

Abstract
Type II topoisomerases are essential enzymes that regulate DNA under- and overwinding and remove knots and tangles from the genetic material. In order to carry out their critical physiological functions, these enzymes utilize a double-stranded DNA passage mechanism that requires them to generate a transient double-stranded break. Consequently, while necessary for cell survival, type II topoisomerases also have the capacity to fragment the genome. This feature of the prokaryotic and eukaryotic enzymes, respectively, is exploited to treat a variety of bacterial infections and cancers in humans. All type II topoisomerases require divalent metal ions for catalytic function. These metal ions function in two separate active sites and are necessary for the ATPase and DNA cleavage/ligation activities of the enzymes. ATPase activity is required for the strand passage process and utilizes the metal-dependent binding and hydrolysis of ATP to drive structural rearrangements in the protein. Both the DNA cleavage and ligation activities of type II topoisomerases require divalent metal ions and appear to utilize a novel variant of the canonical two-metal-ion phosphotransferase/hydrolase mechanism to facilitate these reactions. This article will focus primarily on eukaryotic type II topoisomerases and the roles of metal ions in the catalytic functions of these enzymes.
Keywords: Topoisomerase IIα, divalent metal ion, divalent cation, ATP hydrolysis, DNA cleavage, DNA ligation, topoisomerase II poisons
1. DNA Topology
DNA is essentially an extremely long double-stranded rope in which the two strands are plectonemically coiled about one another. As a result of these structural features, topological challenges profoundly affect every major nucleic acid process that requires opening or movement of the double helix.1–5
Topological properties of DNA, including under- and overwinding, knotting, and tangling, can only be changed when one or both strands of DNA are broken.2 DNA underwinding (i.e., negative supercoiling) is important because duplex DNA is the storage form for the genetic information. In order to replicate or express this information, the two strands of the double helix must be separated. Since negative supercoiling destabilizes the double-stranded nature of DNA, it greatly facilitates strand separation.2–7 The importance of this attribute is underscored by the fact that genomes ranging from bacteria to humans are globally underwound by ~6%.2, 8–10
While negative supercoiling enhances many nucleic acid processes, DNA overwinding (i.e., positive supercoiling) inhibits them. The linear movement of enzymes such as helicases and polymerases through the double helix induces DNA overwinding ahead of tracking systems.2, 8–10 This positive supercoiling makes it increasingly difficult to open the two strands of the double helix and ultimately blocks critical cellular processes such as DNA replication and transcription.2–5, 11
Essential nucleic acid functions, such as DNA recombination and replication generate knots and tangles within the double helix.2, 4, 11–15 DNA knots impair the ability to separate the two strands of the genetic material and reduce the tensile strength of the double helix. As a result, they prevent the completion of replication, adversely affect transcription, and induce genomic instability.16–20 Intermolecular DNA tangles prevent segregation of chromosomes during mitosis.2, 4, 11–13 Consequently, these two topological structures can be lethal to cells if they are not resolved.16–20
2. Topoisomerase II
The topological state of DNA is regulated in the cell by enzymes known as topoisomerases.1, 11–13, 21–23 These enzymes are required for the survival of all organisms and alter DNA topology by generating transient breaks in the double helix.1, 11–13, 21–23 There are two major classes of topoisomerases, type I and type II, that are distinguished by the number of DNA strands that they cleave.1, 11–13, 21–23
Bacterial and eukaryotic type I topoisomerases (topoisomerase I and III) are monomeric enzymes that require no high-energy cofactor.1, 21, 24 These enzymes alter topology by creating a transient single-stranded break in the DNA, followed by passage of the opposite intact strand through the break or by controlled rotation of the helix around the break.1, 21, 24 As a result of their reaction mechanism, type I topoisomerases can modulate DNA under- and overwinding (removing one supercoil per catalytic event), but cannot remove knots or tangles from duplex DNA. These enzymes play important roles in DNA replication and transcription and the maintenance of genomic stability.1, 11, 21, 24 Type I topoisomerases will not be discussed in detail in this article. However, several excellent review articles on these enzymes have appeared recently if the reader is interested in obtaining additional information.21, 24–25
All type II topoisomerases require ATP for complete catalytic activity.12, 26–29 These enzymes interconvert different topological forms of DNA by passing an intact double helix through a transient double-stranded break that they generate in a separate segment of DNA.12, 26–29 As a result of this double-stranded DNA passage mechanism, type II topoisomerases can modulate DNA super-coiling (removing two supercoils per catalytic event) and also can remove DNA knots and tangles.
There are two bacterial type II enzymes, gyrase and topoisomerase IV.1, 26, 30–31 These enzymes 1, are encoded by separate genes, but are closely related and are heterotetrameric (A2B2) in structure.1, 22, 26, 30–31 Gyrase is composed of two subunits: GyrA contains the active site for DNA cleavage and ligation and GyrB contains the site of ATP binding and hydrolysis. GyrB also contains conserved acidic amino acid residues that coordinate the divalent metal ions that are required for DNA cleavage and ligation. The homologous subunits in topoisomerase IV are ParC and ParE (named as genes required for partitioning in Gram-negative species) or GrlA and GrlB (named as gyrase-like genes in Gram-positive species).1, 26, 30–32 Gyrase is unique among type II topoisomerases, in that it can actively underwind (i.e., negatively supercoil) the double helix.1, 22, 26, 30–31 This enzyme is critical for maintaining the global balance of DNA supercoiling in bacteria and plays important roles in DNA replication and transcription.22, 26, 30–31 Topoisomerase IV helps to alleviate positive DNA supercoiling that occurs during processes such as replication and removes the bulk of knots and tangles from the bacterial genome.22, 26, 30–31
Eukaryotic type II topoisomerases share homology with both gyrase and topoisomerase IV, but the two genes that encode the bacterial enzymes have fused into a single gene in eukaryotes.22, 26, 30, 33 Consequently, the eukaryotic enzymes are homodimeric in nature. The N-terminal domain is homologous to GyrB and the central domain is homologous to GyrA.12, 22, 29, 33 The C-terminal domain of eukaryotic topoisomerase II corresponds to the C-terminal domain of GyrA.22, 33 This portion of the protein is highly variable across species and imparts unique functional and physiological properties to individual family members. For example, the C-terminal domain of gyrase contains elements that allow the enzyme to negatively supercoil DNA.31 The corresponding portion of the eukaryotic type II enzyme contains nuclear localization signals34–36 and allows the α isoform of topoisomerase II (see below) to distinguish the handedness of DNA supercoils and relax (i.e., remove) positive DNA supercoils ~10 times faster than it does negative.37–39
Lower eukaryotes and invertebrates encode only a single type II topoisomerase, called topoisomerase II.40–43 In contrast, vertebrate species encode two closely related isoforms, topoisomerase IIα and topoisomerase IIβ .11–12, 21, 29, 44–51 These isoforms differ in their protomer molecular masses (170 vs. 180 kDa, respectively) and are encoded by separate genes. Topoisomerase IIα and topoisomerase IIβ display a high degree (~70%) of amino acid sequence identity and similar enzymological characteristics.
Similarities notwithstanding, topoisomerase IIα and topoisomerase IIβ have distinct patterns of expression and separate cellular functions. Although topoisomerase IIα is essential for the survival of proliferating cells, topoisomerase IIβ is dispensable at the cellular level.52–54 Furthermore, levels of topoisomerase IIα protein rise dramatically during periods of cell growth,55–58 while expression of the β isoform is independent of proliferative status.34, 59–60 Topoisomerase IIα is associated with replication forks and remains tightly bound to chromosomes during mitosis.58–59, 61–63 Thus, it is believed to be the isoform that functions in growth-related processes, such as DNA replication and chromosome segregation.11, 58 In contrast, topoisomerase IIβ dissociates from chromosomes during mitosis and cannot compensate for the loss of topoisomerase IIα in mammalian cells.58, 60, 64–65 Although the physiological functions of the β isoform have yet to be fully defined, recent evidence indicates involvement in the transcription of hormonally- or developmentally-regulated genes.65–66
Many of the fundamental properties of type II topoisomerases were first established with bacterial systems. Several high quality review articles focus on these enzymes.22, 31, 67–68 The present article will concentrate on eukaryotic type II topoisomerases, but will discuss the prokaryotic enzymes as appropriate.
