

Abstract
A total synthesis of (+)-papulacandin D has been achieved in 31 steps, in a 9.2% overall yield from commercially available materials. The synthetic strategy divided the molecule into two nearly equal sized subunits, the spirocyclic C-arylglycopyranoside and the polyunsaturated fatty acid side chain. The C-arylglycopyranoside was prepared in 11 steps in a 30% overall yield from triacetoxyglucal. The fatty acid side chain was also prepared in 11 steps in a 30% overall yield from geraniol. The key strategic transformations in the synthesis are: (1) a palladium-catalyzed, organosilanolate-based cross-coupling reaction of a dimethylglucal-silanol with an electron rich and sterically hindered aromatic iodide and (2) a Lewis base-catalyzed, enantioselective allylation reaction of a dienal and allyltrichlorosilane. A critical element in the successful execution of the synthesis was the development of a suitable protecting group strategy that satisfied a number of stringent criteria.
Keywords: C-arylglycosides, silicon-based cross-coupling, asymmetric allylation
1. Introduction and Background
The papulacandins are a family of antifungal agents, isolated from the deuteromycetous fungus Papularia sphaerosperma1 that have demonstrated potent in vitro antifungal activity against such pathogens as: Candida albicans, Geotrichum lactis, Saccharomyes cerevisiae, Pneumocytis carinii, etc.2 Interestingly, the papulacandins are inactive against filamentous fungi, bacteria, and protozoa.1a Compounds effective in treating fungal infections in mammals by mechanisms that do not interfere with metabolic pathways are rare and so the discovery and synthesis of such new antifungal agents is of great importance to human health.2g, 3
All of the members of the papulacandin family target (1,3)-beta-D-glucan synthase,4 thereby preventing uptake of glucose in the biosynthesis of glucan, one of the most abundant polysaccharide components of the cell wall.2a As with many secondary metabolites displaying potential medical applications, the papulacandins have stimulated interest in their isolation, structural elucidation, investigation into their biological activity, and chemical synthesis.1b–c,4,5 Specifically, the interest in the biological activity of these compounds has focused on their value as potential therapeutic agents to combat human fungal infections; which are linked to the high mortality of immuno compromised hosts such as AIDS patients.6
This potential has stimulated the search for new papulacandin derivatives and other inhibitors of fungal cell wall synthesis. All of the papulacandins are amphipathic molecules; each contain a hydrophilic domain of a different glycoside nature, and a hydrophobic domain characterized by the presence of modified fatty acids. This hydrophobic domain is thought to be essential for activity.1c Papulacandins A–D are complex molecules, composed of an aromatic moiety resembling terrestrial fungal polyketides: 6-methylsalicylic acid or orsellininc acid, linked via a spirocyclic structure to a lactose moiety with two different aliphatic acyl side-chains, one shorter fatty acid chain at the O-(6') position of the β-galactoside and a second longer side chain at the O-(3) position of the glucose moiety.1b The simplest member of the papulacandin family, papulacandin D, lacks the O-(6'-acyl-β-galactoside) at the O-(4) position of the glucose residue (Figure 1).
Figure 1.

Representative papulacandins from the fermentation of Papularia sphaerosperma
Several new compounds structurally related to the papulacandins have also been isolated.4 These papulacandin congeners vary with respect to the degree of oxidation and saturation of the shorter acyl side-chain; however, some analogs display more drastic modifications to the overall papulacandin structure. Some of these analogs exhibit an inhibitory effect of glucan synthase similar to that of papulacandin B, the most potent papulacandin.4a–d,f–h This behavior indicates that slight modifications to the acyl chains do not significantly retard the antifungal activity of the papulacandins. Yet, the more drastically modified analogs display markedly reduced activity. For example, saricandin, a papulacandin-related analog possessing a cinnamyl ester in place of the C(6') acyl chain, displays significantly less potent antifungal activity.4e Papulacandins devoid of their fatty acid side-chain(s) are found to be ineffective as inhibitors.1b
The antibiotic and antifungal properties, together with the structural complexity of these unique spiro C-arylated glycopyranoside derivatives have stimulated the development of creative solutions for the construction of the tricyclic spiro ketal ring system. Representative examples include a synthesis of a racemic spiro ketal unit via a hetero-Diels-Alder5a reaction and dihydroxylation of 5-aryl-2-vinylfurans followed by Achmatowicz rearrangement.5b–d The majority of work has focused on the addition of functionalized organolithium reagents with cyclic or acyclic derivatives of D-glucolactone.5e–j, v These methods provide rapid access to the spiro ketal core, but suffer from moderate to low yields. Alternatively, nucleophilic 1,2-addition of a lithiated hexenopyranose to a functionalized quinone has been utilized to access the aryl-β-D-C-glycopyranoside.5k Furthermore, a (tributyl)stannylhexenopyranose has been employed in a palladium(0)-catalyzed cross-coupling reaction with sterically hindered aryl bromides. Unfortunately, excess amounts of the tin reagent are required because of dimerization of the organotin donor.5l–r Although these methods provide access to the arylglycopyranoside core of the papulacandins, there has been only one total synthesis of one the members of the papulacandin family, that of papulacandin D by Barrett and co-workers in 1996.5s They accomplished the first total synthesis and assigned the absolute configuration of the C(7") and C(14") stereogenic centers of papulacandin D. Barrett's approach employed combination of an aryllithium reagent and a protected D-gluconolactone to assemble the spiro ketal moiety. The C(14") center in the carboxylic acid side-chain was derived from L-isoleucine, and kinetic resolution was employed to separate the C(7") epimers. The two fragments were then coupled via acylation using a mixed anhydride of the side-chain.
As part of our program on the development of new silicon-based, cross-coupling reactions, we have recently demonstrated the synthetic power of fluoride-free activation for a variety of silanol containing reagents.7 Our plan was to amalgamate this new technology with the previous success in the cross-coupling reaction of 2-pyranylsilanols with aryl iodides.8 We felt that the total synthesis of papulacandin D was well suited to highlight the synthetic potential of silanols in complex molecule synthesis.
The synthetic plan for papulacandin D makes the obvious disconnection at the O-C(3) ester linkage to acid 2 and glycopyranoside 1 (Scheme 1). The major challenges in the synthesis resided in these independent units, namely: (1) the construction of the arylglycoside bond and (2) control of the C(7") and C(14") stereogenic centers. Moreover, potential solutions to both of these problems could be identified in ongoing methodological studies in these laboratories. First, the C-spirocyclic arylglycopyranoside could be reduced to arylhexenopyranose 3, where the C(2) hydroxyl group and C(1) spiro ketal could be installed through an oxidative spiroketalization event. Disconnection of 3 at C(1) reduced the problem to a palladium-catalyzed cross-coupling reaction of glucal silanol 5 and aryl iodide 6. Although this approach makes rational disconnects it provides for challenging reaction sequences. Namely, in the cross-coupling reaction the aromatic iodide is both electron-rich and 2,6-disubstituted. Both of these features lead to problematic cross-coupling reactions. In addition, the cross-coupling reaction conditions need to be tolerant of the array of protecting groups on 5 and 6.
Scheme 1.

Second, disconnection of side-chain 2 at the C(6")-C(7") bond essentially divides the molecule in half. A routine carbonyl addition reaction (aldol or allylation) to the unsaturated aldehyde 4 would set the configuration of the C(7") hydroxyl group, concurrently providing a locus for further elaboration to 2. The dienyl aldehyde 4 could arise from a vinylogous Horner-Wadsworth-Emmons olefination reaction of substituted hexenal 7. Finally, the C(14") stereogenic center could be set through an asymmetric hydrogenation of geraniol.
Therefore, the crucial components to this strategy would be the fluoride-free, silicon-based, cross-coupling reaction of a glucal silanol with a sterically hindered, electron-rich aromatic iodide, as well as the diastereoselective installation of the C(7") hydroxyl group in key intermediate 4. We report herein a full account for the efficient, enantioselective total synthesis of (+)-papulacandin D that highlights our recently developed methods as key strategic steps.5t,u
2. Results
2.1. Construction of the C-Spirocyclic Arylglycoside
2.1.1. Silicon-Based, Cross-Coupling of Glucal Silanols. Orienting Experiments
Previous studies from these laboratories have described the synthesis of C-aryl-2-H-pyrans from 2-pyranylsilanol 8, under fluoride activation.8 These conditions were successfully applied to the cross-coupling reaction of 8 with electron-rich and sterically hindered aromatic iodides such as 9 (vide infra) that are similar to those that would be required for the total synthesis of papulacandin D (Scheme 2, eq. 1). However, extension of these conditions to the cross-coupling reaction of glucal silanol 11 and 9 led to protiodesilylation of the silanol and concomitant deprotection of the C(3)-triisopropylsilyl (TIPS) ether (Scheme 2, eq. 2). These results demonstrated that tetrabutylammonium fluoride (TBAF) activation would not be a suitable activator for the desired cross-coupling reaction. Thus, one of the first requirements in the total synthesis was the development of fluoride-free conditions that could be adapted for the desired glucal silanol.
Scheme 2.

Preliminary results for the cross-coupling reaction of 11 with 4-iodoanisole (13) under activation by tetrabutylammonium hydroxide (TBAOH) were encouraging (Scheme 3, eq. 1). Once again for more complex substrates, such as 9, protiodesilylation was a major problem (Scheme 3, eq. 2). The results suggested that a complete survey of reaction variables (solvent, temperature, activator, etc.) was needed to develop a general and robust cross-coupling reaction.
Scheme 3.

