


Abstract
Objective:
Episodic ataxia type 1 (EA1) is a monogenic channelopathy caused by mutations of the potassium channel gene KCNA1. Affected individuals carrying the same mutation can exhibit considerable variability in the severity of ataxia, neuromyotonia, and other associated features. We investigated the phenotypic heterogeneity of EA1 in 2 sets of identical twins to determine the contribution of environmental factors to disease severity. One of the mutations was also found in a distantly related family, providing evidence of the influence of genetic background on the EA1 phenotype.
Methods:
We evaluated 3 families with an EA1 phenotype, 2 of which included monozygotic twins. We sequenced the KCNA1 gene and studied the biophysical consequences of the mutations in HEK cells.
Results:
We identified a new KCNA1 mutation in each pair of twins. Both pairs reported striking differences in the clinical severity of symptoms. The F414S mutation identified in one set of twins also occurred in a distantly related family in which seizures complicated the EA1 phenotype. The other twins had an R307C mutation, the first EA1 mutation to affect an arginine residue in the voltage-sensor domain. Both mutants when expressed exerted a dominant-negative effect on wild-type channels.
Conclusion:
These results broaden the range of KCNA1 mutations and reveal an unexpectedly large contribution of nongenetic factors to phenotypic variability in EA1. The occurrence of epilepsy in 1 of 2 families with the F414S mutation suggests an interplay of KCNA1 with other genetic factors.
GLOSSARY
- EA1
= episodic ataxia type 1.
Episodic ataxia type 1 (EA1) is characterized by brief paroxysms of ataxia and interictal myokymia.1 It is caused by heterozygous point mutations in the voltage-gated KV1.1 potassium channel α-subunit, encoded by KCNA1.2 Scrutiny of genetically confirmed kindreds reveals considerable phenotypic heterogeneity with respect to severity of ataxia and presence of additional features such as epilepsy.1 Hitherto, attempts to account for phenotypic variability have concentrated on differences among distinct KCNA1 mutations with respect to their functional consequences when expressed in vitro. All EA1 mutations cause a loss of function, typically measured as a decrease in current density.1 Mutations associated with a severe phenotype have a dominant negative effect on wild-type channel function.3–5 A recent study in neurons has shown that KCNA1 mutations differentially affect action potential generation and neurotransmitter release.3 Despite attempts to correlate phenotypic severity with functional studies, even within affected kindreds there is frequently considerable heterogeneity in symptom severity. Monozygotic twins potentially provide an insight into the relative importance of modifier genes compared to lifestyle or environmental factors. Here we report 2 families where EA1 affects identical twins, and observe a surprising degree of discordance in symptom severity.
METHODS
See e-Methods on the Neurology® Web site at www.neurology.org.
Standard protocol approvals, registrations, and patient consents.
Ethical approval was obtained from the UCLH ethics committee. Written informed consent was obtained from all patients participating in the study. Consent to disclose was obtained for the video.
RESULTS
Clinical.
Family A.
Affected members of this pedigree (figure 1A) show a classical EA1 phenotype (summarized in the table). In the more severe cases, dysarthria and gait disturbance are also present during an attack. Attack precipitants include sudden movements or fever, although several patients have also had unprovoked attacks. The proband, who experiences up to 30 attacks a day, is the most severely affected, and has experienced no benefit from carbamazepine, in contrast to her mother, who reports a marked effect on attack frequency. Interictal myokymia has been observed in affected individuals. The proband's mother (II-4) has an identical twin (II-6; figure 1A). Although case II-4 is not available for objective evaluation, she and her twin sister report that their symptoms emerged at similar ages. However, their phenotypes differ with respect to attack frequency, duration, and severity. Indeed, while case II-4 has been maintained on carbamazepine, her identical sister, II-6, has not required medication.

Figure 1 Families A and B
(A) Pedigrees of families. Filled boxes represent affected individuals. Arrow denotes proband. (B) Electropherogram showing T-to-C transversion at nucleotide 1,241. Upper panel, proband; lower panel, control. (C) Filled boxes represent affected individuals. Arrow denotes proband. (D) Electropherogram showing T-to-C transversion at nucleotide 919. Upper panel, proband; lower panel, control. (E) Conservation of F414 in KCNA1. Arrow denotes mutation position, yellow box represents S6 segment. (F) Conservation of R307 in KCNA1. Arrow denotes mutation position, yellow box represents S4 segment, positively charged voltage-sensor residues shown in bold.