Finally, much of what we understand regarding the mechanism of action of the eukaryotic type II enzymes comes from experiments with topoisomerase II from species that express only a single form of the protein. Furthermore, the double-stranded DNA passage mechanism utilized by all type II topoisomerases is fundamentally the same. Consequently, eukaryotic type II topoisomerases will be referred to collectively as topoisomerase II, unless the properties being discussed are specific to either the α or β isoform.
3. Catalytic Cycle
As described above, type II topoisomerases alter nucleic acid topology using a double-stranded DNA passage reaction. This reaction can be separated into at least six discrete steps.26–28 One round of catalysis is depicted in Fig. 1.
Figure 1.

Catalytic cycle of topoisomerase II. The homodimeric enzyme is shown in blue, the DNA double helix that is cleaved is shown in green, and the double helix that is passed through the DNA gate is shown in yellow. The double-stranded DNA passage reaction of topoisomerase II can be separated into six discrete steps. 1) DNA binding to regions of helix-helix juxtaposition. 2) Double-stranded DNA cleavage, which requires the presence of Mg2+ (physiologically) or other divalent metal ions. 3) Binding of 2 ATP molecules, which promotes double-stranded DNA passage through the DNA gate generated by cleavage. Strand passage proceeds more rapidly if one of the two ATP molecules is hydrolyzed. 4) Ligation of the cleaved DNA. 5) Hydrolysis of the second ATP molecule, which allows release of the DNA through a C-terminal gate in the protein and promotes 6) enzyme turnover, which allows the enzyme to initiate a new round of catalysis.
1) DNA Binding
Topoisomerase II binds two segments of DNA to initiate a round of catalysis. Evidence suggests that the enzyme preferentially binds to regions of helix-helix juxtaposition formed by DNA supercoiling, knotting, or tangling.69–70 The recognition of DNA crossovers and the subsequent binding of topoisomerase II requires no cofactor.71
2) DNA Cleavage
All topoisomerases utilize active site tyrosine residues to mediate DNA cleavage (Tyr805 and Tyr821 in human topoisomerase IIα and topoisomerase IIβ , respectively).1, 12–13, 21–23, 26–28 Each protomer subunit cleaves one strand of the double helix in a coordinated fashion.72–73 DNA cleavage requires the presence of a divalent metal ion (detailed below),71, 74–75 and takes place via a transesterification reaction that results in the formation of a covalent phosphotyrosyl bond between the protein and the newly generated 5’-terminus of the DNA chain.74–76 Scission also generates a hydroxyl moiety on the 3’-terminus of the cleaved strand. The scissile bonds on the two strands of the double helix are staggered and located across the major groove from one another. Thus, topoisomerase II generates cleaved DNA molecules with 4-base 5’-single-stranded cohesive ends, each of which is covalently linked to a separate protomer subunit of the enzyme.74–76
Although topoisomerase II acts globally, it cleaves DNA at preferred sites.77 The consensus sequence for cleavage is weak,77 and the mechanism by which the enzyme selects its DNA sites is not apparent. Most likely, the specificity of topoisomerase II-mediated cleavage is determined by the local structure, flexibility, or malleability of the DNA that accompanies the sequence, as opposed to a direct recognition of the bases that comprise that sequence.78
It is notable that the scission reaction is reversible and that the enzyme establishes a DNA cleavage-ligation equilibrium. The transient covalent enzyme-DNA cleavage intermediate (known as the “cleavage complex”) that is formed during this process plays two important roles in the topoisomerase II reaction mechanism.1, 22, 26–28 First, it conserves the bond energy of the sugar-phosphate DNA backbone. Second, because it does not allow the cleaved DNA chains to dissociate from the enzyme, the protein-DNA linkage maintains the integrity of the genetic material during the cleavageevent.
The topoisomerase II-DNA cleavage complex is also important because it is the target for a number of clinically relevant drugs. Bacterial gyrase and topoisomerase IV are the targets of quinolone-based drugs such as ciprofloxacin and levofloxacin.30, 79–80 These agents are the most active and broad-spectrum oral antibacterials currently available. Human topoisomerase IIα and topoisomerase IIβ are the targets of some of the most active anticancer agents used in the clinic.12–13, 28–29, 81–83 Drugs such as etoposide, doxorubicin, and mitoxantrone are used routinely to treat a wide variety of hematological and solid tumors. All of these drugs kill cells by increasing the concentration of cleavage complexes, thereby generating high concentrations of DNA strand breaks.12, 28, 81–83 Agents that act by this mechanism are known as topoisomerase II poisons.84
3) DNA Strand Passage
Topoisomerase II binds two ATP molecules, which induce critical conformation changes in the enzyme.85–86 These structural rearrangements are transduced through an interaction between the γ-phosphate group of the bound ATP and a conserved lysine residue (Lys378 in human topoisomerase IIα ) in the QTK loop of the protein.87–89 Ultimately, the rearrangements close an N-terminal gate in the protein, open a gate in the cleaved double helix, and trigger the passage of an intact DNA segment through the nucleic acid gate.85–89 DNA strand passage appears to be faster if one of the two ATP molecules is hydrolyzed.85–86 The transported DNA is passed into a central cavity in topoisomerase II, such that the enzyme forms a protein clamp on the double helix.85, 90 The functional interaction of topoisomerase II and ATP requires a divalent metal ion (discussed below).
4) DNA Ligation
Following DNA translocation, topoisomerase II reseals the original nucleic acid break, and the enzyme establishes a post-strand passage DNA cleavage-ligation equilibrium.91 Ligation requires a divalent metal ion and is assumed to represent the reverse of the transesterificationreaction usedtocleave the DNA.92–93 Onceligation has takenplace, topoisomerase II and the DNA are no longer covalently linked. However, since the transported DNA segment is still engulfed between protein gates, the enzyme and DNA remain topologically complexed.85, 90 It is notable that most clinically relevant topoisomerase II poisons increase the concentration of cleavage complexes primarily by inhibiting the ability of the enzyme to ligate cleaved DNA molecules.12–13
5) ATP Hydrolysis
Topoisomerase II hydrolyzes the second ATP molecule, which opens a C-terminal gate on the protein and allows the transported DNA segment to be released by the enzyme.85–86 ATP hydrolysis requires the presence of a divalent metal ion.
6) Enzyme Turnover
The enzyme returns to its original conformation and regains the ability to begin a new round of catalysis. This process is not well understood.
4. Use of Divalent Metal Ions by Topoisomerase II
As discussed above, type II topoisomerases have two active sites: one that binds and hydrolyzes ATP and another that contains the catalytic tyrosine residue and mediates DNA cleavage and ligation. The use of metal ions in both active sites of topoisomerase II is discussed below. Additional information regarding the interactions of divalent metal ions with both type I and II topoisomerases can be found in an excellent recent review article by Sissi and Palumbo.68
4.1 ATPase Active Site
The ATPase domain of type II topoisomerases is a member of the GHKL superfamily (named for the founding members gyrase, Hsp90, histidine kinase, and MutL), which is characterized by an unconventional Bergerat ATP-binding fold.87–89 It is located in the N-terminal, or GyrB homology domain of topoisomerase II (residues 29–264 in human topoisomerase IIα ).87–89, 94 The key catalytic residue, Glu87, activates a water molecule that hydrolyzes the β-γ phosphodiester bond of the ATP moiety.87–89, 94 The ATPase activity of topoisomerase II increases when DNA is bound.95 Like other enzymes that hydrolyze ATP, the ATPase activity of topoisomerase II has an absolute requirement for a divalent metal ion.71 In the absence of the metal ion, neither the conformational changes in topoisomerase II that accompany ATP binding, nor hydrolysis of the ATP cofactor are observed.71, 87–89, 94 The ATP-related functions of topoisomerase II appear to have a preference for Mg2+.71 Presumably, it is a Mg2+-ATP complex that binds to the protein. Although Mn2+ and Ca2+ support the DNA cleavage/ligation activities of topoisomerase II,93, 96 they do not allow the overall catalytic activity of the enzyme. In contrast, Co2+ supports DNA relaxation and decatenation catalyzed by human topoisomerase IIα , suggesting that this divalent metal ion must also support the ATPase activity of the enzyme.96
The crystal structures of the ATPase domain of Escherichia coli gyrase and topoisomerase IV, Saccharomyces cerevisiae topoisomerase II, and human topoisomerase IIα have been solved in the presence of a bound nucleotide triphosphate (Fig. 2).87–88, 94 In all cases, the Mg2+ ion is octahedrally coordinated to all three phosphate groups of ATP, two water molecules, and the δ1-carbonyl oxygen of a conserved asparagine residue (Asn91 in human topoisomerase IIα ). Following hydrolysis of the high-energy cofactor, a water molecule replaces the leaving γ-phosphate group that contacted the Mg2+ ion. It is notable that the catalytic residue, Glu87 in human topoisomerase IIα , interacts with one of the water molecules that is coordinated by the metal ion.87–88, 94
Figure 2.