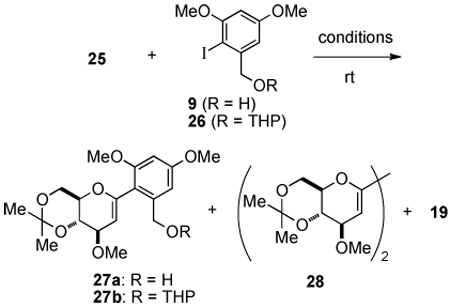
To carry out a systematic evaluation of different reaction parameters, a model study was conducted using aromatic iodide 9 and acetonide-protected glucal silanol 25. Aromatic iodide 9 could be prepared in bulk following a straightforward synthetic sequence from 3,5-dihydroxybenzoic acid.9
The preparation of glucal silanol 26 began with peracetylation of α-D-glucose with Ac2O and NaOAc to provide pentaacetylglucose 16 selectively as its β-isomer in 62% yield after recrystallization (Scheme 4). The β-anomer 16 was treated with thiophenol in the presence of BF3•OEt2 to afford an 82% yield of β-phenylthioglycoside 17.10 The acetyl groups of 17 were removed using LiOH in MeOH to provide the corresponding tetrol, which was immediately protected as the bis-acetonide 18 in 82% yield over the two steps.11 The bis-acetonide 18 was subjected to reductive cleavage using lithium naphthalenide in THF10 providing glucal 12 quantitatively. Finally, the C(3) hydroxy group of glucal acetonide 12 was protected as its methyl ether using NaH and MeI to give an 81% yield of protected glucal 19. Attempts to selectively introduce a silyl moiety at the C(1) position of glucal 19 proved to be difficult as lithiation/silylation occurred preferentially at the C(3) position to afford 20. 12
Scheme 4a.

aConditions: (a) Ac2O, NaOAc, reflux, 1 h, 62%; (b) PhSH, BF3·Et2O, CHCl3, rt, 5 h, 82%; (c) LiOH, MeOH, rt, 1 h; NH4Cl, rt, 20 min; (d) 2-methoxypropene, CSA, DMF, rt, 3 h, 82% (2 steps); (e) lithium naphthalenide, THF, −78°C, 30 min, >99%; (f) NaH, MeI, THF, rt, 2 h, 81%; (g) t-BuLi, THF, −78 °C to 0 °C, 30 min then Me2SiHCl, −78°C to rt, 1 h, 44%.
To circumvent this problem, an alternative route for the preparation of the C(1) silane was considered (Scheme 5). First, protection of the C(3) hydroxyl group of glucal 12 as its TIPS ether 15 was followed by selective metalation at the C(1) position and halogenation with diiodoethane to provide iodo-glucal 21 in 93% yield. The TIPS ether was cleaved with TBAF to afford a 96% yield of 22. The C(3)-hydroxyl group was then reprotected as a methyl ether with NaH and MeI, followed by iodine-lithium exchange and trapping the pyranyllithium reagent with chlorodimethylsilane to provide the desired C(1) silane 24 in 65% yield over the two steps. Silane 24 was subjected to oxidative hydrolysis catalyzed by bis[chloro(p-cymene)ruthenium(II)] in the presence of 2.0 equiv of water to afford the desired silanol 25 in 81% yield.13
Scheme 5a.

aConditions: (a) TIPSCl, imidazole, DMF, 90 °C, 10 h, 89%; (b) t-BuLi, THF, −78 °C to 0 °C; then ICH2CH2I, −78 °C to rt, >93%; (c) TBAF, THF, rt, 30 min, 96%; (d) MeI, NaH, THF, 92%; (e) n-BuLi, THF, −78 °C; then Me2SiHCl, −78 °C to rt, 71%; (f) [RuCl2(p-cymene)]2 (2 mol %) CH3CN-H2O, rt, 1 h, 81%.
The critical palladium-catalyzed cross-coupling of 9 with 25 could now be tested in the presence of Brønsted base activators (Table 1). Unfortunately, the combination of 9 and 25 under standard Brønsted base activation conditions,7d failed to give the desired product (Table 1, entries 1–3). In these cases, protiodesilylation was the major pathway. Tetrahydropyran (THP) protected aromatic iodide 26 was then synthesized14 and subjected to the cross-coupling reaction under NaOt-Bu activation. Gratifyingly, cross-coupling of 25 and 26, promoted by 2.0 equiv of NaOt-Bu in toluene at ambient temperature for 12 h afforded a 69% yield of the desired product 27b along with trace amounts of 19 (Table 1, entry 4).
Table 1.
Cross-Coupling of 25 with 9 and 26.
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | conditionsa | yield, %b |
|||
| 27 | 28 | 19 | Aryl-I | |||
| [allylPdCl]2 | ||||||
| 1 | H | TBAOH | - | - | 38 | 62 |
| MeOH, 5h | ||||||
| APC | ||||||
| 2 | H | KOSiMe3 | - | - | 12 | 44 |
| THF, 5 h | ||||||
| Pd2(dba)3•CHCl3 | ||||||
| 3 | H | NaOt-Bu toluene, 4 h |
- | 40 | - | 17 |
| Pd2(dba)3•CHCl3 | ||||||
| 4 | THP | NaOt-Bu toluene, 12 h |
69 | 20 | trace | 27 |
Pd catalyst (5 mol%) and activator (2 equiv) were used in all cases.
Yield of isolated material.
2.1.2. Protecting Group Strategy
With successful cross-coupling reaction conditions in hand, the focus shifted to designing the global protection / deprotection strategy for the C-spirocyclic arylglycopyranoside hydroxyl groups. The protecting group strategy needs to meet the following criteria (Figure 2): (1) the conditions for the final global deprotection must be mild because of the acid-, base-, and photo-sensitivity of the pendant unsaturated side-chain in i, as well as its obvious incompatibility toward hydrogenolysis and oxidation, (2) the protecting group for the C(3) hydroxyl group of v (P7) has to be complimentary with P2, P3, P4, P5, and P6 to allow for selective acylation at the C(3) position of the arylglycopyranoside, (3) the C(3) protecting group (P7) needs to be bulky to direct the metalation to the C(1) position of glucal viii, (as was the case for OTIPS ether in 15) but not so sterically demanding as to prevent introduction of P2 and, (4) the protecting group for the benzylic alcohol of the aromatic iodide vii has to be cleaved in the presence of P4, P5, P6, P7 prior to oxidative spiroketalization. Taken together, it was decided that fluoride deprotection conditions should be sufficiently mild and selective to allow a late stage global deprotection. This decision mandated that the protecting groups for the C-spirocyclic arylglycopyranoside must be fluoride cleavable (i.e. silyl-derived groups) and be carried through the synthesis.
Figure 2.

Outline for the protecting group strategy.
2.1.3. Optimization of Protecting Groups for the Glucal Silanol
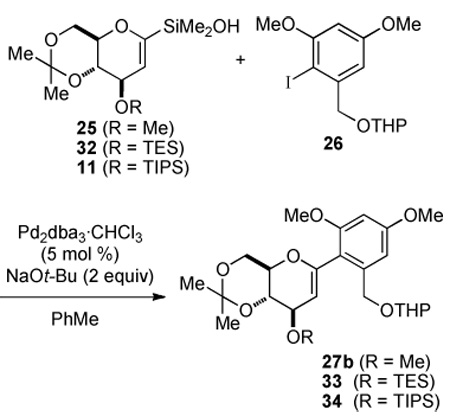
The protecting group optimization strategy focused initially on the C(3) hydroxyl group. The C(3)-O-methyl group was replaced with a bulky silyl protecting group such as TIPS or triethylsilyl (TES). The silanols for the following study were prepared from glucal 12 as shown in Scheme 7. Unlike the methyl ether derivative 19, direct metalation of the protected glucal occurred selectively at the C(1) position thus allowing installation of the silane in excellent yield for either TIPS or TES ether derivative (Scheme 6). Oxidative hydrolysis proceeded smoothly to afford the corresponding silanol 11 or 32 in good yield.
Scheme 7a.

aConditions: (a) K2CO3 (0.01 equiv), MeOH, rt, 1 h; (b) t-Bu2Si(OTf)2, 2,6-lutidine, DMF, 0 °C to rt, 2 h, 89%; (c) TES-Cl, pyridine, CH2Cl2, rt, 4 h, 92%; (d) t-BuLi, Me2SiHCl, THF, −78 °C to rt, 1 h, 89%.
Scheme 6a.