Table Clinical summary of patients
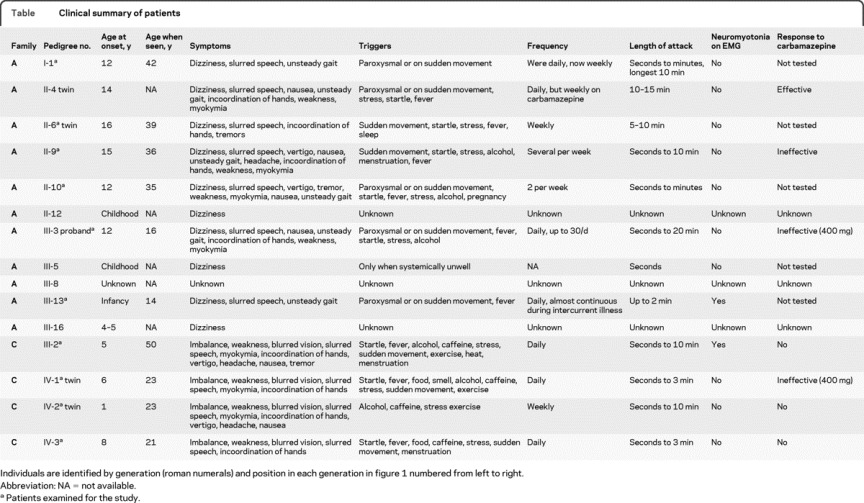
Family B.
Another family with EA1 from the same geographic region is included here because the same mutation was identified. From 6 months to 5 years of age the proband had brief focal epileptic seizures with impairment of awareness and apnea. At age 5, he developed episodic ataxia. In addition, he has secondary generalized seizures once per month; these were not controlled by carbamazepine or lamotrigine, but have responded to acetazolamide, which has also reduced the frequency of episodic ataxia attacks. His father, paternal uncle, and paternal grandmother all have episodic ataxia and interictal myokymia.
Family C.
Affected members of this pedigree have classic EA1 attacks (figure 1C, summarized in the table). The proband is one of confirmed identical twins (IV-1 and IV-2, figure 1C). His attacks began at the age of 6 and last up to 10 minutes. They consist of dizziness, imbalance, weakness, blurred vision, slurred speech, myokymia, and upper limb incoordination. His brother has less frequent, briefer, and less disabling attacks, which are associated with headache, vertigo, and nausea. The proband's mother (III-2) has a progressive interictal cerebellar syndrome, manifest as ataxia and dysarthria (see the video).
Genetics.
A novel T-to-C substitution was identified at position nt1241 of KCNA1 (figure 1B) in families A and B. This was not present in 128 control chromosomes. It leads to the substitution of a conserved phenylalanine (figure 1E) by serine at amino acid 414 (F414S) in the S6 transmembrane segment of the channel. Another mutation affecting the same residue (F414C) was recently identified in a large EA1 kindred.4 Haplotype analysis between members of families A and B demonstrated that all except 1 marker was shared by 6 individuals (figure e-1), covering a chromosomal region of approximately 5.77 Mb. These results suggest a shared ancestral haplotype on which the mutation has arisen and that the 2 families are related.
A novel T-to-C substitution was identified at position nt919 of KCNA1 in family C (figure 1D). This was not present in 228 control chromosomes, and leads to the substitution of a highly conserved arginine (figure 1F), by cysteine at position 307 in the S4 segment of the channel (R307C).
Functional characterization.
When expressed in HEK cells, both F414S and R307C were detected with anti-KV1.1 antibodies with similar distribution to WT KV1.1, suggesting the mutant subunits were present in the cells (figure e-2). However, no potassium currents could be detected in cells transfected with only mutant subunits (figure 2), suggesting that they are not functional. Coexpression of each mutant with WT KV1.1 produced potassium currents with a markedly reduced maximum current density compared to WT Kv1.1 channels alone (WT, 86.8 ± 7.5 pA/pF, n = 7; WT:R307C, 22.1 ± 5.4 pA/pF, n = 7, p = 0.003; WT:F414S, 6.9 ± 0.9 pA/pF, n = 7, p = 0.002). Both mutants also conferred a significant positive shift in the V1/2MAX of the voltage dependence of activation of Kv1.1 when coexpressed with WT channels (WT, V1/2MAX = −41.9 ± 0.5; WT:R307C, V1/2MAX = −30.5 ± 0.9, p < 0.0001; WT:F414S, V1/2MAX = −27.0 ± 1.4, p < 0.0001). These findings are consistent with both mutants exerting a dominant-negative effect on WT subunits.

Figure 2 Electrophysiologic characterization of R307C and F414S KV1.1
(A) Voltage protocol and current traces obtained from representative HEK cells expressing WT, homomeric mutant (R307C and F414S), and WT coexpressed with mutants (WT:R307C and WT:F414S). (B) Maximum tail current density (±SEM) measured at +60 mV in low intracellular potassium solutions (see Methods). (C) Normalized voltage dependence of activation (±SEM) for cells expressing WT, WT:R307C, and WT:F414S. Tail currents were sampled 1 msec after return to −80 mV. For all cells, n = 7.
DISCUSSION
Both mutations were identified in families with symptoms that fall within the spectrum previously reported for KCNA1 mutations. Carbamazepine, where tested, was not effective in all individuals. The occurrence of epilepsy in the proband of family B adds to the list of mutations associated with EA1 and epilepsy.1 The absence of epilepsy in family A, who are related to family B, is consistent with a role for other genes interacting with KCNA1 to determine seizure risk. An unusual feature of the R307C mutation identified in family C is the progressive ataxia in the proband's mother, which has not previously been reported in EA1.