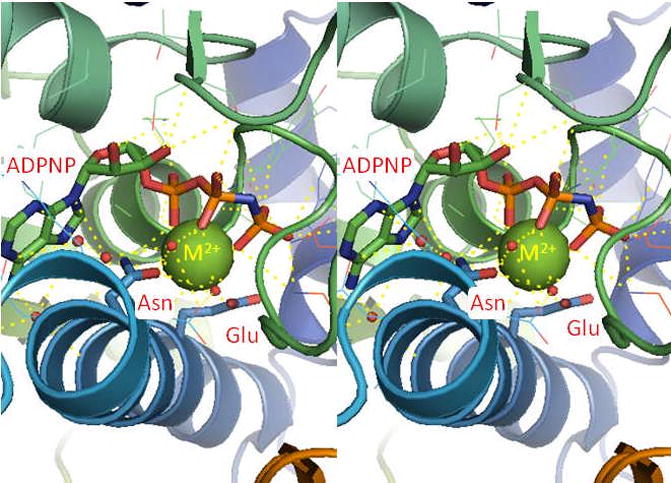
Crystal structure of the ATPase domain of yeast topoisomerase II (PDB ID 1QZR) with ADPNP bound.87 The bound metal ion (Mg2+, green) is coordinated by non-bridging oxygen atoms of the phosphate groups and by an asparagine residue (Asn91 in human topoisomerase IIα ). The glutamic acid residue (Glu87 in human topoisomerase IIα ) is thought to activate a water molecule (shown as red spheres) to initiate hydrolysis of the phosphate group. Yellow dashes represent ligand interactions. The structure is shown in stereo view.
4.2 DNA Cleavage/Ligation Active Site
The ability of topoisomerase II to cleave and ligate DNA is central to all of its catalytic functions.1, 12–13, 27–28 While many of the amino acids involved in catalysis other than the active site tyrosine residues have not been elucidated, it is believed that the enzyme utilizes a general acid–base mechanism for DNA cleavage. Scission is initiated when a general base deprotonates the active site tyrosine hydroxyl moiety, allowing the oxyanion to attack the scissile phosphate. The base has not been identified but is believed to be a conserved histidine (potentially His758 in human topoisomerase IIα ) residue or a metal-associated water molecule.97
The DNA cleavage reaction has an absolute requirement for a divalent metal ion.28, 33, 71, 98–99 Although Mg2+ appears to fulfill this function in vivo,28 other metal ions, such as Mn2+, Ca2+, and Co2+ often support even higher levels of double-stranded DNA cleavage in vitro.93, 96, 99 While divalent metal ions are necessary to promote the chemistry of DNA scission, they do not appear to be in involved in site selection. To a large extent, cleavage maps generated in the presence of all of the above divalent cations are similar.93, 96
Topoisomerase II cuts the two strands of the double helix in a coordinated fashion.72, 76, 100 It is believed that cleavage of the first stand introduces flexibility in the DNA that allows the substrate to attain an acutely bent transition state that is required for efficient cleavage.72–73, 101 Thus, after the enzyme cleaves the first strand in the presence of Mg2+, Mn2+, or Co2+ (average level of cleavage ≈1%), it goes on to cut the second strand ≥50% of the time. In marked contrast, the two protomer subunits of topoisomerase II are considerably less coordinated when Ca2+ is utilized as the divalent metal ion.93 Consequently, much higher levels of single-stranded DNA nicks are generated in the presence of this metal ion. The reason for the decreased coordination seen with Ca2+ is not known.
Commonly, enzymes that cleave nucleic acids utilize divalent metal ions in their active sites in order to stabilize both attacking (i.e., amino acids and/or water) and leaving groups (i.e., DNA backbone oxygen atoms).97, 102–120 However, the number of metal ions per active site and the specific roles played by these cations in stabilizing leaving groups (bridging vs. non-bridging oxygen) varies. A series of enzymological, mutagenesis, and structural studies with prokaryotic and mammalian type II topoisomerases (discussed below) indicate that these enzymes cleave DNA using a two-metal-ion mechanism,97, 99, 121–125 albeit a novel variant125 of the canonical two-metal-ion mechanism described for phosphotransferases and hydrolases such as DNA pol I and RNaseH.126–127
Many topoisomerases (type I and type II) and primases share a conserved region known as the TOPRIM domain.98 This region has been implicated in coordinating divalent metal ions that are used during nucleic acid cleavage, ligation, and polymerization reactions.33, 98, 128 The TOPRIM domain consists of ~125 amino acids (residues ~455–580 in human topoisomerase IIα ) that is characterized by a compact β /α fold. It includes a conserved glutamic acid and two (DXD) or three (DXDXD) conserved aspartic acid residues that are juxtaposed (Fig. 3). Structural studies indicate that the conserved glutamic acid and two of the aspartic acids interact with divalent metal ions during catalysis (E461, D541, and D543 in human topoisomerase IIα ) (Figs. 4 and 5).99, 101, 123–124 A recent crystallographic study suggests that the third aspartic acid residue (D545 in human topoisomerase IIα ) does not play a role in metal ion binding.125 Rather, it is involved in a salt bridge with a highly conserved arginine residue (R713 in human topoisomerase IIα ) (Figs. 4 and 5).125
Figure 3.

Amino acids postulated to coordinate divalent metal ions in the active site of nucleic acid enzymes that contain a TOPRIM domain. Amino acid sequences are shown for human topoisomerase IIα (HsTop2a), topoisomerase IIβ (HsTop2b), S. cerevisiae topoisomerase II (ScTop2), and E. coli gyrase B subunit (EcGyrB). Conserved glutamic and aspartic acid residues that are proposed to bind divalent metal ions are highlighted in blue. Sequences are from Aravind et al. and Caron and Wang.98, 129
Figure 4.
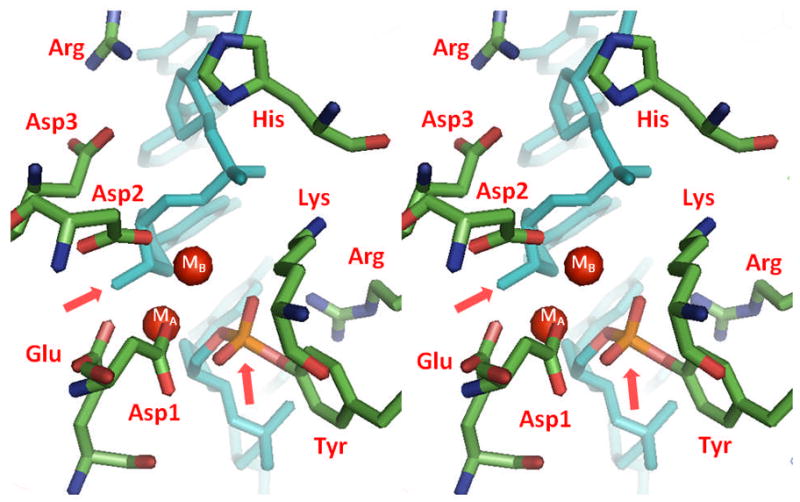
The DNA cleavage/ligation active site of topoisomerase II. Using the crystal structure of yeast topoisomerase II (PDB ID 3L4K) as a docked dimer, the key residues of the cleavage/ligation active site are shown and are labeled in red.125 The residues include three aspartic acid residues (labeled 1, 2, and 3 corresponding to their sequence order), one glutamic acid, one histidine, the active site tyrosine, and two arginine residues. The DNA in the crystal structure (shown in blue) is covalently bound and has been cleaved by the enzyme. The 3’-terminal group and 5’-phosphate of the cleaved DNA are denoted by pink arrows. The red spheres represent the locations of divalent metal ions that are visible in the structure (labeled MA and MB). The structure is shown in stereo view.