aConditions: (a) TES-Cl, pyridine, CH2Cl2, rt, 10 h, 84% or TIPS-Cl, imidazole, DMF, 90 °C, 10 h, 89%; (b) t-BuLi, THF, −78 °C to 0 °C, 30 min then Me2SiHCl, −78°C to rt, 1 h, 84−95% for 30 and 99% for 31; (c) [RuCl2(p-cymene)]2, CH3CN/H2O, 1 h, 70% for 32, and 2h, 61% for 11.
The steric bulk of the C(3) protecting groups in 11 and 32 had a significant impact on the yields of the cross-coupling reactions at ambient temperature (Table 2, entries 2–3). However, if the reactions were carried out at slightly elevated temperatures, 50 °C for 4 h, the effect of the protecting groups was negligible (Table 2, entries 4–5). Therefore, it was decided that the C(3) hydroxyl group would be protected as the TES-ether, which should provide for a more selective deprotection prior to acylation. In addition, the C(4) and C(6) hydroxyl groups were converted to the di-t-butylsilylene acetal15 because the acidic deprotection conditions needed for the acetonide group would not be compatible with the side chain in a late stage global deprotection (Scheme 7).
Table 2.
Optimization of C(3) Protecting Group.
 | ||||
|---|---|---|---|---|
| entrya | R | temp., °C | time, h | yield, %b |
| 1 | Me | rt | 12 | 27b (69) |
| 2 | TES | rt | 12 | 33 (50) |
| 3 | TIPS | rt | 12 | 34 (20) |
| 4 | Me | 50 | 4 | 27b (71) |
| 5 | TES | 50 | 4 | 33 (67) |
| 6 | TIPS | 50 | 4 | 34 (70)c |
Silanol (1.0 equiv) and aromatic iodide (1.0 equiv) were used in all cases.
Correspond to isolated yields.
Product was impure.
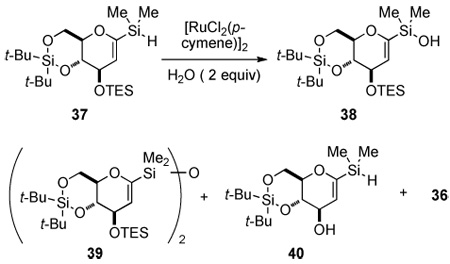
The preparation of silane 37 began by saponification of triacetate 35,16 to form the free hexenopyranose which was immediately protected as its di(tert-butyl)silylene acetal. Protection of the C(3) hydroxyl group with TES-Cl provided fully protected hexenopyranose 36 in good yield (91%, 2 steps). Glucal 36 was metalated at the C(1) position with t-BuLi and the 1-lithiopyran was trapped with chlorodimethylsilane to provide silane 37 in 89% yield.17
Silane 37 was subjected to oxidative hydrolysis catalyzed by bis[chloro(p-cymene)-ruthenium(II)]13 in the presence of 2.0 equiv of water to afford silanol 38 in 73% yield on small scale (Table 3, entry 1). However, silane 37 was insoluble in CH3CN and therefore not amenable to larger scale hydrolyses (Table 3, entries 2–3). Hence, finding scalable oxidative hydrolysis conditions of 37 was important for the efficient synthesis of the spirocyclic C-arylglycopyranoside.
Table 3.
Survey of Oxidative Hydrolysis Conditions for Hydrosilane 37.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| yield, %g |
||||||||
| entrya | catalyst, mol % |
solvent | time, h | 37 | 38 | 39 | 40 | 36 |
| 1b | 4 | CH3CN | 4 | - | 73 | - | - | - |
| 2c | 4 | CH3CN | 1 | - | 70 | - | - | - |
| 3d | 8 | CH3CN | 2 | - | 47 | - | - | - |
| 4e,f | 3 | CH3CN | 3 | 19 | 52 | 3 | 23 | - |
| 5 | 8 | n-BuCN | 2 | - | 87 | 2 | - | - |
| 6 | 3 | n-BuCN | 4 | - | 84 | 4 | - | - |
| 7 | 3 | THF | 4 | - | 54 | 5 | - | 27 |
| 8 | 4 | PhCN | 4 | - | 90 | 4 | - | - |
| C6H6/ | ||||||||
| 9 | 3 | CH3CN | 1 | - | 86 | 7 | - | - |
| (1:1) | ||||||||
| C6H6/ | ||||||||
| 10h | 3 | CH3CN | 1 | - | 84 | 8 | - | - |
| (1:1) | ||||||||
All the reactions were carried out on 100 mg scale, unless otherwise stated.
110 mg scale
564 mg scale.
950 mg scale.
[Ir(cod)Cl]2, H2O (2.0 equiv) were used.
TES ether cleavage was observed.
Yield of isolated product.
The reaction was run on 1.6 mmol scale and the yield of 38 is of analytically pure material.
To this end, several conditions and catalysts were surveyed (Table 3). Hydrolysis of 37 with 3.0 mol % of [IrCl(C8H12)]2,18 gave 38 in moderate yield but cleavage of the TES ether was a major side process (Table 3, entry 4).19 In a less polar solvent, n-BuCN, the reaction proceeded cleanly to the desired silanol in 87% and 84% yield (Table 3, entries 5–6). However, in THF, a significant amount of protiodesilylated product was observed (Table 3, entry 7).19 Switching the solvent to benzonitrile gave an excellent yield of 38. Unfortunately, the higher boiling point of PhCN made purification tedious. Finally, after extensive optimization of mixed solvent systems it was found that a 1:1 mixture of CH3CN/benzene afforded 38 in 86% yield (Table 3, entry 9). To our delight, these conditions were amenable to larger scale preparations (>2 g) of 38 in excellent yield (Table 3, entry 10).
2.1.4. Optimization of Protecting Groups for the Aromatic Iodide
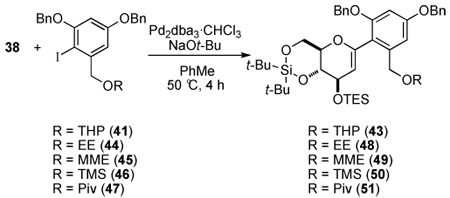
In the next stage of the synthesis, suitable protecting groups for the resorcinol moiety were surveyed. The cross-coupling reaction of glucal silanol 38 and iodide 26 proceeded very smoothly to afford the aryl glucal 42 in 77% yield (Scheme 8). Employing benzyl protecting groups gave the desired product 43 in a 72% yield; however, the benzyl ethers would obviously need to be replaced prior to global silyl deprotection. When the phenolic protecting groups were changed from methyl ethers to tert-butyldimethylsilyl (TBS) or triisopropylsilyl (TIPS) ethers, the cross-coupling reaction did not provide any of the desired product.
Scheme 8.

Finally, various options for the benzylic alcohol protecting group were surveyed (Table 4).14 Interestingly, protection of the benzylic alcohol had a significant impact on the yield of the cross-coupling reaction. The aromatic iodides bearing a 1-ethoxyethyl (EE), 1-methoxy-1-methyl-ethyl (MME) or trimethylsilyl (TMS) ether all afforded the desired product, albeit in low yields (Table 4, entries 2–5). Use of the pivaloyl protected iodide 47 in the cross-coupling reaction provided the aryl glucal 51 in good yield (Table 4, entry 6). The pivaloyl ester could be cleaved quantitatively with DIBAL-H (Scheme 9, eq. 1), whereas the acidic conditions for hydrolysis of the THP ether would lead to spiroketalization and TES-ether cleavage (Scheme 9, eq. 2).20 Clearly, the pivaloyl protecting group is the most suitable for the synthesis.
Table 4.
Survey of Protecting Groups for the Benzylic Alcohol of the Aromatic Iodide.
 | ||
|---|---|---|
| entry | R | product (yield, %) |
| 1 | THP | 43 (72) |
| 2 | EE | 48 (28) |
| 3 | MME | 49 (26) |
| 4b | TMS | 50 (36) |
| 5c | TMS | 50 (52) |
| 6 | Piv | 51 (75) |
Yield corresponds to isolated product.
The reaction was heated to 80 °C for 2 h.
The reaction was heated to 110 °C for 1 h.
Scheme 9.

2.1.5. Implementation of the Key Cross-Coupling Sequence
The next objective in the synthesis was to secure large quantities of the 1-arylhexenopyranose 52 which required the cross-coupling reaction to be scaleable (i.e. > 1.0 mmol scale). Gratifyingly, the key cross-coupling reaction using stoichiometric amounts of aryl iodide 47 and silanol 38 in the presence of 5 mol % of Pd2(dba)3•CHCl3 and 2.0 equiv of NaOt-Bu at 50 °C for 5 h proceeded smoothly and reproducibly to afford 51 in 82% yield (Scheme 10).21 In addition, the pivaloyl ester was selectively cleaved by DIBAL-H reduction to give desired benzylic alcohol 52 in 98% yield (Scheme 10).
Scheme 10a.

aConditions: (a) Pd2(dba)3•CHCl3, NaOt-Bu (2.0 equiv), Toluene, 50 °C, 5 h, 82%; (b) DIBAL-H, CH2Cl2, −78 °C to rt, 1 h, 98%.
2.1.6. Oxidative Spiroketalization and C(2)-Hydroxyl Group Protection
To assemble the carbon framework of the C-spirocyclic arylglycopyranoside of papulacandin D, the next challenge was to effect both stereocontrolled installation of the C(2)-hydroxyl moiety and oxidative spiroketalization. The conditions for oxidative spiroketalization must be basic to prevent the undesired Brønsted acid catalyzed spiroketalization (Scheme 10, eq 2). Selective formation of α-glycosyl anhydrides from 1,2-anhydrosugars using dimethyldioxirane (DMDO) is well known.22 For the current study, m-chloroperoxybenzoic acid (m-CPBA) under nonequilibrating conditions was found to be optimal.5p Hence, treatment of a mixture of 52 and m-CPBA at 0 °C in the presence of NaHCO3 in CH2Cl2, smoothly provided the chromatographically separable spiro ketals 54-α and 54-β in a 5:1 ratio (Scheme 11).
Scheme 11a.

aConditions: (a) m-CPBA, NaHCO3, CH2Cl2, 0 °C, 2 h (dr 5:1, 77% α, 15% β). (b) 0.1% HCl in CHCl3, 20 °C, 12 h, 96% recovery
The configuration of the C(2)-hydroxyl group was confirmed by inspection of the 1H-NMR spectra of each anomer as well as more extensive NMR analyses. The 1H-NMR spectrum of 54-α in dry, neutralized CDCl3 displayed diagnostic signals at δ 4.32 ppm and δ 3.84 ppm corresponding to the HC(2) and HC(3) proton resonances, respectively. The structure of α-anomer was confirmed by analysis of the coupling constants of the vicinal protons in the glucose ring in the spiro ketal 54-α. The key constants (J2,3 = 10.0 Hz, J3,4 = 10.0 Hz, J4,5 = 10.2 Hz) were consistent with a chair conformation, where the large coupling constants suggest that mutual trans-diaxial relationships exits between HC(2)/HC(3) and HC(3)/HC(4). The distorted chair conformation of the β-anomer was also confirmed by vicinal coupling constants (J2,3 = 8.2 Hz, J3,4 = 8.2 Hz, J4,5 = 9.4 Hz), which are consistent with the observations of Dubois and Beau for a similar system.5r
Finally, the β-anomer could be isomerized to the α-anomer with a 96% recovery with a solution of chloroform containing with 0.1% dry HCl. The isomerization experiment also aided in the assignment and determination of the absolute configuration determination of the 54-β C(2) hydroxyl group (Scheme 11).
To complete the synthesis of the C-spirocyclic arylglycopyranoside, the following protecting group manipulations were required: (1) the aromatic benzyl ethers must be switched to silicon-based protecting groups for the late stage global deprotection, (2) the C(2)-hydroxyl group must be replaced with a protecting group complimentary to the C(3) TES ether, thus allowing for selective deprotection and acylation of the C(3) hydroxyl group.
Hydrogenolytic debenzylation of 54-α at 1 atm of H2 with Pd/C in the presence of NaHCO3 proceeded smoothly to afford triol 55 quantitatively (Scheme 12). Initially, bulky silicon protecting groups such as tert-butyldiphenylsilyl (TBDPS) or TIPS ethers for the phenolic hydroxyl groups were planned. However, treatment of triol 55 with either TBDPS chloride or TIPS chloride in the presence of lutidine did not afford any of the desired silyl ethers. Therefore, silylation with a more reactive silylating agent, TIPSOTf, was attempted. In this case, only the mono-protected alcohol 56 was isolated and characterized by 1H-NMR analyses.
Scheme 12a.

aConditions: (a) Pd/C, H2, NaHCO3, THF, rt, 5 h, >99%; (b) TIPS-OTf, 2,6-lutidine, CH2Cl2, 1 h, 69%.
A less sterically-demanding protecting group such as 2-(trimethylsilyl)-ethoxymethyl (SEM) ether was considered for the protection of the all three hydroxyl groups of triol 55 (Scheme 13).23 Unfortunately only two SEM groups were introduced to afford 57 as the major product, presumably due to a slow alkylation of the C(2)-hydroxyl group. To access the C(2) protected hydroxyl group, additives such as tetrabutylammonium iodide (TBAI) and AgOTf24 were tested but with only moderate success, see Supplementary Data for the conditions surveyed. Resubjecting 57 to SEMCl in DMF, and in the presence of TBAI, again did not afford any of the tris-SEM ether 58. Ultimately, 58 could be obtained in good yield using a combination of 3 equiv of TBAI and 9 equiv of AgOTf in DMF.
Scheme 13.

