

Abstract
Background:
Detection of aquaporin-4–specific immunoglobulin G (IgG) has expanded the spectrum of neuromyelitis optica (NMO). Rare reports of familial aggregation have suggested a component of genetic susceptibility but these reports mostly antedated the discovery of the NMO-IgG biomarker and recently updated diagnostic criteria.
Methods:
We report a case series describing the demographic, clinical, neuroimaging, and NMO-IgG serologic status of 12 multiplex NMO pedigrees with a total of 25 affected individuals.
Results:
Twenty-one patients (84%) were women. Families were Asian (n = 5), Latino (n = 4), white (n = 1), or African (n = 2). Apparent transmission was either maternal (n = 5) or paternal (n = 2). In 1 family, 3 individuals had NMO; in the others, 2 individuals were affected. Sibling pairs (n = 6), parent-child (n = 4), and aunt-niece (n = 3) pairs were observed. Nineteen patients (76%) were NMO-IgG positive. Twelve (48%) had clinical or serologic evidence of another autoimmune disease. Familial occurrence of NMO occurs in approximately 3% of patients with well-established diagnosis of NMO.
Conclusions:
A small proportion of patients with NMO have relatives with this condition, but familial occurrence is more common than would be expected from its frequency in the general population. Familial NMO is indistinguishable from sporadic NMO based on clinical symptoms, age at onset, sex distribution, and frequency of NMO-IgG detection. One or 2 generations were affected and affected individuals represented a small fraction of family members. Taken together, these data suggest complex genetic susceptibility in NMO.
GLOSSARY
- AQP4
= aquaporin-4;
- CI
= confidence interval;
- HLA
= human leukocyte antigen;
- IgG
= immunoglobulin G;
- LETM
= longitudinally extensive transverse myelitis;
- MS
= multiple sclerosis;
- NMO
= neuromyelitis optica;
- ON
= optic neuritis;
- OR
= odds ratio.
Neuromyelitis optica (NMO) and the NMO spectrum disorders are autoimmune inflammatory diseases of the CNS associated with severe relapses of optic neuritis (ON) and myelitis. A serum IgG autoantibody marker, NMO-IgG,1 which targets the astrocyte water channel aquaporin-4 (AQP4),2 and validated clinical diagnostic criteria3,4 allow sensitive and specific diagnosis of NMO. An NMO spectrum disorder can now be confidently diagnosed even in the face of previously unrecognized clinical phenomena (e.g., symptomatic brain lesions) that precluded a diagnosis of NMO in the past. Furthermore, NMO can now be distinguished from multiple sclerosis (MS) with an initial presentation of ON and myelitis.5,6
Although there are few population-based studies, the prevalence of NMO has been estimated by its relative frequency to MS, and a consensus estimate of NMO prevalence is approximately 1 per 100,000.7,8 Ethnic predilection is controversial; the previously held belief that it is a disease predominantly of Asians and perhaps Africans has not been supported in some studies.7,9 The disproportionate occurrence of NMO in Asians, Latinos, and Africans in some regions may represent a dearth of MS, to which its frequency is compared, in these ethnic groups rather than a true excess of NMO.
NMO-IgG is pathogenic in vitro,10,11 and can passively transfer NMO-specific brain lesions,12 but the etiology of NMO remains unknown. We describe the demographic, clinical, neuroimaging, and laboratory profiles of 12 families with NMO, totaling 25 affected individuals, and compare the findings in these NMO cases with those in a series of sporadic NMO cases. The number of multiplex families is unexpectedly large relative to the number expected from the estimated frequency of NMO in the general population, suggesting that heritable factors are involved in susceptibility, although common exposure to an exogenous causative factor within families cannot be excluded. We estimate the frequency of familial aggregation in this disease and evaluated metrics that are potential indicators of the relative importance of genetic susceptibility vs common exposures.
METHODS
Standard protocol approvals, registrations, and patient consents.
The institutional review board or ethics committee of Mayo Clinic, Rochester, MN; the National Cancer Center, Goyang, South Korea; the University of São Paulo, Brazil; the Legacy Emanuel Hospital, Portland, OR; and the National Hospital for Neurology and Neurosurgery Queen Square, London, UK, approved this study. All patients provided informed consent.