Figure 5.

Proposed mechanism of DNA cleavage and ligation by topoisomerase II. The type II enzyme utilizes a novel variant of the canonical two-metal-ion mechanism employed by primases and polymerases.98–99, 102, 105, 123–124 Amino acids in the active site that are postulated to function in catalysis by topoisomerase IIα and topoisomerase IIβ are indicated. Metal ions (MA2+ and MB2+) are highlighted in red. Interactions between metal ions and nucleic acid or acidic amino acid residues are denoted in green or blue, respectively. In the proposed model, MA2+ makes critical contacts with the 3’-bridging atom (red) and a non-bridging atom of the scissile phosphate. These interactions are likely to play a role in the transition chemistry and stabilizing the leaving 3’-terminal oxygen. MB2+ also is required for DNA scission and contacts the non-bridging oxygen of the phosphate that connects the -1 and -2 bases upstream from the scissile bond. This metal ion appears to play a structural role in anchoring the DNA during scission. Cleavage is initiated when a general base deprotonates the active site tyrosine hydroxyl, allowing the oxyanion to attack the scissile phosphate. The base has not been identified but is believed to be a conserved histidine residue or a metal-associated water molecule. Ligation is initiated when a general acid extracts the hydrogen from the 3’-terminal hydroxyl group. The acid may be a water molecule or an unidentified amino acid in the active site of topoisomerase II. Figure is adapted from Schmidt, et al.125
The first evidence for a two-metal-ion mechanism for DNA cleavage by type II enzymes came from work on Eschericia coli gyrase carried out by Noble and Maxwell.97 These authors were able to distinguish the two metal ion sites in the DNA cleavage/ligation domain by mutating acidic amino acid residues that were proposed to bind divalent cations and found higher levels of DNA scission in the presence of Mg2+ and Ca2+ than with either metal ion alone. This study strongly suggested that type II topoisomerases utilized a two-metal-ion mechanism and provided a working model for the field. However it did not elucidate the specific roles played by divalent metal ions in the DNA cleavage reaction of type II enzymes.
Later studies by Deweese et al. extended the two-metal-ion mechanism to eukaryotic type II topoisomerases and began to define the roles of divalent cations in DNA cleavage.99, 122 Two lines of evidence support the use of two metal ions by human topoisomerase IIα and topoisomerase IIβ . First, titration experiments with Mg2+, Mn2+, and Ca2+ all indicate a biphasic dependence on divalent cations for DNA cleavage. When Mg2+ is utilized, the high affinity metal ion site (referred to alternatively as site A or 1) appears to have a KD value of ~100 μM, while the lower affinity site (referred to alternatively as site B or 2) appears to have a KD that is approximately an order of magnitude higher (sites are shown in Figs. 4 and 5). There is an absolute requirement for both metal ion sites to be filled in order for DNA cleavage to take place.99, 122
Second, slightly higher rates and levels of scission are observed in the presence of two metal ions (Mn2+ and Ca2+) than predicted by calculated levels derived from the sum of cleavage in the presence of the individual cations.99, 122 More dramatic enhancements (in some cases, >20 – fold) are seen when modified DNA substrates are used to distinguish the two metal ions. These substrates contain a sulfur atom in place of the bridging or non-bridging oxygen atoms of the scissile phosphate (phosphorothiolate or phosphorothioate, respectively).99, 122 Experiments take advantage of the fact that soft (or type B) metals (which tend to be larger and more polarisable), such as Mn2+ often prefer sulfur over oxygen as an inner-sphere ligand, while hard (or type A) metals (which tend to have a smaller ionic radius and are non-polarisable), such as Ca2+, usually coordinate more readily with oxygen.105, 111, 130–135
Modified phosphorothiolate and phosphorothioate substrates also have been used to define interactions between metal ions and the scissile phosphate atoms.99–100, 122 Relative levels and rates of cleavage determined for reactions mediated by human topoisomerase IIα and topoisomerase IIβ in Ca2+-supported reactions (relative to Mn2+-supported reactions) decrease significantly when the bridging oxygen atom at the scissile bond is substituted by a sulfur atom. 99–100, 122
Based on these findings, it was concluded that both type II enzymes utilize an interaction between the high affinity metal ion A and the 3’-bridging atom of the scissile phosphate to accelerate rates of enzyme-mediated DNA cleavage, most likely by stabilizing the leaving 3’-oxygen (Fig. 5).122, 125 Based on similar experiments that examined cleavage on substrates that substituted a non-bridging sulfur atom for oxygen, there also appears to be an important contact between one of the metal ions and a non-bridging atom of the scissile phosphate in the active site of topoisomerase IIβ (Fig. 5).122 This latter interaction plays a significant role in DNA cleavage mediated by the β isoform and greatly stimulates scission.97, 99, 122, 124 As proposed previously, this metal ion-DNA contact is believed to stabilize the DNA transition state. While a similar interaction between the metal ion and the non-bridging oxygen in the active site of topoisomerase IIα cannot be ruled out, if it exists, its effects on rates of DNA cleavage appear to be equivocal.99, 124
Critical evidence for the roles of metal ions in topoisomerase II-mediated DNA cleavage comes from structural studies with the catalytic core of yeast topoisomerase II complexed with DNA substrates.101, 125 The first study solved the structure of the DNA cleavage/ligation active site of the enzyme in the presence of non-covalently bound DNA.101 While only one divalent metal ion was observed in the structure, its location was consistent with the assigned TOPRIM amino acid residues shown in Fig. 3. It should be noted, however, that the position of the active site tyrosine residue in this structure was not in a cleavage competent configuration.
A more recent study with the catalytic core of yeast topoisomerase II provided the first structure of a covalent enzyme-DNA complex for a eukaryotic type II topoisomerase.125 This structure shows the coordination of two metal ions in the cleavage/ligation active site of the enzyme and reveals the constellation of amino acids required to form a cleavage-competent active site (Fig. 4). Metal ion A appears to interact with both the bridging oxygen and a non-bridging oxygen of the scissile bond in the enzyme-DNA cleavage complex.125 Thus, it is well positioned to assist with transition state chemistry. In contrast, metal ion B does not contact either the active site tyrosine or any DNA atoms at the scissile bond.125 Rather, it is located adjacent to the active site and appears to contact a non-bridging oxygen of the phosphate linking the (−1) and (−2) nucleotides upstream of the scissile bond.
Both metal ions in the covalent enzyme-DNAstructure are coordinated bythe highly-conserved acidic residues in the TOPRIM domain of yeast topoisomerase II. Metal ion A is anchored by E449 and D526, and metal ion B is anchored by D526 and D528 (Figs. 4 and 5). The functional importance of the conserved TOPRIM acidic residues in the catalytic activity of type II topoisomerases and their role in metal ion binding is strongly supported by mutagenesis studies that examined gyrase, topoisomerase IIα , and topoisomerase IIβ .97, 123–124, 136 In all cases, substitution of the coordinating glutamic acid or aspartic acid residues with a non-acidic amino acid resulted in an enzyme with dramatically reduced DNA strand passage and cleavage activities. Furthermore, mutant enzymes required higher levels of divalent metal ions for activity than did the corresponding wild-type proteins. Finally, enzymes that substituted cysteine residues for the conserved aspartic acids displayed higher activity in the presence of Mn2+, a thiophilic metal ion, than did the corresponding alanine-containing mutants.136
In the canonical two-metal mechanism, one divalent cation activates a catalytic water or ribose hydroxyl for nucleophilic attack, while the other coordinates a leaving group.137 Both metal ions stabilize the pentavalent transition state. Based on the collective data from the enzymology, mutagenesis, and structural studies discussed above, there appear to be important interactions between divalent metal ions and the bridging atom of the scissile bond, the non-bridging atoms of the scissile bond and the bond one nucleotide upstream, and conserved acidic amino acid residues in type II topoisomerases.97, 123–125, 136 Therefore, it is proposed that type II topoisomerases employ a novel variation of the canonical two-metal-ion mechanism for DNA cleavage (Fig. 5). In this model, metal ion A plays a direct role in the transition chemistry, most likely by promoting the leaving of the ribose 3’-OH. In contrast, metal ion B appears to play a structural role in anchoring the DNA during cleavage.