The final step in the synthesis of the spirocyclic arylglycopyranoside, the selective deprotection of the C(3)-TES ether, was accomplished using PPTS in ethanol albeit in low yield (52%) (Scheme 13).
2.2. Synthesis of Fatty Acid Side-Chain 2 and Acylation
To set the C(7") stereogenic center of acid 2, one of the most convergent routes would employ an asymmetric aldol addition reaction of a doubly vinylogous enolate 60 to aldehyde 4 (Scheme 14).
Scheme 14.

Numerous attempts were made to set the C(7") stereogenic center through Lewis-base catalyzed aldol reactions; unfortunately, a suitable method that provided both high yield and enantiomeric ratio were not found. For further discussion and examples of the Lewis-base catalyzed aldol reactions attempted, see Supplementary Data. Given these undesirable results alternative asymmetric carbonyl addition reactions were explored.
2.2.1. Model Study of an Asymmetric Allylation Reaction
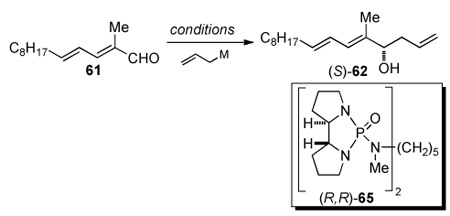
An alternative strategy for the construction of the C(7") stereogenic center would employ an asymmetric allylation reaction.25,26 The allyl group was of interest because it provides a handle for further elaboration of the side-chain of papulacandin D through a sequential cross-metathesis and two-carbon homologation. To vouchsafe this plan, the cross-metathesis reaction of model homoallylic alcohol 62 was tested, before starting the investigation into asymmetric allylation reaction.
The cross metathesis of racemic homoallylic alcohol 62 (prepared by the addition of allylmagnesium bromide into aldehyde 61) was tested with both methyl acrylate and acrolein using 5 mol % of Grubbs' second-generation catalyst (Scheme 15).27 The reactions afforded the desired α,β-unsaturated ester 63 and aldehyde 64 in 87% and 81% yield respectively (Scheme 15, eqs. 2 and 3).
Scheme 15a.

aConditions: (a) allylMgBr, Et2O, 0 °C, 90%. (b) Grubb's 2nd gen. cat (5.0 mol %), methyl acrylate (13 equiv), rt, 1 h, 87%.; (c) Grubb's 2nd gen. cat (5.0 mol %), acrolein (13 equiv), rt, 1 h, 81%.
Next, a variety of asymmetric allylation reactions were evaluated. The use of allyltributyltin in the presence of 0.2 equiv BINOL/Ti(Oi-Pr)4 and 4 Å molecular sieves as described by Keck28 provided excellent enantioselectivity (99.4 : 0.6), albeit in only 30% yield (Table 5, entry 1). The chiral borane reagent, allyl(Ipc)2B, developed by Brown,29a afforded an improved yield (Table 5, entry 2), and the enantiomeric ratio reflected the purity of the reagent. The enantiomeric purity of the borane reagent can be enriched by synthesizing methoxy(Ipc)2B rather than using chloro(Ipc)2B.29 Therefore, (−)-methoxy(Ipc)2B was prepared from (+)-α-pinene. The allylation using (−)-methoxy(Ipc)2B at −78 °C afforded homoallylic alcohol (S)-62 in 73% yield with a 96:4 enantiomeric ratio (Table 5, entry 3). Lower temperature (−100 °C) did not improve the enantioselectivity (Table 5, entry 4). Although successful, the Brown-type allylation was not ideal because it required the synthesis and stoichiometric amounts of a chiral reagent.
Table 5.
Survey of Asymmetric Allylation Reactions.
 | ||||
|---|---|---|---|---|
| entry | conditions | yield, %a |
erb | |
| 1 | 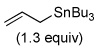 |
(S)-BINOL (24 mol %) Ti(Oi-Pr)4 (20 mol %) −78 to −20 °C |
30 | 99:1 |
| 2 | (+)-Ipc2B-Cl (1 equiv) −78 °C |
70 | 90:10 | |
| 3 | (+)- Ipc2B-OMe (1 equiv) −78 °C |
73 | 96:4 | |
| 4 | (+)- Ipc2B-OMe (1 equiv) −100 °C |
71 | 96:4 | |
| 5 | (R,R)-65 (10 mol %) i-Pr2NEt / CH2Cl2 −78 °C |
76 | 95:5 | |
Yield corresponds to isolated product.
The enantiomeric ratio was determined by CSP-SFC.
Fortunately, an enantioselective allylation method developed in these laboratories, namely the chiral bisphosphoramide-catalyzed allylation reaction of allyltrichlorosilanes and aldehydes, was successful.30 Thus, the addition of allyltrichlorosilane (2.0 equiv) to 61 using 0.05 equiv of chiral bisphosphoramide (R,R)-65 at −78 °C for 8 h afforded the desired homoallylic alcohol (S)-62 in 76% yield with an enantiomeric ratio of 95:5 (Table 5, entry 5). Because the Lewis-base catalyzed reaction provided excellent enantioselectivity and yield, in the model system it was applied in the synthesis of the side-chain of papulacandin D.
2.2.2. Preparation of Side-Chain Acid 2
Synthesis of the unsaturated acid 2 began by asymmetric hydrogenation of geraniol31 with Ru(OAc)2[(S)-BINAP]32 to provide (S)-citronellol in excellent yield and enantiomeric purity (99%, 97:3 er) (Scheme 16). Tosylation of the alcohol followed by deoxygenation with LiHBEt3 afforded hydrocarbon 67 in 88% yield for the two steps. Alkene 67 was then subjected to ozonolysis and vinylogous Horner-Wadsworth-Emmons olefination to afford unsaturated ester 68 as an inseparable mixture of isomers ((Δ8",9" E/Z, 91:9)). Subsequently, DIBAL-H reduction of the ester followed by chromatographic separation of the geometrical isomers of the allylic alcohol and oxidation afforded aldehyde 4 in 84% yield (four steps). Now, the enantioselective allylation of the substrate could be tested. Gratifyingly, allylation of 4 using chiral bisphosphoramide (R,R)-65 smoothly provided 70 in good yield and excellent diastereomeric selectivity (88%, dr 96:4). The expected C(7") S configuration30 was confirmed by analysis of the corresponding Mosher esters.33 Olefin metathesis with acrolein using Grubb's 2nd generation catalyst, and protection of the C(7") hydroxyl group with TESCl gave 72 in 91% yield for the two steps. The synthesis of 2 was completed by Wittig olefination and saponification with potassium trimethylsilanoate34 in excellent 90% yield (2 steps).
Scheme 16a.

aConditions: (a) Ru(OAc)2[(S)-BINAP] (0.7 mol %), H2 (1500 psi), 95% MeOH, rt, 20 h, 99% (97:3 er); (b) Ts-Cl, pyridine, rt, 10 h, 89%; (c) LiHBEt3, THF, NaOH-H2O2, rt, 1 h, 86%; (d) O3, NaHCO3, CH2Cl2-MeOH, −78 °C to rt, DMS, 2.5 h; (e) (EtO)2POCH2C=C(CH3)CO2Et, LiOH, MS 4Å, THF, reflux, 2 h, 78% (2 steps) (E,E : E,Z 91:9); (f) DIBAL-H, THF, 0 °C, 0.5 h, 85% (pure E,E); (g) MnO2, CHCl3, reflux, 4 h, 89%; (h) (R,R)-65 (10 mol%), allyltrichlorosilane, CH2Cl2, i-Pr2EtN, −78 °C, 8 h, 88% (96:4 dr); (i) Grubb’s 2nd gen. catalysts (5.0 mol %), acrolein (13 equiv), CH2Cl2, 90%; (j) TES-Cl, 2,6-lutidine, CH2Cl2, rt, 4 h, 92%; Ph3P=CCO2Me, ClCH2CH2Cl, reflux, 18 h, 90% (E:Z 90:10); (k) TMSOK, THF, rt, 4 h, 94%.
2.3. Model Acylation with Tris-SEM Spirocyclic Arylglycoside and Sorbic Acid
The next challenge in the synthesis was the union of fragments 59 and 2, through an acylation reaction. Because the sensitivity of the side chain acid 2, the Yamaguchi mixed anhydride protocol was first tested.35 Barrett5s reported the use of such a mixed anhydride of 2 in combination with O-4,O-6-di-tert-butylsilylene acetal and TIPS protecting groups on the spirocyclic arylglycopyranoside to improve the site selectivity of the acylation. In the present study the C(2) hydroxyl group is protected and therefore, the acylation should be selective for the C(3) hydroxyl group. However, the secondary hydroxyl group is in significantly more hindered environment compared to Barrett's substrate. Hence, the acylation was tested with sorbic acid before committing precious acid 2.
Sorbic acid was activated via a mixed anhydride using 2,4,6-trichlorobenzoyl chloride in the presence of DMAP and DMF and then a solution of 59 and DMAP in DMF was added to the mixed anhydride at ambient temperature. The acylated product 73 was isolated in 31% yield, where the remaining mass corresponded to unreacted 59 (Scheme 17). Although the yield of the reaction was poor, it demonstrated that acylation can proceed at the C(3) position. Applying the above conditions to the crucial coupling of fragments 2 and 59; however, in THF, for 4 h afforded the desired ester 74 in good yield (88%) (Scheme 18).
Scheme 17.