Pedigree ascertainment and assessment.
Five families (11 patients) were identified through Mayo Clinic neurology practice. The remaining 7 families (14 patients) were identified by collaborators in South Korea (n = 3), Brazil (n = 3), and the United Kingdom (n = 1). Each index case in this report fulfilled clinical criteria for NMO or an NMO spectrum disorder (e.g., longitudinally extensive transverse myelitis [LETM] or recurrent ON and seropositive for NMO-IgG).13 The median duration of disease was 4.8 years (range 1–52 years). All patients' sera were tested for NMO-IgG by indirect immunofluorescence on a mouse composite tissue substrate.1
We abstracted the following data from the medical record: demographic information, disease duration, age at and year of clinical onset, number and severity of NMO episodes, history and serologic evaluation for evidence of other autoimmune diseases, and MRI results.
We compared intrapair differences in age at onset and calendar year of onset. We used linear regression analysis (age and year of onset) and intraclass correlation of age at onset to evaluate putative common exposures to environmental factors and genetic effects. If susceptibility to NMO was primarily determined by heritable factors, differences in onset age might be smaller between members of pairs relative to the differences between the pairs. If a common exposure to a triggering factor was present, and there was a similar incubation period, affected relatives within pairs might be expected to develop NMO in approximately the same calendar year.
We estimated the frequency of familial aggregation of NMO in families where familial aggregation was not the basis for referral and the family history was determined based on systematic inquiry in patients referred for a diagnosis of NMO at each participating center. We compared the familial cases to a series of patients with sporadic NMO.3
RESULTS
Demographic characteristics and pedigree structure.
Demographic characteristics and pedigree structure are illustrated in table 1. Twenty-one patients (84%) were women. Five families were Asian, 4 Latino, 1 African American, 1 African Caribbean, and 1 white. In 6 families, the affected individuals lived in the same house when they developed initial symptoms of NMO.
Table 1 Demographic and clinical characteristics of familial NMO patients
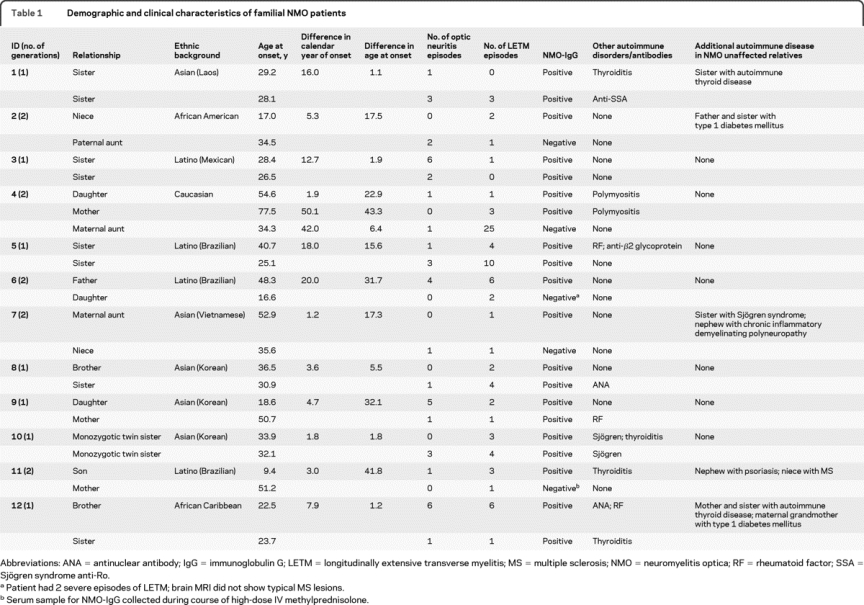
In 6 families, 2 generations were involved. One affected pair consisted of half siblings with a common mother. Of the 7 families in which we inferred transmission, 5 (71.4%) had maternal and 2 (28.6%) had paternal line transmission. This difference is likely due to the overrepresentation of women in NMO rather than a sex bias in transmission.