The fate of the metal ions when the cleaved DNA gate is opened is not known. Metal ion B could remain complexed with the phosophate of the upstream nucleotide during gate opening. However, since metal ion A is complexed to oxygen atoms that become the 3’-terminal hydroxyl and 5’-terminal phosphate moieties, it is clear that it cannot remain in contact with both atoms during this process. The resolution of this important issue likely will have to wait for future structural studies that capture the cleavage complex in a “gate-opened” form.
It is not obvious why type II topoisomerases have lost the need for a second metal in the transition chemistry required for DNA cleavage. However, it may be due to the relatively low pKa (~10–11) of the active site tyrosine nucleophile, which makes it easier to deprotonate than either water or an attacking nucleic acid 3’-hydroxyl group (pKa ~13–15). Alternatively, the presence of the positively–charged arginine immediately adjacent to the active site tyrosine residue may take the place of the second metal ion to assist with transition state stabilization or to further depress the pKa of the tyrosine hydroxyl moiety.125
The DNA ligation reaction of type II topoisomerases is considerably less well understood than cleavage. Ligation has an absolute requirement for a divalent cation and is supported by a range of metal ions, including Mg2+, Mn2+, and Ca2+.92, 138–139 The reaction is believed to represent the reverse of DNA cleavage/transesterification. However, Co2+, which efficiently supports DNA scission mediated by human topoisomerase IIα , does not support rapid rates of ligation.96 Thus, there may be subtle, but important differences in the use of divalent metal ions during these two processes.
An important role of metal ions during ligation may be to properly align the DNA termini for resealing. To this point, the 3’-terminal ribose moiety in a metal-free structure adopts a C2’-endo (B-form DNA) sugar pucker that is not optimally oriented for ligation.125 In contrast, the ribose adopts a C3’-endo state (A form) similar to its neighboring nucleotides in a structure that includes two divalent metal ions, and is positioned in line with the phosphotyrosyl linkage.125
5. Divalent Metal Ions as Probes of Topoisomerase II Function
The finding that type II topoisomerases are able to use a variety of divalent metal ions for DNA cleavage and ligation has played an important role in understanding these critical enzyme processes. For example, it has been known for more than 20 years that substituting Ca2+ for Mg2+ significantly raises levels of DNA cleavage mediated by bacterial gyrase and eukaryotic topoisomerase II (including yeast, Drosophila, bovine, and human).93, 96, 138, 140–141 Prior to this finding, the study of gyrase-generated DNA scission was confined to reactions that contained quinolone antibacterials. The use of Ca2+ in DNA cleavage reactions greatly facilitated the ability to map sites of enzyme action and monitor DNA scission in a multitude of studies.93, 138, 140, 142
Topoisomerase II-mediated DNA cleavage and ligation normally exist in a tightly coupled equilibrium. Consequently, until the advent of suicide- or activated-substrates,143–148 it was impossible to carry out mechanistic studies of ligation. However, a unique property of Ca2+ allowed topoisomerase II-mediated ligation to be studied in isolation.93 In contrast to Mg2+, which appears to be inaccessible to chelating agents when involved in a cleavage complex, Ca2+ can be removed from the covalent topoisomerase II-cleaved DNA complex with EDTA or similar chelating agents. Thus, chelation of Ca2+ traps the cleavage complex in a kinetically competent form.93 The subsequent addition of excess Mg2+ reactivates the trapped enzyme and allows a unidirectional ligation event. The use of this assay permitted the first analysis of ligation and demonstrated that topoisomerase II ligates double-stranded breaks one strand at a time.76, 93 It also provided the first direct evidence that etoposide and several other anticancer drugs increase levels of topoisomerase II-generated DNA strand breaks primarily by inhibiting ligation.91, 149–150
Finally, the use of divalent metal ions with varying degrees of thiophilicity in conjunction with sulfur-containing DNA substrates provided important information regarding the role of divalent cations in the DNA cleavage reaction mediated by type II topoisomerases.99, 122, 136 These studies demonstrated that human topoisomerase IIα and topoisomerase IIβ cleave DNA using a two-metal-ion mechanism and defined interactions between divalent metal ions and the scissile phosphate oxygen atoms.99, 122, 136
6. Summary
All type II topoisomerases require divalent metal ions to support their catalytic activities. Although we understand many aspects of the interactions between the metal ions, the enzymes, and their DNA substrates, a number of important issues have yet to be investigated. For example, what are the specific role(s) of Mg2+ in the chemistry of DNA cleavage and ligation? Do metal ions interact with anticancer drugs in the topoisomerase II-DNA cleavage complex? Can the finding that divalent metal ions affect levels of enzyme-mediated DNA scission be exploited in the design of novel topoisomerase II-targeted anticancer drugs? It is clear that future studies of metal ions and type II topoisomerases still hold considerable promise for important discoveries.
Acknowledgments
We are grateful to Adam C. Ketron and Dr. Qasim A. Khan for critical reading of the manuscript. We would like to thank Dr. Ken Dong for providing the Pymol file from which Fig. 4 was generated. Work in the senior author’s laboratory is supported by National Institutes of Health grants GM33944 and GM53960. J.E.D. was a trainee under National Institutes of Health grant T32 CA09592.
Biographies

Neil Osheroff holds a B.A. in Chemistry from Hobart College (1974) and a Ph.D. in Biochemistry and Molecular Biology from Northwestern University (1979). He was a Helen Hay Whitney Foundation postdoctoral fellow in the Department of Biochemistry at the Stanford University School of Medicine from 1980–1983. In 1983, Dr. Osheroff moved to the Vanderbilt University School of Medicine where he currently holds Professorships in the Departments of Biochemistry and Medicine and was endowed with the John G. Conglio Chair in Biochemistry in 2003. Research in Dr. Osheroff’s laboratory focuses on topoisomerases, enzymes that remove knots, tangles, and torsional stress from the genetic material, and are the targets for several widely prescribed antibacterial and anticancer drugs. Among his work on these enzymes, he has investigated the role of metal ions in topoisomerase function for more than twenty years.

Joe Deweese holds a B.S. in Biochemistry from Freed-Hardeman University (2004) and a Ph.D. in Biochemistry from Vanderbilt University School of Medicine (2009). Dr. Deweese performed his graduate research in the laboratory of Dr. Neil Osheroff where he studied the involvement of metal ions in the cleavage reaction of mammalian type II topoisomerases. He currently is an Assistant Professor of Pharmaceutical Sciences at Lipscomb University College of Pharmacy and an Adjunct Assistant Professor of Biochemistry at Vanderbilt University School of Medicine.