Scheme 18.

2.4. Global Deprotection and Completion of the Synthesis of Papulacandin D
2.4.1. Model Study of SEM Deprotection
The last step in the synthesis is the global deprotection of all the silicon-protecting groups in 74. Standard fluoride sources such as TASF, TBAF, HF•Et3N, etc. were expected cleave the protecting groups in one step, thus revealing the natural product. To find optimal conditions for the cleavage of the SEM protecting groups, it was decided to work with a simple model compound, 5-(methoxymethyl)resorcinol, prepared from methyl 3,5-dihydroxybenzoate.
A variety of conditions were surveyed that are known for silicon- and SEM ether deprotection,36 see (Supplementary Data Table S4) for a full survey of deprotection conditions. Only a combination of MgBr2 and n-BuSH provided the fully deprotected resorcinol derivative. These SEM-deprotection conditions were then applied to a more appropriate model compound, acyl spirocyclic arylglycopyranoside 73. Because of the stability of SEM-ethers to fluoride treatment (vide infra), a two-step deprotection sequence was employed. First, to cleave the silyl-ether protecting groups, 73 was treated with 10 equiv of a 1 M solution of TBAF in THF at room temperature for 1.5 h (Scheme 19). Unfortunately, the product (isolated in low yield) was a mixture of C(3) and C(4) acylated isomers 75 and 76, respectively.
Scheme 19a.

aConditions: (a) TBAF (10 equiv), THF, rt, 1.5 h, <40%; b. MgBr2 (25 equiv), n-BuSH (23 equiv), Et2O, rt, 1 h, trace product. (c) MgBr2 (60 equiv), n-BuSH (60 equiv), MeNO2, Et2O, 0°C to rt, 3 h, >99%.
Nevertheless, sufficient material was secured to investigate the SEM-ether cleavage. Therefore, the mixture of 75 and 76 was subjected to the action of 25 equiv of MgBr2 and 23 equiv n-BuSH in Et2O at room temperature for 1 h. The reaction became biphasic and only traces of the desired SEM-deprotected products 77 were isolated. Next, the inverse of the above sequence was employed, 73 was first treated with 60 equiv MgBr2 and 60 equiv n-BuSH in the presence of MeNO2 as a co-solvent to provide the desired product 78 quantitatively. Treatment of 78 with a HF•Et3N solution should cleave the silyl-ether protecting groups to afford the fully deprotected product.
These results encouraged attempts at the global-deprotection of 74 (Scheme 20). The SEM-protected papulacandin D precursor 74 was treated with 60 equiv MgBr2 and 60 equiv n-BuSH in Et2O / MeNO2 (100 : 1 mixture) from 0 °C to room temperature. After 3 h consumption of 74 was observed by TLC analysis; however, upon isolation, a mixture of butanethiol-incorporated products were observed tentatively assigned as 79.
Scheme 20.

The failure of the deprotection strategy at the last step in the synthesis was very disappointing. Clearly, the removal of the SEM protecting group required more forcing conditions than anticipated. Thus, the search for a new protecting group that could be removed without damage to the sensitive C(7") hydroxyl group was needed. Therefore, a number of different protecting groups were examined to increase the stability of the C(7") hydroxyl group under the SEM deprotection conditions (Scheme 21). Regardless of the C(7") hydroxyl protecting group, elimination was always the major pathway. Thus the identification of milder deprotection conditions became paramount. The stability of the C(7") TES-ether 80 was tested using a solution of 5 equiv TASF in THF. Surprising, TASF induced the elimination of the silyl ether, even though Barrett and co-workers used similar conditions for their global silyl deprotection.5s In previous studies, the TES-ether was stable in the presence of a buffered hydrofluoric acid solution in CH3CN (HF•NEt3 (46:54)). Unfortunately, it was observed that neither HF•NEt3 (70:30) or HF•pyridine (60:40) were strong enough to induce SEM ether cleavage.
Scheme 21a.

aConditions: (a) HF·NEt3 (46:54), CH3CN, rt, 1 h, >99%; (b) TIPSOTf, lutidine, CH2Cl2, −78 to 0 °C, 2 h, >99%; (c) MgBr2, n-BuSH, MeNO2, Et2O, 0 °C to rt, 1–3 h.
2.4.2. New Protecting Group Strategy
The failure to cleave the SEM groups shifted the focus to revising the initial protecting group strategy that would accommodate the sensitivity of the pendant side chain. The study focused on silicon-based protecting groups that could be easily cleaved with HF•NEt3 or HF•pyridine, because of the observed stability of the side chain to these conditions. The survey began with trialkylsiloxymethyl ethers (TBSCH2OR and TIPSCH2OR) using methyl 3,5-dihydroxybenzoate as a model compound.37 However, it was observed that the protection afforded a mixture of trialkylsilyl and trialksilyloxymethyl ethers. Because the introduction of a trialkylsiloxymethyl protecting group was unsuccessful, 2-(trimethylethylsilylethoxycarbonyl) (TEOC) was investigated.38 It was anticipated that the TEOC protecting group could be easily cleaved under HF•Et3N conditions, and was small enough to protect the sterically encumbered C(2) hydroxyl group. The 5-(methoxymethyl)resorcinol was then protected as the bis TEOC carbonate quantitatively using 3 equiv TEOC-Cl, in the presence of 6 equiv i-Pr2NEt. To our delight, the TEOC groups introduced could be quantitatively removed using a mixture of HF•NEt3 (40:60) in CH3CN at 40 °C for 9 h.39
The TEOC protection of all three hydroxyl groups in triol 55 was attempted first. However, only the aromatic hydroxyl groups could be protected (Scheme 22, eq. 1). Surprisingly, when the aromatic hydroxyl groups were protected as TEOC or the benzyl ethers normal TEOC protection failed at the C(2)-hydroxyl group (Scheme 22, eq. 2). The acylation of 85 or 86 was slow because of the steric congestion at the C(2) hydroxyl group.
Scheme 22.

The C(2) hydroxyl group of 54-α could be acylated, with acetic anhydride and pyridine albeit very slowly. Even, after 12 h the reaction did not go to completion and the acetate was isolated along with some of the starting material. The failure to attach a TEOC group at C(2) most likely arises from insufficient reactivity of TEOC-Cl. To circumvent this problem, the TEOC protection group was affixed in a stepwise manner to allow use of a more reactive acylating agent, namely triphosgene. The resulting chloroformate could then be combined with 2-(trimethylsilyl)ethanol to complete the TEOC protection.
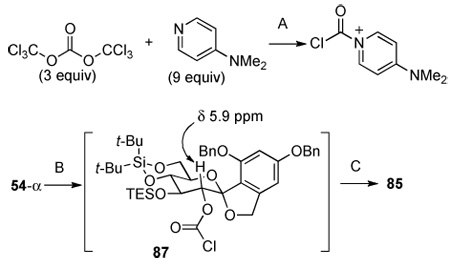
This idea was put into practice by generating a chlorocarbonylpyridinium cation with triphosgene and DMAP in a mixture of i-Pr2NEt and CH2Cl2. To this solution was added alcohol 54-α. However, the reaction did not afford any of the desired product (Table 6, entries 1 and 2). The experiment was repeated in CDCl3 and was monitored by 1H-NMR spectroscopy. In CDCl3, the reaction mixture became biphasic when i-Pr2NEt was added to the solution of DMAP and triphosgene (Table 6, entry 3). Therefore, i-Pr2NEt was added later along with 54-α. When the chlorocarbonylpyridinium cation was observed, this solution was transferred to a solution of 54-α in CDCl3/i-Pr2NEt. To observe the formation of chloroformate 87 the reaction mixture was diluted with CDCl3 (conc. 0.0014 M). Once complete conversion of 54-α to 87 was confirmed, the solution was treated with 2-(trimethylsilyl)ethanol. At this the low concentration, the rate of reaction was very slow and required ca. 70 h for complete conversion (Table 6, entry 4). However, at 0.043 M, complete conversion was observed within 12 h to afford 85 in 92% yield (Table 6, entry 5). It is of note that all the attempts listed in Table 6 were accomplished using a total of 2.4 mg of 54-α demonstrating the power of 1H-NMR for reaction monitoring.
Table 6.
TEOC Protection of 54-α.
 | ||
|---|---|---|
| entry | solvent | Result |
| 1a | CH2Cl2 | 54-α (>99%) |
| 2b | CH2Cl2 | 54-α (>99%) |
| 3c | CDCl3 | biphasic mixture |
| 24 h; 87:85 = 52:48 | ||
| 4d | CDCl3 | 48 h; 87:85 = 16:84 |
| 72 h; 87:85 = 0:100 | ||
| 5e | CDCl3 | 85 (92%) |
A: i-Pr2NEt (1055 equiv), −78 °C, 20 min; B: −78 °C, 1 h, (0.002 M); C: TMS-ethanol (18 equiv), −78 °C to rt, 4 h (0.0012 M).
A: i-Pr2NEt (1055 equiv), −78 °C, 20 min; B: −78 °C to rt, 1 h, (0.002 M); C: TMS-ethanol (72 equiv), −78 °C to rt, 9 h (0.0012 M).
A: i-Pr2NEt (28 equiv), −78 °C to rt, 30 min, (DMAP 0.6 M); B: −78 °C to rt, 1 h, (0.002 M); C: TMS-ethanol (72 equiv), −78 °C to rt, 9 h (0.0012 M).
A: −78 °C to rt, 1 h, (DMAP, 1.2 M); B; i-Pr2NEt (28 eq), −78 °C to rt, 1 h, (0.043 M); C: TMS-ethanol (26 equiv), rt, (0.0041 M).
A: −78 °C to rt, 30 min, (DMAP 1.2 M); B: i-Pr2NEt (28 equiv), −78 °C to rt, 1 h, (0.043 M); C: TMS-ethanol (26 equiv), rt, 12 h (0.043 M).
Gratifyingly, only with minor modification to the reagent stoichiometry, treatment of 54-α with a preformed acylpyridinium species followed by the addition of 2- (trimethylsilyl)ethanol gave the 2-TEOC-protected spiro ketal in 92% yield, on preparative scale (Scheme 23). Next, debenzylation of 85 with hydrogen and Pd/C proceeded quantitatively. Subsequent protection of the resorcinol portion of 88 was achieved under standard TEOC protection conditions to produce the fully silylated spiro ketal 89 in 96% yield overall. Finally, selective removal of the O-C(3) TES ether with PPTS/ETOH afforded 90 in 93% yield.
Scheme 23a.

