 129.40, 129.21, 129.14, 128.53, 128.34, 127.37, 68.23, 67.60, 62.55, 59.55, 59.43, 39.20, 38.31, 24.52, 23.23, 22.67.
(RS)-N-(3-Benzyl-4-isobutyl-5-oxoimidazolidin-1-yl)-N-(methylsulfonyl)methanesulfon-amide 25
White solid (38% yield), mp 88.0-89.5 °C. 1H NMR (DMSO): δ 0.77-1.03 (m, 6H), 1.48-1.73 (m, 2H), 1.83-2.03 (m, 1H), 3.24-3.35 (m, 1H), 3.36-3.50 (d, 6H, J = 9 Hz), 3.70-3.80 (d, 1H, J = 18 Hz), 3.88-3.97 (d, 1H, J = 18 Hz), 4.07-4.14 (d, 1H, J = 9 Hz), 4.45-4.53 (d, 1H, J = 9 Hz), 7.23-7.47 (m, 5H); 13C NMR (CDCl3): δ 174.67, 137.33, 129.35, 128.90, 128.15, 69.25, 62.08, 59.77, 45.60, 45.05, 38.03, 24.95, 23.00, 22.26.
(RS)-N-(3-Benzyl-4-isobutyl-5-oxoimidazolidin-1-yl)-4-methyl-N-tosylbenzenesulfon amide 26
White solid (53% yield), mp 83.0-85.0 °C. 1H NMR (DMSO): δ 0.73-0.95 (m, 6H), 1.34-1.71 (m, 2H), 1.82-2.02 (m, 1H), 2.42 (s, 6H), 3.12-3.22 (m, 1H), 3.67-3.77 (d, 1H, J = 12 Hz), 3.84-3.93 (d, 1H, J = 12 Hz), 4.13-4.18 (d, 1H, J = 6 Hz), 4.44-4.56 (d, 1H, J = 6 Hz), 7.20-7.45 (m, 9H), 7.70-7.92 (m, 4H); 13C NMR (CDCl3): δ 174.02, 146.06, 145.99, 137.37, 135.37, 135.27, 129.82, 129.79, 129.42, 129.30, 129.22, 128.78, 127.97, 69.03, 62.18, 59.77, 37.86, 24.78, 23.21, 22.26, 22.03.
Enzyme assay and inhibitory studies
Human neutrophil elastase (HNE)
In a typical inhibition experiment, 10 µL of 7.0 µM HNE was incubated with 10 µL of 3.5 mM solution of inhibitor in DMSO ([inhibitor]/[enzyme]=500), 970 µL 0.1 M HEPES/0.5 M NaCl buffer, pH 7.25, at 25 °C. After 30 min, 10 µL of 70 mM methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide was added and the amount of active enzyme was determined by monitoring the release of p-nitroaniline at 410 nm for 2 min. A control (hydrolysis) containing 970 µL 0.1 M HEPES/0.5 M NaCl buffer, pH 7.25, 10 µL of 7.0 µM HNE, and 10 µL DMSO was run under the same conditions. The remaining activity of HNE was expressed as %remaining activity = (v/v0) × 100 and is the average of duplicate or triplicate determinations.
Determination of inhibition constant (KI) for compound 24 against HNE
The KI of inhibitor 24 was determined using a Dixon plot [18]. Briefly, 970 µL 0.1 M HEPES buffer, pH 7.25, containing 0.5 M NaCl, was added in six cuvettes, followed by the sequential addition of 0, 2, 4, 6, 8 and 10 µL of 5 mM solution of inhibitor 24 in DMSO and 10, 8, 6, 4, 2, 0 µL DMSO for final inhibitor concentrations of 0–50 µM. Ten µL of 7.0 µM HNE and 10 µL of 35 mM substrate (methoxysuccinyl Ala-Ala-Pro-Val p-nitroanilide) were added. The amount of active enzyme was determined by monitoring the release of p-nitroaniline at 410 nm for 2 minutes. The experiment was repeated at a second substrate concentration (70 mM) and a Dixon plot was constructed by plotting 1/v versus [I] (Figure 3).
Human neutrophil proteinase 3 (Pr 3)
Ten microliters of a 1.73 mM inhibitor solution in DMSO ([inhibitor]/[enzyme]=500) and 20 µL of 12.98 mM Boc-Ala-Ala-Nva-SBzl in DMSO were added to a cuvette containing 940 µL 0.1 M HEPES/0.5 M NaCl buffer, pH 7.25. After the solution was well mixed, 20 µL of 32 mM 5,5’-dithio-bis(2-nitrobenzioc acid) in DMSO and 10 µL of 3.45 µM solution of proteinase 3 in 0.1 M phosphate/0.25 M NaCl buffer, pH 6.50, were added and the change in absorbance was monitored at 410 nm for 2 min. A control (hydrolysis) containing 940 µL 0.1 M HEPES/0.5 M NaCl, pH 7.25, 10 µL 3.40 µM solution of proteinase 3, 20 µL DMSO, 20 µL of 16 mM 5,5’-dithio-bis(2-nitrobenzioc acid) in DMSO, and 20 µL of 6.49 mM Boc-Ala-Ala-Nva-SBzl in DMSO was also run under the same condition. Determinations were performed in duplicate or triplicate and remaining activity was calculated as described for HNE.