References
- 1.Wang JC. Annu Rev Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 2.Bates AD, Maxwell A. DNA Topology. Oxford University Press; New York: 2005. [Google Scholar]
- 3.Espeli O, Marians KJ. Mol Microbiol. 2004;52:925–931. doi: 10.1111/j.1365-2958.2004.04047.x. [DOI] [PubMed] [Google Scholar]
- 4.Falaschi A, Abdurashidova G, Sandoval O, Radulescu S, Biamonti G, Riva S. Cell Cycle. 2007;6:1705–1712. doi: 10.4161/cc.6.14.4495. [DOI] [PubMed] [Google Scholar]
- 5.Travers A, Muskhelishvili G. EMBO reports. 2007;8:147–151. doi: 10.1038/sj.embor.7400898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allemand JF, Bensimon D, Lavery R, Croquette V. Proc Natl Acad Sci USA. 1998;95:14152–14157. doi: 10.1073/pnas.95.24.14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Randall GL, Zechiedrich L, Pettitt BM. Nuc Acids Res. 2009;37:5568–5577. doi: 10.1093/nar/gkp556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauer WR, Crick FH, White JH. Sci Am. 1980;243:100–113. [PubMed] [Google Scholar]
- 9.White JH, Cozzarelli NR. Proc Natl Acad Sci USA. 1984;81:3322–3326. doi: 10.1073/pnas.81.11.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vologodskii AV, Cozzarelli NR. Annu Rev Biophys Biomol Struct. 1994;23:609–643. doi: 10.1146/annurev.bb.23.060194.003141. [DOI] [PubMed] [Google Scholar]
- 11.Wang JC. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 12.McClendon AK, Osheroff N. Mutat Res. 2007;623:83–97. doi: 10.1016/j.mrfmmm.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deweese JE, Osheroff N. Nucleic Acids Res. 2009;37:738–749. doi: 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kouzine F, Liu J, Sanford S, Chung HJ, Levens D. Nat Struct Mol Biol. 2004;11:1092–1100. doi: 10.1038/nsmb848. [DOI] [PubMed] [Google Scholar]
- 15.Kouzine F, Sanford S, Elisha-Feil Z, Levens D. Nat Struct Mol Biol. 2008;15:146–154. doi: 10.1038/nsmb.1372. [DOI] [PubMed] [Google Scholar]
- 16.Portugal J, Rodriguez-Campos A. Nucleic Acids Res. 1996;24:4890–4894. doi: 10.1093/nar/24.24.4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arai Y, Yasuda R, Akashi K, Harada Y, Miyata H, Kinosita K, Jr, Itoh H. Nature. 1999;399:446–448. doi: 10.1038/20894. [DOI] [PubMed] [Google Scholar]
- 18.Deibler RW, Mann JK, Sumners de WL, Zechiedrich L. BMC Mol Biol. 2007;8(44):1–14. doi: 10.1186/1471-2199-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baxter J, Diffley JF. Mol Cell. 2008;30:790–802. doi: 10.1016/j.molcel.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 20.Liu Z, Deibler RW, Chan HS, Zechiedrich L. Nuc Acids Res. 2009;37:661–671. doi: 10.1093/nar/gkp041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Champoux JJ. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 22.Schoeffler AJ, Berger JM. Biochem Soc Trans. 2005;33:1465–1470. doi: 10.1042/BST0331465. [DOI] [PubMed] [Google Scholar]
- 23.Schoeffler AJ, Berger JM. Q Rev Biophys. 2008;41:41–101. doi: 10.1017/S003358350800468X. [DOI] [PubMed] [Google Scholar]
- 24.Leppard JB, Champoux JJ. Chromosoma. 2005;114:75–85. doi: 10.1007/s00412-005-0345-5. [DOI] [PubMed] [Google Scholar]
- 25.Pommier Y. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 26.Wang JC. Quart Rev Biophys. 1998;31:107–144. doi: 10.1017/s0033583598003424. [DOI] [PubMed] [Google Scholar]
- 27.Berger JM, Gamblin SJ, Harrison SC, Wang JC. Nature. 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 28.Fortune JM, Osheroff N. Prog Nucleic Acid Res Mol Biol. 2000;64:221–253. doi: 10.1016/s0079-6603(00)64006-0. [DOI] [PubMed] [Google Scholar]
- 29.Velez-Cruz R, Osheroff N. In: Encyclopedia of Biological Chemistry. Editon. Lennarz W, Lane DM, editors. Vol. 1. Elsevier Science; San Diego: 2004. pp. 806–811. [Google Scholar]
- 30.Levine C, Hiasa H, Marians KJ. Biochim Biophys Acta. 1998;1400:29–43. doi: 10.1016/s0167-4781(98)00126-2. [DOI] [PubMed] [Google Scholar]
- 31.Nollmann M, Crisona NJ, Arimondo PB. Biochimie. 2007;89:490–499. doi: 10.1016/j.biochi.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 32.Ferrero L, Cameron B, Manse B, Lagneaux D, Crouzet J, Famechon A, Blanche F. Molec Microbiol. 1994;13:641–653. doi: 10.1111/j.1365-2958.1994.tb00458.x. [DOI] [PubMed] [Google Scholar]
- 33.Berger JM, Fass D, Wang JC, Harrison SC. Proc Natl Acad Sci USA. 1998;95:7876–7881. doi: 10.1073/pnas.95.14.7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isaacs RJ, Davies SL, Sandri MI, Redwood C, Wells NJ, Hickson ID. Biochim Biophys Acta. 1998;1400:121–137. doi: 10.1016/s0167-4781(98)00131-6. [DOI] [PubMed] [Google Scholar]
- 35.Jensen S, Andersen AH, Kjeldsen E, Biersack H, Olsen EH, Andersen TB, Westergaard O, Jakobsen BK. Mol Cell Biol. 1996;16:3866–3877. doi: 10.1128/mcb.16.7.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mirski SE, Gerlach JH, Cummings HJ, Zirngibl R, Greer PA, Cole SP. Exp Cell Res. 1997;237:452–455. doi: 10.1006/excr.1997.3805. [DOI] [PubMed] [Google Scholar]
- 37.McClendon AK, Rodriguez AC, Osheroff N. J Biol Chem. 2005;280:39337–39345. doi: 10.1074/jbc.M503320200. [DOI] [PubMed] [Google Scholar]
- 38.McClendon AK, Dickey JS, Osheroff N. Biochemistry. 2006;45:11674–11680. doi: 10.1021/bi0520838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McClendon AK, Gentry AC, Dickey JS, Brinch M, Bendsen S, Andersen AH, Osheroff N. Biochemistry. 2008;47:13169–13178. doi: 10.1021/bi800453h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goto T, Wang JC. J Biol Chem. 1982;257:5866–5872. [PubMed] [Google Scholar]
- 41.Goto T, Laipis P, Wang JC. J Biol Chem. 1984;259:10422–10429. [PubMed] [Google Scholar]
- 42.Goto T, Wang JC. Cell. 1984;36:1073–1080. doi: 10.1016/0092-8674(84)90057-6. [DOI] [PubMed] [Google Scholar]
- 43.Nolan JM, Lee MP, Wyckoff E, Hsieh TS. Proc Natl Acad Sci USA. 1986;83:3664–3668. doi: 10.1073/pnas.83.11.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drake FH, Zimmerman JP, McCabe FL, Bartus HF, Per SR, Sullivan DM, Ross WE, Mattern MR, Johnson RK, Crooke ST. J Biol Chem. 1987;262:16739–16747. [PubMed] [Google Scholar]
- 45.Tsai-Pflugfelder M, Liu LF, Liu AA, Tewey KM, Whang-Peng J, Knutsen T, Huebner K, Croce CM, Wang JC. Proc Natl Acad Sci USA. 1988;85:7177–7181. doi: 10.1073/pnas.85.19.7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drake FH, Hofmann GA, Bartus HF, Mattern MR, Crooke ST, Mirabelli CK. Biochemistry. 1989;28:8154–8160. doi: 10.1021/bi00446a029. [DOI] [PubMed] [Google Scholar]
- 47.Chung TD, Drake FH, Tan KB, Per SR, Crooke ST, Mirabelli CK. Proc Natl Acad Sci USA. 1989;86:9431–9435. doi: 10.1073/pnas.86.23.9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Austin CA, Fisher LM. FEBS Lett. 1990;266:115–117. doi: 10.1016/0014-5793(90)81520-x. [DOI] [PubMed] [Google Scholar]