aConditions: (a) triphosgene, DMAP, CHCl3, −56 °C to rt; i-Pr2EtN, 54-α, CHCl3, −56 °C to rt, 2-(trimethylsilyl)ethanol, rt, 12 h, 92%; (b) Pd/C, NaHCO3, H2, THF, rt, 5 h, >99%; (c) TEOC-Cl, i-Pr2EtN, CH2Cl2, rt, 9 h, 96%; (d) PPTS, EtOH, rt, 4 h, 93%.
2.4.3. Studies on the Removal of the TEOC Groups
To evaluate if the sterically congested C(2)-TEOC protecting group could be removed using HF•NEt3 solution, TEOC protected menthol was used as a test substrate. A variety of reaction variables were systematically surveyed (time, concentration of fluoride, HF•NEt3 ratio, temperature and solvent) and some representative examples are presented in Table S5 in the Supplementary Data. These studies concluded that the rate of TEOC cleavage in DMSO was significantly faster than the other solvents (Table S5, entries 7 and 8, SI). Applying these conditions to the TEOC cleavage of a sorbyl ester of 90 demonstrated that at 40 °C in DMSO the cleavage was not complete, but at 60 °C after 12 h complete deprotection to 91 was observed. Finally, to establish that acyl migration did not occur (a problem observed during the initial SEM deprotection studies of 73), structure 91 was confirmed (and structures A and B were eliminated) through a series of NMR experiments (Figure 3).
Figure 3.

Assignment of esterification site in 91.
Finally, the side chain must not degrade under the deprotection reaction conditions. Before subjecting the very precious, fully protected papulacandin D to the HF•NEt3 deprotection conditions, the stability of the side chain was once again tested. In a Teflon flask a biphasic solution of DMSO and HF•NEt3 (40:60) was added to methyl ester 80 (Scheme 21) and the mixture was heated to 60 °C. Deprotection of the C(7")-silyl ether was rapid under these conditions. However, heating for 15 h is was required to remove all the TEOC groups of the 1-arylglycopyranoside. Therefore, the stability of the unprotected side chain, upon extended heating, needed to be tested. Fortunately, it was determined that the side chain was stable under prolonged heating and alcohol 81 was isolated in 93% yield.
2.5. Completion of the Synthesis of (+)-Papulacandin D
With routes for the side-chain 2 and the protected spirocyclic arylglycopyranoside 81 secured, the penultimate challenge was the coupling of the two fragments via the mixed anhydride of 2 from 2,4,6-trichlorobenzoyl chloride. The two fragments were united (1.3 equiv 2, toluene, rt, 3 h) in 87% yield, with only slight modification from the original acylation protocol (Scheme 24).
Scheme 24a.

aConditions: (a) 2,4,6-Cl3C6H2COCl, Et3N, rt, 1 h, 90, toluene, rt, 3 h, 87%; (b) HF• Et3N (40:60 ratio), DMSO, 60 °C, 15 h, 89%.
Finally, the most daunting step in the synthesis could be attempted, namely deprotection of all the silicon protecting groups in protected papulacandin D, 92. Gratifyingly, the extensive optimization of protecting groups and deprotection conditions proved worthwhile. Global deprotection of 92 proceeded smoothly on >80 mg scale using a buffered HF•Et3N (40:60 ratio) in DMSO at 60 °C for 15 h to afford an 89% yield of synthetic (+)-papulacandin D.
The physical and spectroscopic data for the synthetic sample were nearly identical in all respects [m.p. 126−128 °C (lit. 127~130 °C), 1H-NMR, 13C-NMR, IR, UV-Vis, [α]24D +8.78 (c = 0.21, MeOH)]; lit. 7±1 (MeOH) to those reported for natural (+)-papulacandin D and that of Barrett's synthetic material.
3. Discussion
The synthesis of (+)-papulacandin D was undertaken to extend the applicability of fluoride-free, silicon-based, cross-coupling for a challenging synthetic target. This key step proceeded after minor optimization, in gratifyingly high yield. Once it was established that a glucal dimethylsilanol could be activated using a simple Brønsted base (NaOt-Bu), the optimized conditions for this key step could be directly applied to the cross-coupling reaction of a variety of protected glucal and aromatic iodides with excellent results. Therefore, discussion of the key strategic maneuver shall receive only minor comment; instead the influence of the protecting groups on the cross-coupling reaction will be discussed more thoroughly.
3.1. Synthesis of Fragment 52
To overcome the obvious limitations associated with fluoride activation of a glucal 2-dimethylsilanol, we sought to develop a cross-coupling reaction of a glucal 2-dimethylsilanol with a sterically hindered and electron rich and sterically hindered aromatic iodide. The major questions that needed to be addressed were: (1) how is a silicon donor, such as a silanol, activated for productive cross-coupling reaction and (2) would these conditions be compatible with the protecting groups on the silanol and aromatic iodide.
In the initial studies on the cross-coupling of 25 with 9, both TBAOH and KOSiMe3 were unsuitable activators as they afforded the protiodesilylated glycal 19 exclusively. This outcome is not surprising, because these types of activators have been problematic in the cross-coupling reaction of substituted enol silanols and heteroarylsilanols.7c,8 It is believed that the co-existence of enol or heteroarylsilanol and silanolate causes protiodesilylation. Therefore, a strong, soluble Brønsted base, such as NaOt-Bu, was needed to bring about rapid formation of the sodium silanolate in situ and thus suppress protiodesilylation. This idea in combination with protection of 9 as the corresponding THP acetal, finally provided for successful cross-coupling.
Protection of the benzylic alcohol may have two benefits: (1) removal a proton source for potential protiodesilylation and (2) to suppress coordination of the free benzylic alcohol to the Pd(II) intermediate after oxidative addition. It is believed that the rate of displacement of this alkoxide palladium intermediate by silanolate may be slow, and thus significantly decrease the rate of cross-coupling relative to undesirable side processes. Subsequently, it was found that the protecting group for the benzylic alcohol had a dramatic impact on the rate and yield of the cross-coupling reaction (Table 4). This behavior could be rationalized in terms of the coordinating ability of the protecting group (41, 44, 45 and 47, Table 4) to a Pd(II) intermediate. The pivaloyl protecting group was ultimately chosen for the following reasons: (1) the reduced ability of the carbonyl group to coordinate to the Pd(II) intermediate, compared to the free alkoxide, MME, and EE protecting groups and (2) the near quantitative deprotection with DIBAL-H that also did not induce nonoxidative spirocyclization of the corresponding arylglucal.
3.2. Oxidative Spiroketalization of 52
The oxidative spiroketalization of 1-arylglucal 52, most likely proceeded via: (1) α-selective epoxidation of the enolic double bond, generating a glycosyl anhydride 85 or Brigl's intermediate,40 (2) opening of the internal epoxide to generate an oxocarbenium ion 86, and (3) trapping of the oxocarbenium ion with the pendant hydroxyl group of the C(1) aromatic moiety to provide the desired C-spirocyclic arylglycopyranoside (Scheme 25). Crucial to the success of this transformation is the selective epoxidation of the glucal, by m-CPBA from the α-face of the enol ether. This process sets the configuration of the C(2)-hydroxyl group of the spirocyclic arylglycopyranoside 54-α. The epoxidation of 52 relies upon substrate-controlled diastereoselectivity in the initial oxidation event. The use of sterically demanding protecting groups on the C(3)-, C(4)-, and C(6)-hydroxyl groups, shielded the β-face, allowing epoxidation to proceed exclusively from the α-face. The diastereoselectivity of the oxidation was supported by determination of the absolute configuration of the C(2)-hydroxyl group in both α- and β-anomers.
Scheme 25.

The 5:1 mixture of α- and β-anomers represented the kinetic ratio, as no isomerization was observed when the β-anomer was exposed to m-CPBA and NaHCO3 in CH2Cl2.41 However, the use of CHCl3 containing 0.1% of dry HCl provided a source of activation for either anomeric hydroxyl groups; therefore, inducing complete isomerization to the desired α-anomer.
3.3. Allylation of Aldehyde 4
Despite the excellent 97:3 selectivity observed in the Brown allylation, the low chemical yield, tedious reagent preparation, and use of a stoichiometric amount of the chiral reagent, warranted investigation of another allylation protocol. Our attention focused on the catalytic, enantioselective, Lewis-based catalyzed allylation reaction of allyltrichlorosilanes developed in these laboratories. Initial attempts using 0.05 equiv of (R,R)-65 with aldehyde 61 at −78 °C provided good enantioselectivity (93:7 er), unfortunately only in 11% yield. It was determined that the Lewis-base catalyst (R,R)-65 was not efficiently turning over. Therefore, it was anticipated that switching to a more Lewis-basic co-solvent such as i-Pr2NEt instead of Et3N would facilitate a more efficient catalyst turnover.30 With this simple modification the highest yield (76%) and a respectable enantiomeric selectivity (94:6 er) were observed for the desired homoallylic alcohol (S)-62. Gratifyingly, when these conditions were applied to the synthesis of the papulacandin side chain, an excellent yield (88%) and diastereomeric selectivity (96:4 dr) were observed.
On the basis of our stereochemical analysis of the allyltrichlorosilane allylation the (R,R)-65 catalyst should produce the S-configuration at the homoallylic position (Figure 4).30 To confirm this stereochemical outcome, the allylated product 70 was subjected to Mosher ester analysis (Figure 5). The Mosher ester was prepared by the reaction of 70 with 1-methoxy-1-triflouoromethyl-phenylacetyl (MTPA) chloride in pyridine/CDCl3 and was analyzed by 1H NMR of the crude reaction mixture. Both diastereomers of the S- and R-MTPA esters (S-MTPA-70 (95) and R-MTPA-70 (96), respectively) were analyzed, and their chemical shift differences were measured. (Figure 5).33 From the Mosher ester analysis it was determined that the configuration at the homoallylic position was indeed of the S-configuration, which correlated with the above model in Figure 4.
Figure 4.