Several observations point to causal roles for the F414S and R307C mutations in the disease. First, they lead to radical amino acid substitutions in the ion-conducting pore and voltage sensor, respectively. Second, the mutations were not detected in control chromosomes. Third, both the nucleotides and the amino acids are highly conserved through evolution. Fourth, functional expression revealed loss of function with a dominant-negative effect when coexpressed with wild type channels, consistent with previous functional studies on EA1 mutations. And finally, the residue mutated in families A and B is also affected in another family with EA1, who carry the F414C mutation, which decreases potassium currents.4
Both sets of identical twins reported unexpectedly large differences in severity and frequency of EA attacks. In both sets of twins, one has sought treatment, whereas the less severely affected twin has not required medication. The self-reported variability was as large among the identical twins as among other affected members of each family.
Few channelopathies have been studied in twins. One pair of identical twins with severe myoclonic epilepsy of infancy sharing an SCNA1 mutation was reported to be concordant for phenotype.5 However, discordant monozygotic twin pairs have also been reported in familial long QT syndrome6 and in episodic ataxia type 2.7 Although the numbers remain small, these findings suggest that symptom heterogeneity among individuals harboring the same mutation reflects the interplay not only of modifier genes, but also of nongenetic factors.
ACKNOWLEDGMENT
The authors thank M. Sweeney for carrying out the monozygosity testing and V. Gibbons and P. Sleiman for technical assistance.
DISCLOSURE
Dr. Graves held an Action Medical Research Training Fellowship and was previously funded by the Guarantors of Brain. Dr Rajakulendran receives research support from a Wellcome Trust Research Training Fellowship. Dr. Zuberi serves as an Associate Editor of the European Journal of Paediatric Neurology and receives research support from the Muir Maxwell Trust. Dr. Morris served on scientific advisory boards for Solvay Pharmaceuticals, Inc. and Boehringer Ingelheim; has received funding for travel or speaker honoraria from GlaxoSmithKline, UCB, Boehringer Ingelheim, Teva Pharmaceutical Industries Ltd., and Wellcome Trust; serves on the editorial board of Parkinson's Disease; and receives research support from the Ipsen Fund Research Fellowship, the Welsh Assembly Government, the Medical Research Council (UK), the Parkinson's Disease Society, the Progressive Supranuclear Palsy Association, and the Motor Neuron Disease Association. Dr. Schorge receives salary funding from a charitable contribution from the Worshipful Company of Pewterers and receives research support from the Medical Research Council (UK) and the Wellcome Trust. Dr. Hanna served as Deputy Editor of the Journal of Neurology, Neurosurgery & Psychiatry; and receives research support from the Medical Research Council and Action Medical Research. Dr. Kullmann receives research support from the Wellcome Trust, Action Medical Research, the Myasthenia Gravis Association, and the Medical Research Council (UK) and serves as an Associate Editor for Brain and a clinical editor for the Journal of Physiology.
Supplementary Material
Address correspondence and reprint requests to Dr. D.M. Kullmann, MRC Centre for Neuromuscular Diseases, UCL Institute of Neurology, Queen Square, London, WC1N 3BG United Kingdom d.kullmann@ion.ucl.ac.uk
Supplemental data at www.neurology.org
*These authors contributed equally to this work.
Study funding: This work was undertaken at UCLH/UCL and was supported by the Department of Health NIHR Biomedical Research Centres funding scheme. T.D.G., S.R., and M.G.H. belong to the Consortium for Clinical Investigation of Neurological Channelopathies (CINCH) supported by NIH RU54 RR019482 (NINDS/ORD).
Disclosure: Author disclosures are provided at the end of the article.
Received November 3, 2009. Accepted in final form April 9, 2010.
REFERENCES
- 1.Rajakulendran S, Schorge S, Kullmann DM, Hanna MG. Episodic ataxia type 1: a neuronal potassium channelopathy. Neurotherapeutics 2007;4:258–266. [DOI] [PubMed] [Google Scholar]
- 2.Browne DL, Gancher ST, Nutt JG, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 1994;8:136–140. [DOI] [PubMed] [Google Scholar]
- 3.Heeroma JH, Henneberger C, Rajakulendran S, Hanna MG, Schorge S, Kullmann DM. Episodic ataxia type 1 mutations differentially affect neuronal excitability and transmitter release. Dis Model Mech 2009;2:612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imbrici P, Gualandi F, D'Adamo MC, et al. A novel KCNA1 mutation identified in an Italian family affected by episodic ataxia type 1. Neuroscience 2008;157:577–587. [DOI] [PubMed] [Google Scholar]
- 5.Sugawara T, Mazaki-Miyazaki E, Fukushima K, et al. Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology 2002;58:1122–1124. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi K, Shimizu M, Ino H, et al. Identical twins with long QT syndrome associated with a missense mutation in the S4 region of the HERG. Jpn Heart J 2000;41:399–404. [DOI] [PubMed] [Google Scholar]
- 7.Anderson JH, Christova PS, Xie T, Schott KS, Ward K, Gomez CM. Spinocerebellar ataxia in monozygotic twins. Arch Neurol 2002;59:1945–1951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.