In 11 families, 2 individuals were affected, and in 1 family, 3 individuals were affected. We observed the following pairings: sibling pairs (n = 6), parent-child pairs (n = 3), aunt-niece pairs (n = 2), and a mother-daughter-maternal aunt trio (n = 1). One pair of monozygotic twins was identified. Consanguineous marriages were not reported in any family.
Clinical features.
The mean (SD) age at onset was 34.3 years (15.0). The mean difference in age at onset between familial pairs was 17.9 (15.4) years and the mean difference in calendar year of onset was 11.2 (13.4) years. Pearson linear correlation coefficient between the intrapair difference in ages with differences in age at onset was r = 0.82 (p = 0.01) and with differences in year of onset was r = 0.16 (p = 0.60) (figure e-1 on the Neurology® Web site at www.neurology.org). The intraclass correlation estimate for differences of age at onset was 0, indicating greater variability within relative pairs than across subjects.
The initial syndrome was ON in 14 (51.8%) individuals and LETM in 11 (40.7%) individuals. Intrafamilial concordance for the presenting syndrome (ON or LETM) was seen in 5/12 families (41.6%), which was as expected based on the frequency of these 2 presentations. Two individuals from the same family had both ON and myelitis within a 1-month interval. The median relapse count was 3 (range 1–26) and the median annualized relapse rate was 0.81 (range 0.12–4.8).
NMO-IgG was positive in 19 (76%), including all 12 index cases. We tested 3 nonaffected relatives: the sister of the index case in family 1 and the father and sister of the index case in family 2. All 3 were NMO-IgG seronegative.
Other autoimmune diseases were documented in 7 affected individuals (autoimmune thyroiditis, n = 3; Sjögren syndrome, n = 2; polymyositis, n = 2). In 6 families, unaffected relatives had other autoimmune diseases (autoimmune thyroiditis, n = 2; type 1 diabetes, n = 2; psoriasis, n = 1; Sjögren syndrome, n = 1; chronic inflammatory demyelinating polyradiculoneuropathy, n = 1).
In family 12, besides the mother and son with NMO, a maternal niece had a diagnosis of MS, with 12 relapses since 1994 involving sensory, cerebellar, and pyramidal systems. Brain and spinal cord MRI were compatible with MS, oligoclonal bands were present in the CSF, and NMO-IgG was negative.
Three additional families with NMO index cases were excluded because it could not be determined with confidence that the relative had NMO or NMO spectrum disorder. In the first family, the mother had relapses of ON (n = 3) and LETM (n = 6) over 15 years of disease and was seropositive for NMO-IgG. Her daughter had 2 episodes of ON over 16 years of disease and was seronegative for NMO-IgG on 2 occasions. In the second family, the NMO-IgG seropositive index case had 2 episodes of ON and 1 of myelitis over 15 months of disease. Her sister died at age 39 due to complications of presumed MS (details not available) and her niece (daughter of a different sister) had multiple episodes of ON and one of transverse myelitis but was seronegative for NMO-IgG and LETM was not documented by MRI. In the third family, the index case had 3 episodes of LETM and was seropositive for NMO-IgG. Her maternal cousin presented with severe bilateral ON and had an episode of hearing loss associated with a lesion in the brainstem 2 years after being documented as seronegative for NMO-IgG.
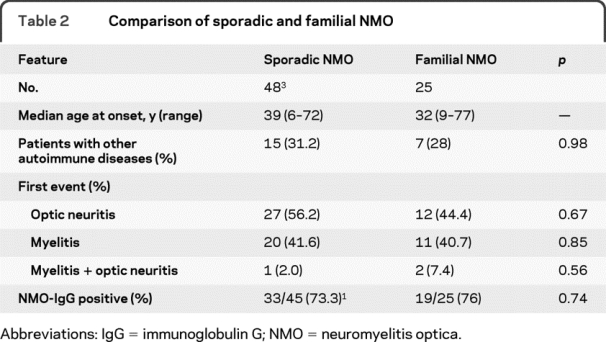
Familial NMO cases were similar, if not indistinguishable, from sporadic cases, both individually and at a group level in comparison to a reference series of sporadic NMO cases with regard to age at onset, sex distribution, frequency of other coexisting autoimmune diseases, presenting symptoms, and relapsing clinical course (table 2).2,3
Table 2 Comparison of sporadic and familial NMO
The frequency of familial aggregation in NMO is 2.8% based on the number of familial index cases in a combined series of 386 consecutive patients with NMO seen at Mayo Clinic, Rochester, MN (n = 5/175; 2.9%), National Cancer Center, Goyang, South Korea (3/88; 3.4%), Medical School of University Hospital of Riberão Preto, São Paulo, Brazil (2/33; 6.0%), National Hospital for Neurology and Neurosurgery Queen Square, London, UK (1/16; 6.3%), and University of São Paulo School of Medicine, São Paulo, Brazil (1/74; 1.4%).