Hypothetical model for the reaction promoted by (R,R)-65.
Figure 5.

Mosher ester analysis of MTPA-70.
3.4. Summary of Global Protecting Group Strategy
One of the most challenging aspects of the total synthesis was the successful implementation of the protecting group strategy. In the previous section the criteria for the protecting group strategy was discussed; however, this proved to be much more challenging in practice that initially anticipated. Early in the synthesis it was decided that the hydroxyl groups of papulacandin D would be protected with silicon-based protecting groups to facilitate a global silicon deprotection as the last step in the synthesis. This strategy limited the choice of protecting groups to those that are easily cleaved in the presence of fluoride and thus required a very delicate balance between the protecting groups chosen. For instance, when the resorcinol portion of the aromatic iodide contained TIPS or TBS silyl ethers, the cross-coupling reaction did not proceed. However, when the hydroxyl groups were protected as the benzyl ethers productive cross-coupling was achieved. Although the benzyl protecting groups were optimal for cross-coupling, the reductive deprotection conditions would not be compatible with the unsaturated side chain 2. Therefore, these groups needed to be removed and replaced before the introduction of the side chain.
The first attempt at the synthesis of papulacandin D employed SEM protection because, at the time, SEM was the only protecting group that would efficiently protect the aromatic hydroxyl groups and the sterically hindered C(2) hydroxyl group. Unfortunately, SEM ethers could not be cleaved in the presence of those fluoride sources that would also allow the side chains to emerge intact. Thus, the related TEOC group was selected as a slightly more labile alternative.
The sequence for the introduction of the TEOC groups was crucial for the successful preparation of the tris-TEOC arylglycopyranoside 89. If the phenolic hydroxyl groups were protected first, then C(2)-hydroxyl group protection was very difficult, because of the additional steric encumbrance. Therefore, the following sequence was employed: (1) the C(2)-hydroxyl group was protected first, through an in situ formation of a chlorocarbonate 85 and trapping with 2-(trimethylsilyl)ethanol, (2) the benzyl groups were removed under hydrogenolysis conditions, and (3) the phenolic hydroxyl groups were protected using TEOC-Cl in the presence of i-Pr2NEt. This three step sequence proceeded in 88% overall yield compared to the 38% yield for the 3 step sequence for the SEM protection of 55.
Finally, the successful global deprotection required extensive optimization for the cleavage of all the TEOC groups. The C(2)-TEOC was the most difficult to remove of all the silicon protecting groups in 92. Manipulation of the fluoride concentration, ratio of HF to Et3N, changing the solvent from CH3CN to DMSO, and raising the temperature from 40 °C to 60 °C were all required to effect complete desilylation and provide excellent yield (89%) of the natural product.
4. Conclusion
The total synthesis of (+)-papulacandin D has been accomplished in a convergent approach (31 steps overall, 9.2%, over 80 mg) from commercially available triacetoxyglucal and geraniol. The synthetic strategy breaks the molecule into two nearly equal subunits, the C-spirocyclic arylglycoside 90 and polyunsaturated fatty acid side chain 2. The key significant features of the synthetic strategy are (1) the palladium catalyzed, organosilanolate-based, cross-coupling of a protected glucal silanol and (2) a catalytic enantioselective allylation reaction using chiral bisphosphoramide (R,R)-65 for the construction of the C(7") stereogenic center. Further extension of organosilanolate-based, cross-coupling reactions for the synthesis of other complex molecules is underway.
Supplementary Material
Acknowledgment
Funding was provided by the National Institutes of Health (Grant R01 GM63167). CSR thanks Eli Lilly research laboratories and Johnson & Johnson PRI for graduate fellowships. We are also grateful to Prof. A. G. M. Barrett for providing spectra for synthetic (+)-papulacandin D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data: Detailed procedures and full characterization of all synthetic intermediates and products are provided, along with spectral comparison of natural and synthetic (+)-papulacandin D are provided (215 pages).
References
- 1.Isolation: Traxler P, Gruner J, Auden JA. J. Antibiot. 1977;30:289–296. doi: 10.7164/antibiotics.30.289. Structural characterization: Traxler P, Frittz H, Richter WJ. Helv. Chim. Acta. 1977;60:578–584. doi: 10.1002/hlca.19770600230. Traxler P, Fritz H, Fuhrer H, Richter W. J. Antibiot. 1980;33:967–978. doi: 10.7164/antibiotics.33.967. Traxler P, Gruner J, Nuesch J. 4,278,665. U. S. Patent. 1981
- 2.(a) Pérez P, Varona R, Garcia-Acha I, Durán A. FEBS Lett. 1981;129:249–252. [Google Scholar]; (b) Varona R, Pérez P, Durán A. FEMS Microbiol. Lett. 1983;20:243–247. [Google Scholar]; (c) Baguley BC, Rommele G, Gruner J, Wehrli W. Eur. J. Biochem. 1979;97:345–351. doi: 10.1111/j.1432-1033.1979.tb13120.x. [DOI] [PubMed] [Google Scholar]; (d) Murgui A, Victoria Elorza M, Sentandreu R. Biochem. Biophys. Acta. 1985;841:215–222. doi: 10.1016/0304-4165(85)90024-8. [DOI] [PubMed] [Google Scholar]; (e) Pérez P, Garcia-Acha I, Durán A. J. Gen. Microbiol. 1983;129:245–250. doi: 10.1099/00221287-129-1-245. [DOI] [PubMed] [Google Scholar]; (f) Kopecka M. Folia Microbiol. 1984;29:441–449. doi: 10.1007/BF02873157. [DOI] [PubMed] [Google Scholar]; (g) Debono M, Gordee RS. Annu. Rev. Microbiol. 1994;48:471–497. doi: 10.1146/annurev.mi.48.100194.002351. [DOI] [PubMed] [Google Scholar]; (h) Schmatz DM, Romancheck MA, Pittarelli LA, Schwartz RE, Fromtling RA, Nollstadt KH, Vanmiddlesworth FL, Wilson KE, Turner M. J. Proc. Natl. Acad. Sci. USA. 1990;87:5950–5954. doi: 10.1073/pnas.87.15.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Traxler P, Tosch W, Zak O. J. Antibiot. 1987;40:1146–1164. doi: 10.7164/antibiotics.40.1146. [DOI] [PubMed] [Google Scholar]; (i) Schmatz DM. 5,089,478. U.S. Patent. 1992
- 3.(a) Bartizal K, Abruzzo G, Trainor C, Krupa D, Nollstadt K, Schmartz D, Schwartz R, Hammond M, Balkovec J, Vanmiddlesworth F. Antimicrob. Agents Chemother. 1992;36:1648–1657. doi: 10.1128/aac.36.8.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Traxler P. 4,374,129. U. S. Patent. 1983
- 4.For L-687, 781 see VanMiddlesworth F, Olmstead MN, Schmatz D, Bartizal K, Fromling R, Bills G, Nollstadt K, Honeycutt S, Zweerink M, Garrity G, Wilson K. J. Antibiot. 1991;44:45–51. doi: 10.7164/antibiotics.44.45. For Mer-WF3010 see Kaneto R, Chiba H, Agematu H, Shibamoto N, Yoshioka T, Nishida H, Okamoto R. J. Antibiot. 1993;46:247–250. doi: 10.7164/antibiotics.46.247. Chiba H, Kaneto R, Agematu H, Shibamoto N, Yoshioka T, Nishida H, Okamoto R. J. Antibiot. 1993;46:356–358. doi: 10.7164/antibiotics.46.356. For BU-4794F see Aoki M, Andoh T, Ueki T, Masuyoshi S, Sugawara K, Oki T. J. Antibiot. 1993;46:952–960. doi: 10.7164/antibiotics.46.952. For Saricandin see Chen RH, Tennant S, Frost D, O'Beirne MJ, Karwowski JP, Humphrey PE, Malmberg LH, Choi W, Brandt KD, West P, Kadam SK, Clement JJ, McAlpine JB. J. Antibiot. 1996;49:596–598. doi: 10.7164/antibiotics.49.596. For chaetiacandin see: Komori T, Itoh Y. J. Antibiot. 1985;38:544–546. doi: 10.7164/antibiotics.38.544. Komori T, Yamashita M, Tsurumi Y, Kohsaka M. J. Antibiot. 1985;38:455–459. doi: 10.7164/antibiotics.38.455. For F-10748 A1, A2, B1, B2, C1, C2, D1 and D2 see: Ohyama T, Iwadate-Kurihara Y, Hosoya T, Ishikawa T, Miyakoshi S, Hamano K, Inukai M. J. Antibiot. 2002;55:758–763. doi: 10.7164/antibiotics.55.758. Traxler P. 4,251,517. U. S. Patent. 1981