- 49.Jenkins JR, Ayton P, Jones T, Davies SL, Simmons DL, Harris AL, Sheer D, Hickson ID. Nucleic Acids Res. 1992;20:5587–5592. doi: 10.1093/nar/20.21.5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan KB, Dorman TE, Falls KM, Chung TD, Mirabelli CK, Crooke ST, Mao J. Cancer Res. 1992;52:231–234. [PubMed] [Google Scholar]
- 51.Wilstermann AM, Osheroff N. Curr Top Med Chem. 2003;3:321–338. doi: 10.2174/1568026033452519. [DOI] [PubMed] [Google Scholar]
- 52.Chen M, Beck WT. Oncol Res. 1995;7:103–111. [PubMed] [Google Scholar]
- 53.Dereuddre S, Delaporte C, Jacquemin-Sablon A. Cancer Res. 1997;57:4301–4308. [PubMed] [Google Scholar]
- 54.Yang X, Li W, Prescott ED, Burden SJ, Wang JC. Science. 2000;287:131–134. doi: 10.1126/science.287.5450.131. [DOI] [PubMed] [Google Scholar]
- 55.Heck MM, Earnshaw WC. J Cell Biol. 1986;103:2569–2581. doi: 10.1083/jcb.103.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hsiang YH, Wu HY, Liu LF. Cancer Res. 1988;48:3230–3235. [PubMed] [Google Scholar]
- 57.Woessner RD, Mattern MR, Mirabelli CK, Johnson RK, Drake FH. Cell Growth Differ. 1991;2:209–214. [PubMed] [Google Scholar]
- 58.Grue P, Grasser A, Sehested M, Jensen PB, Uhse A, Straub T, Ness W, Boege F. J Biol Chem. 1998;273:33660–33666. doi: 10.1074/jbc.273.50.33660. [DOI] [PubMed] [Google Scholar]
- 59.Linka RM, Porter AC, Volkov A, Mielke C, Boege F, Christensen MO. Nucleic Acids Res. 2007;35:3810–3822. doi: 10.1093/nar/gkm102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Austin CA, Marsh KL. BioEssays. 1998;20:215–226. doi: 10.1002/(SICI)1521-1878(199803)20:3<215::AID-BIES5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 61.Bauman ME, Holden JA, Brown KA, Harker WG, Perkins SL. Mod Pathol. 1997;10:168–175. [PubMed] [Google Scholar]
- 62.Nitiss JL. Biochim Biophys Acta. 1998;1400:63–81. doi: 10.1016/s0167-4781(98)00128-6. [DOI] [PubMed] [Google Scholar]
- 63.Christensen MO, Larsen MK, Barthelmes HU, Hock R, Andersen CL, Kjeldsen E, Knudsen BR, Westergaard O, Boege F, Mielke C. J Cell Biol. 2002;157:31–44. doi: 10.1083/jcb.200112023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sakaguchi A, Kikuchi A. J Cell Sci. 2004;117:1047–1054. doi: 10.1242/jcs.00977. [DOI] [PubMed] [Google Scholar]
- 65.Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 66.Haince JF, Rouleau M, Poirier GG. Science. 2006;312:1752–1753. doi: 10.1126/science.1129808. [DOI] [PubMed] [Google Scholar]
- 67.Maxwell A, Costenaro L, Mitelheiser S, Bates AD. Biochem Soc Trans. 2005;33:1460–1464. doi: 10.1042/BST0331460. [DOI] [PubMed] [Google Scholar]
- 68.Sissi C, Palumbo M. Nucleic Acids Res. 2009;37:702–711. doi: 10.1093/nar/gkp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zechiedrich EL, Osheroff N. EMBO J. 1990;9:4555–4562. doi: 10.1002/j.1460-2075.1990.tb07908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roca J, Berger JM, Wang JC. J Biol Chem. 1993;268:14250–14255. [PubMed] [Google Scholar]
- 71.Osheroff N. Biochemistry. 1987;26:6402–6406. doi: 10.1021/bi00394a015. [DOI] [PubMed] [Google Scholar]
- 72.Mueller-Planitz F, Herschlag D. J Biol Chem. 2008;283:17463–17476. doi: 10.1074/jbc.M710014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deweese JE, Osheroff N. Biochemistry. 2009;48:1439–1441. doi: 10.1021/bi8021679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu LF, Rowe TC, Yang L, Tewey KM, Chen GL. J Biol Chem. 1983;258:15365–15370. [PubMed] [Google Scholar]
- 75.Sander M, Hsieh T. J Biol Chem. 1983;258:8421–8428. [PubMed] [Google Scholar]
- 76.Zechiedrich EL, Christiansen K, Andersen AH, Westergaard O, Osheroff N. Biochemistry. 1989;28:6229–6236. doi: 10.1021/bi00441a014. [DOI] [PubMed] [Google Scholar]
- 77.Capranico G, Binaschi M. Biochim Biophys Acta. 1998;1400:185–194. doi: 10.1016/s0167-4781(98)00135-3. [DOI] [PubMed] [Google Scholar]
- 78.Velez-Cruz R, Riggins JN, Daniels JS, Cai H, Guengerich FP, Marnett LJ, Osheroff N. Biochemistry. 2005;44:3972–3981. doi: 10.1021/bi0478289. [DOI] [PubMed] [Google Scholar]
- 79.Anderson VE, Osheroff N. Curr Pharm Des. 2001;7:337–353. doi: 10.2174/1381612013398013. [DOI] [PubMed] [Google Scholar]
- 80.Drlica K, Malik M. Curr Top Med Chem. 2003;3:249–282. doi: 10.2174/1568026033452537. [DOI] [PubMed] [Google Scholar]
- 81.Martincic D, Hande KR. Cancer Chemother Biol Response Modif. 2005;22:101–121. doi: 10.1016/s0921-4410(04)22005-1. [DOI] [PubMed] [Google Scholar]
- 82.Baldwin EL, Osheroff N. Curr Med Chem Anticancer Agents. 2005;5:363–372. doi: 10.2174/1568011054222364. [DOI] [PubMed] [Google Scholar]
- 83.Bender RP, Osheroff N. In: Checkpoint Responses in Cancer Therapy. Dai W, editor. Humana Press; Totowa, New Jersey: 2008. pp. 57–91. [Google Scholar]
- 84.Kreuzer KN, Cozzarelli NR. J Bacteriol. 1979;140:424–435. doi: 10.1128/jb.140.2.424-435.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roca J, Wang JC. Cell. 1992;71:833–840. doi: 10.1016/0092-8674(92)90558-t. [DOI] [PubMed] [Google Scholar]
- 86.Lindsley JE, Wang JC. J Biol Chem. 1993;268:8096–8104. [PubMed] [Google Scholar]
- 87.Classen S, Olland S, Berger JM. Proc Natl Acad Sci USA. 2003;100:10629–10634. doi: 10.1073/pnas.1832879100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei H, Ruthenburg AJ, Bechis SK, Verdine GL. J Biol Chem. 2005;280:37041–37047. doi: 10.1074/jbc.M506520200. [DOI] [PubMed] [Google Scholar]
- 89.Bendsen S, Oestergaard VH, Skouboe C, Brinch M, Knudsen BR, Andersen AH. Biochemistry. 2009;48:6508–6515. doi: 10.1021/bi9005978. [DOI] [PubMed] [Google Scholar]
- 90.Osheroff N. J Biol Chem. 1986;261:9944–9950. [PubMed] [Google Scholar]
- 91.Robinson MJ, Osheroff N. Biochemistry. 1991;30:1807–1813. doi: 10.1021/bi00221a012. [DOI] [PubMed] [Google Scholar]
- 92.Bromberg KD, Hendricks C, Burgin AB, Osheroff N. J Biol Chem. 2002;277:31201–31206. doi: 10.1074/jbc.M204741200. [DOI] [PubMed] [Google Scholar]
- 93.Osheroff N, Zechiedrich EL. Biochemistry. 1987;26:4303–4309. doi: 10.1021/bi00388a018. [DOI] [PubMed] [Google Scholar]
- 94.Wigley DB, Davies GJ, Dodson EJ, Maxwell A, Dodson G. Nature. 1991;351:624–629. doi: 10.1038/351624a0. [DOI] [PubMed] [Google Scholar]
- 95.Maxwell A, Gellert M. J Biol Chem. 1984;259:14472–14480. [PubMed] [Google Scholar]
- 96.Baldwin EL, Byl JA, Osheroff N. Biochemistry. 2004;43:728–735. doi: 10.1021/bi035472f. [DOI] [PubMed] [Google Scholar]
- 97.Noble CG, Maxwell A. J Mol Biol. 2002;318:361–371. doi: 10.1016/S0022-2836(02)00049-9. [DOI] [PubMed] [Google Scholar]
- 98.Aravind L, Leipe DD, Koonin EV. Nucleic Acids Res. 1998;26:4205–4213. doi: 10.1093/nar/26.18.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deweese JE, Burgin AB, Osheroff N. Nucleic Acids Res. 2008;36:4883–4893. doi: 10.1093/nar/gkn466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Deweese JE, Burgin AB, Osheroff N. Biochemistry. 2008;47:4129–4140. doi: 10.1021/bi702194x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dong KC, Berger JM. Nature. 2007;450:1201–1205. doi: 10.1038/nature06396. [DOI] [PubMed] [Google Scholar]