DISCUSSION
Analysis of pedigrees of multiplex families with NMO may be helpful in understanding the pattern of transmission and ultimately, combined with molecular genetic analysis, in discovering susceptibility genes. Four multiplex families with NMO (8 affected individuals) have been reported, all but one report from the pre-NMO-IgG1 era and before formal diagnostic criteria4 were proposed. Identical twin sisters (24 and 26 years of age at disease onset) with NMO were reported in 1938. Both had severe episodes of transverse myelitis and bilateral blindness and died of NMO-related complications. The autopsy evaluation showed demyelination of the optic nerve and diffuse inflammation and demyelination extending from the cervical to the lower thoracic cord.14 These reports were followed by those of 2 infant sisters (age 2 and 3 at onset) who had relapses of bilateral ON and thoracic myelitis15; of 2 sisters with late onset of NMO (62 and 59)16; and of a daughter (aged 29 at onset) with myasthenia gravis and NMO and a mother (62 years at onset) with NMO-IgG seropositive NMO.17 Recently, 2 sisters, 1 with NMO and 1 with prototypic MS, were reported.18
The frequency of NMO in family members of our NMO index cases is greater than expected based on the best estimate of its prevalence frequency in the general population of 1/100,000.7,8 If the average pedigree size consisted of 100 first- and second-degree relatives, which is likely an overestimate, one would expect 0.38 affected relatives among the 386 sporadic cases from which familial cases were derived at the collaborating institutions; we detected 12 cases (p < 0.0001; χ2). Familial cases accounted for 3% of NMO but considering the cases that were excluded this is a minimum estimate.
In both the historical sporadic and the current familial series, NMO was characterized by acute relapses. The distribution of presenting symptoms (ON, myelitis, or both) was very similar. In both series, ON and myelitis were the dominant clinical manifestations. We had not recognized the extended spectrum of NMO, which includes patients who have recurrent myelitis or recurrent ON, in the sporadic series published in 1999, and NMO-IgG serology was unavailable at that time; therefore, some differences in the characteristics of cases reflects improved understanding of the spectrum of NMO in the familial series. Comparison of outcome data and response to therapy is difficult, because early diagnosis, consistent early treatment with immunosuppressive drugs, and systematic follow-up are much more typical of the contemporary series than it was for the sporadic series.3
Only 1 or 2 generations were affected. There was no evidence of bias of transmission from either paternal or maternal line; the excess of maternal line transmission reflects the increased predisposition of women for NMO.
Analysis of age and calendar year of onset in the NMO relative pairs did not strongly support either common environmental exposure or a purely genetic basis for susceptibility. The linear regression analysis of the age at onset with age indicated that age, rather than a specific common exposure, was the principal determinant of age at onset. Furthermore, intraclass correlation of age at onset revealed no excess similarity between members of a pair compared to the interpair differences contrary to expectation if genetic factors solely determined susceptibility to NMO. The lack of intraclass correlation of age at onset and the age dependence of onset may largely be explained by the predilection for NMO to occur in late middle-aged individuals that may obscure pedigree-specific effects on age at onset.3,19 There was wide variation between members of pairs in years of onset, suggesting that common exposure was unlikely to be an important etiologic factor.