Acknowledgements
Support of this work by the Heart and Blood Institute of the National Institutes of Health (HL 57788) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Maclay JD, Rabinovich RA, MacNee W. Am. J. Respir. Crit. Care Med. 2009;179:533–541. doi: 10.1164/rccm.200901-0134UP. [DOI] [PubMed] [Google Scholar]
- 2.Senior RW, Shapiro SD. COPD: Epidemiology, Pathophysiology, and Pathogenesis in Fishman’s Pulmonary Diseases and Disorders. 3rd ed. NY: McGrawHill; 1998. pp. 659–628. [Google Scholar]
- 3.Vandivier RM, Voelkel NF. J. Chron. Obstr. Pulmon. Dis. 2005;2:177–184. [Google Scholar]
- 4.Churg A, Wright JL. Proc. Am. Thorac. Soc. 2009;6:550–552. doi: 10.1513/pats.200903-012DS. [DOI] [PubMed] [Google Scholar]
- 5.(a) MacNee W. Proc. Am. Thor. Soc. 2005;2:50–60. doi: 10.1513/pats.200411-056SF. [DOI] [PubMed] [Google Scholar]; (b) Luppi F, Hiemstra PS. Am. J. Respir. Crit. Care Med. 2007;175:527–531. doi: 10.1164/rccm.200612-1821ED. [DOI] [PubMed] [Google Scholar]; (c) Rangasamy T, Misra V, Zhen L, Tankersley CG, Tuder RM, Biswal S. Am. J. Physiol. 2009;296:L888–L900. doi: 10.1152/ajplung.90369.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Aoshiba K, Yokohori N, Nagai A. Am. J. Respir. Cell Mol. Biol. 2003;28:555–562. doi: 10.1165/rcmb.2002-0090OC. [DOI] [PubMed] [Google Scholar]; (b) Tuder RM, Petrache I, Elias JA, Voelkel NF, Henson PM. Am. J. Respir. Cell Mol. Biol. 2003;28:551–554. doi: 10.1165/rcmb.F269. [DOI] [PubMed] [Google Scholar]; (c) Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Respir. Res. 2006;7:53–62. doi: 10.1186/1465-9921-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stockley RA, Mannino D, Barnes PJ. Proc. Am. Thorac. Soc. 2009;6:524–526. doi: 10.1513/pats.200904-016DS. [DOI] [PubMed] [Google Scholar]
- 8.(a) Wiedow O, Meyer-Hoffert UJ. Int. Med. 2005;257:319–328. doi: 10.1111/j.1365-2796.2005.01476.x. [DOI] [PubMed] [Google Scholar]; (b) Pham CT. N. Nat. Rev. Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 9.(a) Shapiro SD, Ingenito EP. Am. J. Respir. Cell Mol. Biol. 2005;32:367–372. doi: 10.1165/rcmb.F296. [DOI] [PubMed] [Google Scholar]; (b) Barnes PJ. Pharmacol. Rev. 2004;56:548–515. doi: 10.1124/pr.56.4.2. [DOI] [PubMed] [Google Scholar]
- 10.Mouded M, Egea EE, Brown MJ, Hanlon SM, Houghton AM, Tsai LW, Ingenito EP, Shapiro SD. Am. J. Respir. Cell Mol. Biol. 2009;41:407–414. doi: 10.1165/rcmb.2008-0137OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Greene CM, McElvaney NG. Brit. J. Pharmacol. 2009;158:1048–1058. doi: 10.1111/j.1476-5381.2009.00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hansel TH, Barnes PJ. Lancet. 2009;374:744–755. doi: 10.1016/S0140-6736(09)61342-8. [DOI] [PubMed] [Google Scholar]
- 12.Nomenclature used is that of Schechter I, Berger A. Biochem. Biophys. Res. Comm. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. where S1, S2, S3,…Sn and S1’, S2’, S3’,…Sn’ correspond to the enzyme subsites on either side of the scissile bond. Each subsite accommodates a corresponding amino acid residue side chain designated P1, P2, P3, …Pn and P1’, P2’, P3’, …Pn’ of the substrate or (inhibitor). S1 is the primary substrate specificity subsite, and P1-P1’ is the scissile bond.