- 102.Beese LS, Friedman JM, Steitz TA. Biochemistry. 1993;32:14095–14101. doi: 10.1021/bi00214a004. [DOI] [PubMed] [Google Scholar]
- 103.Sarnovsky RJ, May EW, Craig NL. EMBO J. 1996;15:6348–6361. [PMC free article] [PubMed] [Google Scholar]
- 104.Peracchi A, Beigelman L, Scott EC, Uhlenbeck OC, Herschlag D. J Biol Chem. 1997;272:26822–26826. doi: 10.1074/jbc.272.43.26822. [DOI] [PubMed] [Google Scholar]
- 105.Curley JF, Joyce CM, Piccirilli JA. J Am Chem Soc. 1997;119:12691–12692. [Google Scholar]
- 106.Viadiu H, Aggarwal AK. Nat Struct Biol. 1998;5:910–916. doi: 10.1038/2352. [DOI] [PubMed] [Google Scholar]
- 107.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 108.Brautigam CA, Steitz TA. J Mol Biol. 1998;277:363–377. doi: 10.1006/jmbi.1997.1586. [DOI] [PubMed] [Google Scholar]
- 109.Brautigam CA, Sun S, Piccirilli JA, Steitz TA. Biochemistry. 1999;38:696–704. doi: 10.1021/bi981537g. [DOI] [PubMed] [Google Scholar]
- 110.Allingham JS, Pribil PA, Haniford DB. J Mol Biol. 1999;289:1195–1206. doi: 10.1006/jmbi.1999.2837. [DOI] [PubMed] [Google Scholar]
- 111.Sontheimer EJ, Gordon PM, Piccirilli JA. Genes Dev. 1999;13:1729–1741. doi: 10.1101/gad.13.13.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Galburt EA, Chevalier B, Tang W, Jurica MS, Flick KE, Monnat RJ, Jr, Stoddard BL. Nat Struct Biol. 1999;6:1096–1099. doi: 10.1038/70027. [DOI] [PubMed] [Google Scholar]
- 113.Lukacs CM, Kucera R, Schildkraut I, Aggarwal AK. Nat Struct Biol. 2000;7:134–140. doi: 10.1038/72405. [DOI] [PubMed] [Google Scholar]
- 114.Horton JR, Cheng X. J Mol Biol. 2000;300:1049–1056. doi: 10.1006/jmbi.2000.3938. [DOI] [PubMed] [Google Scholar]
- 115.Yean SL, Wuenschell G, Termini J, Lin RJ. Nature. 2000;408:881–884. doi: 10.1038/35048617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang L, Arora K, Beard WA, Wilson SH, Schlick T. J Am Chem Soc. 2004;126:8441–8453. doi: 10.1021/ja049412o. [DOI] [PubMed] [Google Scholar]
- 117.Perry JJ, Yannone SM, Holden LG, Hitomi C, Asaithamby A, Han S, Cooper PK, Chen DJ, Tainer JA. Nat Struct Mol Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- 118.Stahley MR, Strobel SA. Science. 2005;309:1587–1590. doi: 10.1126/science.1114994. [DOI] [PubMed] [Google Scholar]
- 119.Steitz TA. EMBO J. 2006;25:3458–3468. doi: 10.1038/sj.emboj.7601211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Radhakrishnan R, Arora K, Wang Y, Beard WA, Wilson SH, Schlick T. Biochemistry. 2006;45:15142–15156. doi: 10.1021/bi061353z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sissi C, Chemello A, Vazquez E, Mitchenall LA, Maxwell A, Palumbo M. Biochemistry. 2008;47:8538–8545. doi: 10.1021/bi800480j. [DOI] [PubMed] [Google Scholar]
- 122.Deweese JE, Burch AM, Burgin AB, Osheroff N. Biochemistry. 2009;48:1862–1869. doi: 10.1021/bi8023256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.West KL, Meczes EL, Thorn R, Turnbull RM, Marshall R, Austin CA. Biochemistry. 2000;39:1223–1233. doi: 10.1021/bi991328b. [DOI] [PubMed] [Google Scholar]
- 124.Leontiou C, Lakey JH, Lightowlers R, Turnbull RM, Austin CA. Mol Pharmacol. 2006;69:130–139. doi: 10.1124/mol.105.015933. [DOI] [PubMed] [Google Scholar]
- 125.Schmidt BH, Burgin AB, Deweese JE, Osheroff N, Berger JM. Nature. 2010 doi: 10.1038/nature08974. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang J, Yu P, Lin TC, Konigsberg WH, Steitz TA. Biochemistry. 1996;35:8110–8119. doi: 10.1021/bi960178r. [DOI] [PubMed] [Google Scholar]
- 127.Nowotny M, Yang W. EMBO J. 2006;25:1924–1933. doi: 10.1038/sj.emboj.7601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhu CX, Tse-Dinh YC. J Biol Chem. 2000;275:5318–5322. doi: 10.1074/jbc.275.8.5318. [DOI] [PubMed] [Google Scholar]
- 129.Caron PR, Wang JC. Adv Pharmacol. 1994;29B:271–297. doi: 10.1016/s1054-3589(08)61143-6. [DOI] [PubMed] [Google Scholar]
- 130.Pearson RG. Science. 1966;151:172–177. doi: 10.1126/science.151.3707.172. [DOI] [PubMed] [Google Scholar]
- 131.Pecoraro VL, Hermes JD, Cleland WW. Biochemistry. 1984;23:5262–5271. doi: 10.1021/bi00317a026. [DOI] [PubMed] [Google Scholar]
- 132.Sigel RKO, Song B, Sigel H. J Am Chem Soc. 1997;119:744–755. [Google Scholar]
- 133.Basu S, Strobel SA. RNA. 1999;5:1399–1407. doi: 10.1017/s135583829999115x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Szewczak AA, Kosek AB, Piccirilli JA, Strobel SA. Biochemistry. 2002;41:2516–2525. doi: 10.1021/bi011973u. [DOI] [PubMed] [Google Scholar]
- 135.Railsback LB. Geology. 2003;31:737–740. [Google Scholar]
- 136.Deweese JE, Guengerich FP, Burgin AB, Osheroff N. Biochemistry. 2009;48:8940–8947. doi: 10.1021/bi900875c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yang W, Lee JY, Nowotny M. Mol Cell. 2006;22:5–13. doi: 10.1016/j.molcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 138.Gmunder H, Kuratli K, Keck W. Antimicrob Agents Chemother. 1995;39:163–169. doi: 10.1128/aac.39.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wilstermann AM, Osheroff N. J Biol Chem. 2001;276:17727–17731. doi: 10.1074/jbc.M100197200. [DOI] [PubMed] [Google Scholar]
- 140.Reece RJ, Maxwell A. J Biol Chem. 1989;264:19648–19653. [PubMed] [Google Scholar]
- 141.Fisher LM, Barot HA, Cullen ME. EMBO J. 1986;5:1411–1418. doi: 10.1002/j.1460-2075.1986.tb04375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Strumberg D, Nitiss JL, Dong J, Kohn KW, Pommier Y. J Biol Chem. 1999;274:28246–28255. doi: 10.1074/jbc.274.40.28246. [DOI] [PubMed] [Google Scholar]
- 143.Gale KC, Osheroff N. Biochemistry. 1990;29:9538–9545. doi: 10.1021/bi00493a007. [DOI] [PubMed] [Google Scholar]
- 144.Lund K, Andersen AH, Christiansen K, Svejstrup JQ, Westergaard O. J Biol Chem. 1990;265:13856–13863. [PubMed] [Google Scholar]
- 145.Andersen AH, Sørensen BS, Christiansen K, Svejstrup JQ, Lund K, Westergaard O. J Biol Chem. 1991;266:9203–9210. [PubMed] [Google Scholar]
- 146.Froelich-Ammon SJ, Gale KC, Osheroff N. J Biol Chem. 1994;269:7719–7725. [PubMed] [Google Scholar]
- 147.Wang Y, Knudsen BR, Bjergbaek L, Westergaard O, Andersen AH. J Biol Chem. 1999;274:22839–22846. doi: 10.1074/jbc.274.32.22839. [DOI] [PubMed] [Google Scholar]
- 148.Wilstermann AM, Osheroff N. J Biol Chem. 2001;276:46290–46296. doi: 10.1074/jbc.M105733200. [DOI] [PubMed] [Google Scholar]
- 149.Osheroff N. Biochemistry. 1989;28:6157–6160. doi: 10.1021/bi00441a005. [DOI] [PubMed] [Google Scholar]
- 150.Robinson MJ, Osheroff N. Biochemistry. 1990;29:2511–2515. doi: 10.1021/bi00462a012. [DOI] [PubMed] [Google Scholar]