Genetic association studies for this disease are limited. Human leukocyte antigen (HLA) associations have been observed in studies of sporadic cases of NMO. HLA-DPB1*0501 allele was more frequent in 38 Japanese patients seropositive for NMO-IgG compared to 52 patients with MS.20 Forty-five French Caucasian NMO cases were compared to healthy controls and patients with MS for HLA class II A and B alleles; no association was found of DRB1*1501 with NMO (odds ratio [OR] 1.74; 95% confidence interval [CI] 0.97–3.11, p = 0.06). HLA-DRB1*03 was associated with NMO-IgG-seropositive NMO (OR 3.08; 95% CI 1.52–6.27, p = 0.001).21 No mutation was found in genes known to harbor the majority of LHON mutations in 32 patients with NMO.22 No association was found in a study of 7 AQP4 SNPs genotyped in 901 MS trio families, including 69 in which the affected offspring had clinical history of optic-spinal disease.23 Recently, genome scan was performed with samples of 53 NMO Korean cases and 240 controls. The study was underpowered but the strongest SNP association signals were tested in a cohort of 93 NMO patients and 368 controls. A common promoter SNP in CYP7A1, a gene that encodes a member of the cytochrome P450 superfamily of enzymes, had a dose-dependent protective effect against NMO (p = 0.0004).24
Although certain infections25 and cancer26 have been reported to co-occur with NMO, no environmental or disease trigger has been rigorously associated with NMO. In the families we report, we did not find clear indicators of common exposure to infections. One patient had breast cancer a year prior to the development of NMO. We are unaware of NMO occurring in unrelated household members. There does not appear to be any geographic or ethnic restriction of familial occurrence of NMO. Several of the NMO families had members with other autoimmune diseases, suggesting that these individuals may share common genetic risk factors for autoimmunity in addition to factors that lead to AQP4-specific autoimmunity.
The small number of cases within affected pedigrees, the lack of multigenerational pedigrees, the high ratio of sporadic to familial cases, and lack of distinctive characteristics of familial cases, taken together, support the hypothesis that NMO is a complex genetic disease.
ACKNOWLEDGMENT
The authors thank Dr. Vanda Lennon (Mayo Clinic) for discussion and review of the manuscript, Dr. Dagoberto Calegaro (University of São Paulo, Brazil) for ascertainment of 1 Brazilian pedigree, and Dr. Mithu Storoni (National Hospital for Neurology and Neurosurgery Queen Square, London, UK) for abstracting data of the UK pedigree.
DISCLOSURE
Dr. Matiello is supported by a postdoctoral fellowship from the National Multiple Sclerosis Society. Dr. H.J. Kim has served as a consultant for and received funding for travel and speaker honoraria from Bayer Schering Pharma and Merck Serono. Dr. W Kim, Dr. Brum, Dr. Barreira, and Dr. Kingsbury report no disclosures. Dr. Plant serves as Editor-in-Chief of Neuro-ophthalmology and as an Associate Editor of Acta Ophthalmologica. Dr. Adoni has received funding for travel and speaker honoraria from Teva Pharmaceutical Industries Ltd. and Biogen Idec. Dr. Weinshenker has served/serves on data safety monitoring boards for Novartis and Biogen Idec; receives research support from Guthy Jackson Charitable Foundation; and has received royalty payments from R.S.R. Ltd. for a patent re: NMO-IgG for diagnosis of neuromyelitis optica.
Supplementary Material
Address correspondence and reprint requests to Dr. Brian Weinshenker, Department of Neurology, Mayo Clinic, 200 First St. SW, Rochester, MN 55905 weinb@mayo.edu
Supplemental data at www.neurology.org
Study funding: Supported by the Guthy-Jackson Charitable Foundation, National Multiple Sclerosis Society, and by the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and the NIH Roadmap for Medical Research (NCRR 1 UL1 RR024150).
Disclosure: Author disclosures are provided at the end of the article.
Received December 8, 2009. Accepted in final form March 29, 2010.