- 13.(a) Korkmaz B, Moreau T, Gauthier F. Biochimie. 2008;90:227–242. doi: 10.1016/j.biochi.2007.10.009. [DOI] [PubMed] [Google Scholar]; (b) Fujinaga M, Chernaia MM, Halenbeck R, Koths K, James MNG. J. Mol. Biol. 1996;261:267–278. doi: 10.1006/jmbi.1996.0458. [DOI] [PubMed] [Google Scholar]; (c) Wysocka M, Lesner A, Guzow K, Mackiewicz L, Legowska A, Wiczk W, Rolka K. Anal. Biochem. 2008;378:208–215. doi: 10.1016/j.ab.2008.04.003. [DOI] [PubMed] [Google Scholar]; (d) Koehl C, Knight CG, Bieth JG. J. Biol. Chem. 2003;278:12609–12612. doi: 10.1074/jbc.M210074200. [DOI] [PubMed] [Google Scholar]; (e) Hajjar E, Korkmaz B, Gauthier F, Brandsdal BO, Witko-Sarsat V, Reuter NJ. Med. Chem. 2006;49:1248–1260. doi: 10.1021/jm051018t. [DOI] [PubMed] [Google Scholar]; (f) Korkmaz B, Attucci S, Hazouard E, Ferrandiere M, Jourdan ML, Brillard-Bourdet M, Juliano L, Gauthier FJ. Biol. Chem. 2002;277:39074–39081. doi: 10.1074/jbc.M202918200. [DOI] [PubMed] [Google Scholar]; (g) Korkmaz B, Attucci S, Moreau T, Godat E, Juliano L, Gauthier F. Am. J. Respir. Cell Mol. Biol. 2004;30:801–807. doi: 10.1165/rcmb.2003-0139OC. [DOI] [PubMed] [Google Scholar]
- 14.Wei L, Gan X, Zhong J, Alliston KR, Groutas WC. Bioorg. Med. Chem. 2003;11:5149–5153. doi: 10.1016/j.bmc.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 15.Abdel-Magid AF, Garson KG, Harris BD, Maryanoff CA, Shah RD. J. Org. Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 16.Apelqvist T, Wensbo D. Tetrahedron Lett. 1996;37:1471–1472. [Google Scholar]
- 17.Li Y, Dou D, He G, Lushington GH, Groutas WC. Bioorg. Med. Chem. 2009;17:3536–3542. doi: 10.1016/j.bmc.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dixon M. Biochem. J. 1953;55:170–171. doi: 10.1042/bj0550170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Groutas WC, Hoidal JR, Brubaker MJ, Stanga MA, Venkataraman R, Gray BH, Rao NV. J. Med. Chem. 1990;33:1085–1087. doi: 10.1021/jm00166a001. [DOI] [PubMed] [Google Scholar]; (b) Groutas WC, Brubaker MJ, Chong LS, Venkataraman R, Huang H, Epp JB, Kuang R, Hoidal JR. Med. Chem. Lett. 1995;3:375–381. doi: 10.1016/0968-0896(95)00024-b. [DOI] [PubMed] [Google Scholar]; (c) Groutas WC, Chong LS, Venkataraman R, Kuang R, Epp JB, Houser-Archield N, Huang H, Hoidal JR. Arch. Biochem. Biophys. 1996;332:335–340. doi: 10.1006/abbi.1996.0350. [DOI] [PubMed] [Google Scholar]; (d) Groutas WC, Kuang R, Ruan S, Epp JB, Venkataraman R, Truong TM. Bioorg. Med. Chem. 1998;6:661–671. doi: 10.1016/s0968-0896(98)00006-6. [DOI] [PubMed] [Google Scholar]; (e) Groutas WC, Epp JB, Ruan S, Yu S, Huang H, He S, Tu J, Schechter NM, Turbov J, Froelisch CJ, Groutas WC. J. Am. Chem. Soc. 1999;121:8128–8129. [Google Scholar]; (f) Kuang R, Epp JB, Ruan S, Chong LS, Venkataraman R, Tu J, He S, Truong TM, Groutas WC. Bioorg. Med. Chem. 2000;8:1005–1016. doi: 10.1016/s0968-0896(00)00038-9. [DOI] [PubMed] [Google Scholar]; (g) He S, Kuang R, Venkataraman R, Tu J, Truong TM, Chan H-K, Groutas WC. Bioorg. Med. Chem. 2000;8:1713–1717. doi: 10.1016/s0968-0896(00)00101-2. [DOI] [PubMed] [Google Scholar]; (h) Groutas WC, He S, Kuang R, Ruan S, Tu J, Chan H-K. Bioorg. Med. Chem. 2001;9:1543–1548. doi: 10.1016/s0968-0896(01)00037-2. [DOI] [PubMed] [Google Scholar]; (i) Groutas WC, Epp JB, Kuang R, Ruan S, Chong LS, Venkataraman R, Tu J, He S, Yu H, Fu Q, Li Y-H, Truong TM, Vu NT. Arch. Biochem. Biophys. 2001;385:162–169. doi: 10.1006/abbi.2000.2139. [DOI] [PubMed] [Google Scholar]; (j) Huang W, Yamamoto Y, Li Y, Dou D, Alliston KR, Hanzlik RP, Groutas WC. J. Med. Chem. 2008;51:2003–2008. doi: 10.1021/jm700966p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Powers JC, Asgian JL, Ekici OD, James KE. Chem. Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]