REFERENCES
- 1.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 2.Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wingerchuk DM, Hogancamp WF, O'Brien PC, et al. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 4.Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 5.Weinshenker BG, Wingerchuk DM, Vukusic S, et al. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 2006;59:566–569. [DOI] [PubMed] [Google Scholar]
- 6.Matiello M, Lennon VA, Jacob A, et al. NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology 2008;70:2197–2200. [DOI] [PubMed] [Google Scholar]
- 7.Cabrera-Gomez JA, Kurtzke JF, Gonzalez-Quevedo A, et al. An epidemiological study of neuromyelitis optica in Cuba. J Neurol 2009;256:35–44. [DOI] [PubMed] [Google Scholar]
- 8.Cabre P. Environmental changes and epidemiology of multiple sclerosis in the French West Indies. J Neurol Sci 2009;286:58–61. [DOI] [PubMed] [Google Scholar]
- 9.Marignier R, de Seze J, Vukusic S, et al. NMO-IgG and Devic's neuromyelitis optica: a French experience. Mult Scler 2008;14:440–445. [DOI] [PubMed] [Google Scholar]
- 10.Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007;69:2221–2231. [DOI] [PubMed] [Google Scholar]
- 11.Kinoshita M, Nakatsuji Y, Moriya M, et al. Astrocytic necrosis is induced by anti-aquaporin-4 antibody-positive serum. Neuroreport 2009;20:508–512. [DOI] [PubMed] [Google Scholar]
- 12.Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: Pathogenicity of patient immunoglobulin in vivo. Ann Neurol 2009;66:630–643. [DOI] [PubMed] [Google Scholar]
- 13.Wingerchuk DM, Lennon VA, Lucchinetti CF, et al. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815. [DOI] [PubMed] [Google Scholar]
- 14.McAlpine D. Familial neuromyelitis optica: its occurrence in identical twins. Brain 1938;61:430–438. [Google Scholar]
- 15.Ch'ien LT, Medeiros MO, Belluomini JJ, et al. Neuromyelitis optica (Devic's syndrome) in two sisters. Clin Electroencephalogr 1982;13:36–39. [DOI] [PubMed] [Google Scholar]
- 16.Yamakawa K, Kuroda H, Fujihara K, et al. Familial neuromyelitis optica (Devic's syndrome) with late onset in Japan. Neurology 2000;55:318–320. [DOI] [PubMed] [Google Scholar]
- 17.Braley T, Mikol DD. Neuromyelitis optica in a mother and daughter. Arch Neurol 2007;64:1189–1192. [DOI] [PubMed] [Google Scholar]
- 18.Cabrera-Gomez JA, Ramon-Perez L, Saiz A, et al. Neuromyelitis optica and multiple sclerosis in sisters. Mult Scler 2009;15:269–271. [DOI] [PubMed] [Google Scholar]
- 19.Matiello M, Jacob A, Wingerchuk DM, et al. Neuromyelitis optica. Curr Opin Neurol 2007;20:255–260. [DOI] [PubMed] [Google Scholar]
- 20.Matsushita T, Matsuoka T, Isobe N, et al. Association of the HLA-DPB1*0501 allele with anti-aquaporin-4 antibody positivity in Japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens 2009;73:171–176. [DOI] [PubMed] [Google Scholar]
- 21.Zephir H, Fajardy I, Outteryck O, et al. Is neuromyelitis optica associated with human leukocyte antigen? Mult Scler 2009;15:571–579. [DOI] [PubMed] [Google Scholar]
- 22.Cock H, Mandler R, Ahmed W, et al. Neuromyelitis optica (Devic's syndrome): no association with the primary mitochondrial DNA mutations found in Leber hereditary optic neuropathy. J Neurol Neurosurg Psychiatry 1997;62:85–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ban M, Walton A, Goris A, et al. Polymorphisms in the neuromyelitis optica auto-antigen AQP4 and susceptibility to multiple sclerosis. J Neurol 2007;254:398–399. [DOI] [PubMed] [Google Scholar]
- 24.Kim HJ, Park HY, Kim E, et al. Common CYP7A1 promoter polymorphism associated with risk of neuromyelitis optica. Neurobiol Dis 2010;37:349–355. [DOI] [PubMed] [Google Scholar]
- 25.Sellner J, Hemmer B, Muhlau M. The clinical spectrum and immunobiology of parainfectious neuromyelitis optica (Devic) syndromes. J Autoimmun 2010;34:371–379. [DOI] [PubMed] [Google Scholar]
- 26.Pittock SJ, Lennon VA. Aquaporin-4 autoantibodies in a paraneoplastic context. Arch Neurol 2008;65:629–632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.