- 5.For arylglycoside synthesis see: Danishefsky S, Phillips G, Ciufolini M. Carbohydrate Res. 1987;171:317–327. doi: 10.1016/s0008-6215(00)90895-4. Balachari D, O’Doherty GA. Org. Lett. 2000;2:863–866. doi: 10.1021/ol0000253. Balachari D, O’Doherty GA. Org. Lett. 2000;2:4033–4036. doi: 10.1021/ol006662a. Ahmed MM, O'Doherty GA. Tetrahedron Lett. 2005;46:4151–4155. McDonald FE, Zhu HYH, Holmquist CR. J. Am. Chem. Soc. 1995;117:6605–6606. Schmidt RR, Frick W. Tetrahedron. 1988;44:7163–7169. Barrett AGM, Peña M, Willardsen JA. J. Chem. Soc., Chem. Commun. 1995:1145–1146. Barrett AGM, Peña M, Willardsen JA. J. Chem. Soc., Chem. Commun. 1995:1147–1148. Rosenblum SB, Bihovsky RJ. Am. Chem. Soc. 1990;112:2746–2748. Czernecki S, Perlat M-C. J. Org. Chem. 1991;56:6289–6292. Parker KA, Georges AT. Org. Lett. 2000;2:497–499. doi: 10.1021/ol991346l. Friesen RW, Sturnio CF. J. Org. Chem. 1990;55:2572–2574. Friesen RW, Sturnio CF. J. Org. Chem. 1990;55:5808–5810. Friesen RW, Sturnio CF, Daljeet AK, Kolaczewska A. J. Org. Chem. 1991;56 1994-1947. Friesen RW, Sturnio CF, Daljeet AK, Kolaczewska A. J. Org. Chem. 1991;56 1994-1947. Dubois E, Beau J-M. J. Chem. Soc. Chem. Commun. 1990:1191–1192. Dubois E, Beau J-M. Tetrahedron Lett. 1990;36:5165–5168. Dubois E, Beau J-M. Carbohydr. Res. 1992;223:157–167. doi: 10.1016/0008-6215(92)80014-r. For the total synthesis of papulacandin D see: Barrett AGM, Peña M, Willardsen JA. J. Org. Chem. 1996;61:1082–1100. (t) Previous communication see: Denmark SE, Regens CS, Kobayashi T. J. Am. Chem. Soc. 2007;129:2774–2776. doi: 10.1021/ja070071z. Kobayashi T, Regens CS, Denmark SE. J. Syn. Org. Chem. Jpn. 2008;66:616–628. Hitchcock SA, Chafig H, Sanchez-Martinez C, Esteban AR. 6,069,238 U. S. Patent.
- 6.Hector RF. Clin. Microbiol. Rev. 1993:1–21. doi: 10.1128/cmr.6.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Denmark SE, Sweis RF. In: Metal-Catalyzed Cross-Coupling Reactions. de Meijere A, Diederich F, editors. Germany: Wiley-VCH Weinheim; 2004. pp. 163–216. [Google Scholar]; (b) Denmark SE, Yang S-M. Chapt. 4. In: Harmata MA, editor. Strategies and Tactics in Organic Synthesis. Volume 6. Amsterdam: Elsevier; 2005. [Google Scholar]; (c) Denmark SE, Baird JD. Chem. Eur. J. 2006;12:4954–4963. doi: 10.1002/chem.200600034. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE, Regens CS. Acc. Chem. Res. 2008;41:1486–1499. doi: 10.1021/ar800037p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Denmark SE. J. Org. Chem. 2009;74:2915–2927. doi: 10.1021/jo900032x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denmark SE, Neuville L. Org. Lett. 2000;2:3221–3224. doi: 10.1021/ol0064112. [DOI] [PubMed] [Google Scholar]
- 9. Swindell CS, Fan W. J. Org. Chem. 1996;61:1109–1118. Niclaou KC, Ramanjulu JM, Natarajan S, Brase S, Li H, Boddy CNC, Rubsam F. Chem. Commun. 1997;19:1899–1900. (c) See Supplementary Data for the synthesis of 9.
- 10.Ferrier RJ, Furneaux RH. Carbohydr. Res. 1976;52:63–68. [Google Scholar]
- 11. Fernandez-Mayoralas A, Marra A, Trumtel M, Veyrieres A, Siney P. Tetrahedron Lett. 1989;30:2537–2540. Idem, Carbohydr. Res. 1989;188:81–95. Jäkel C, Dötz KH. J. Organomet. Chem. 2001;624:172–185. Although the glycal actonide can be prepared from glucose via triacetyl glycal, but is low yielding. Fraser-Reid B, Walker DL, Tam SY-K, Holder NL. Can. J. Chem. 1973;51:3950–3954. Franck RW, John TV. J. Org Chem. 1983;48:3269–3276.
- 12.Direct formation of silanol from metalation of glucals and trapping with hexamethylcyclotrisiloxane the yields and scale were irreproducible. Butler CR, Denmark SE. Encyclopedia of Reagents for Organic Synthesis. 2007 [DOI: 10.1002/047084289X.rn00785].
- 13.Lee M, Ko S, Chang S. J. Am. Chem. Soc. 2000;122:12011–12012. [Google Scholar]
- 14.For the preparation of protected aromatic iodides see the Supplementary Data.
- 15.(a) Corey EJ, Hopkins PB. Tetrahedron Lett. 1982;23:4871–4874. [Google Scholar]; (b) Trost BM, Caldwell CD. Tetrahedron Lett. 1981;22:4999–5002. [Google Scholar]
- 16.Roth W, Pigman W. Methods in Carbohydr. Chem. 1963;2:405–408. [Google Scholar]
- 17.It was found through deuterium incorporation experiments 1.0–1.5 equiv t-BuLi was sufficient to effect the lithiation at C(1), see the Supplementary Data.
- 18.Lee Y, Seomoon D, Kim S, Han H, Chang S, Lee P. J. Org. Chem. 2004;69:1741–1743. doi: 10.1021/jo035647r. [DOI] [PubMed] [Google Scholar]
- 19.Attempts to trap C(1)-lithio 41 with dichlorodimethylsilane, followed by hydrolysis of the corresponding chlorosilane, under pH 5.0 buffered aqueous conditions, gave disiloxane as the sole product.
- 20.This type of nonoxidative spiroketalization was observed with 57 in the presence of acidic media.
- 21.The 82% yield was consistently reproducible on both 1.0 mmol to 5.0 mmol scales.
- 22. Halcomb RL, Danishefsky SJ. J. Am. Chem. Soc. 1989;111:6661–6666. Danishefsky SJ, Bilodeau MT. Angew. Chem. Int. Ed. Engl. 1996;35:1380–1419. (c) For a recent review on DMDO see: Adam W, Saha-Möller CR, Zhao C-G. Org. React. 2002;61:219–516.
- 23.TMS-ethanol Mancini ML, Honek JF. Tetrahedron Lett. 1982;23:3249–3250. SEM-Cl Lipshutz BH, Pegram J. J. Tetrahedron Lett. 1980;21:3343–3346. Lipshutz BH, Moretti R, Crow R. Tetrahedron Lett. 1989;30:15–18.
- 24.Laganis ED, Chenard BL. Tetrahedron Lett. 1984;25:5831–5834. [Google Scholar]
- 25.Denmark SE, Almstead NG. Chapt. 10. In: Otera J, editor. Modern Carbonyl Chemistry. Weinheim: Wiley-VCH; 2000. [Google Scholar]
- 26.Denmark SE, Fu J. Chem. Rev. 2003;123:2208–2216. doi: 10.1021/ja021280g. [DOI] [PubMed] [Google Scholar]
- 27.(a) Grubbs’ 2nd generation catalysts: (1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)dichloro(phenylmethylene)(tricyclohexylphosphine)ruthenium. Lautens M, Maddes ML. Org. Lett. 2004;6:1883–1886. doi: 10.1021/ol049883f. BouzBouz S, Simmons R, Cossy J. Org. Lett. 2004;6:3465–3467. doi: 10.1021/ol049079t. Chatterjee AK, Morgan JP, Scholl M, Grubbs RH. J. Am. Chem. Soc. 2000;122:3783–3784. Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH. J. Am. Chem. Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882.
- 28.(a) Keck GE, Tarbet KH, Geraci LS. J. Am. Chem. Soc. 1993;115:8467–8468. [Google Scholar]; (b) Keck GE, Geraci LS. Tetrahedron Lett. 1993;34:7827–7828. [Google Scholar]; (c) Meng D, Su D-S, Balog A, Bertinato P, Sorensen EJ, Danishefsky SJ, Zheng Y-H, Chou T-C, He L, Horwitz SB. J. Am. Chem. Soc. 1997;119:2733–2734. [Google Scholar]
- 29. Jadhav PK, Bhat KS, Perumal T, Brown HC. J. Org Chem. 1986;51:432–439. Brown HC, Joshi NN. J. Org Chem. 1988;53:4059–4062. Lautens M, Maddes ML, Sauer ELO, Ouellet SG. Org. Lett. 2002;4:83–86. doi: 10.1021/ol016946a. Salt free conditions: Racherla US, Brown HC. J. Org Chem. 1991;56:401–404.
- 30.(a) Denmark SE, Fu J, Coe DM, Su X, Pratt N, Griedel BD. J. Org. Chem. 2006;71:1513–1522. doi: 10.1021/jo052202p. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Fu J, Lawler MJ. J. Org. Chem. 2006;71:1523–1536. doi: 10.1021/jo052203h. [DOI] [PubMed] [Google Scholar]
- 31.Commercially available geraniol was enriched by spinning band distillation, (GC analysis 99.8% geraniol : 0.2% nerol)
- 32.(a) Takaya H, Ohta T, Inoue S-I, Tokunaga M, Kitamura M, Noyori R. Org Synth. 72:169–175. [Google Scholar]; (b) Takaya H, Mashima K, Koyano K, Yagi M, Kumobayashi H, Taketomi T, Akutagawa S, Noyori R. J. Am. Chem. Soc. 1987;109:1596–1597. [Google Scholar]
- 33. Dale JA, Mosher HS. J. Am. Chem. Soc. 1973;95:512–519. Seco JM, Quinoa E, Riguera R. Chem. Rev. 2004;104:17–117. doi: 10.1021/cr2003344. (c) See the Supplementary Data for a complete list of chemical shift data.
- 34.Langanis ED, Chenard BL. Tetrahedron Lett. 1984;25:5831–5834. [Google Scholar]
- 35.Inanaga J, Hirata K, Saeki H, Katsuski T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989–1993. [Google Scholar]
- 36.Removal of SEM from alcohols: CsF HMPA Ireland RE, Norbeck DW. J. Am. Chem. Soc. 1985;107:3279–3285. TFA Schlessinger RH, Poss MA, Richardson S. J. Am. Chem. Soc. 1986;108:3112–3114. Ammonium flouride sources in HMPA or THFKan T, Hashimoto M, Yanagiya M, Shirahama H. Tetrahedron Lett. 1988;29:5417–5418. TBAF, MS4Å, DMPULipshutz BH, Miller TA. Tetrahedron Lett. 1989;30:7149–7152. MgBr2, n-BuSH, Et2O Kim S, Kee IS, Park YH, Park JH. Synlett. 1991:183–184. MgBr2, MeNO2, Et2O Vakalopoulos A, Hoffmann HMR. Org. Lett. 2000;2:1447–1450. doi: 10.1021/ol0057784. ZnBr2, MeOH, Et2O or MgBr2, MeNO2, Et2O Vakalopoulos A, Hoffmann HMR. Org. Lett. 2001;3:2185–2188. doi: 10.1021/ol016037l. ZnX2 in CH2Cl2 Kolb HC, Hoffmann HMR. Tetrahedron: Asymmetry. 1990;1:237–250. MgBr2, EtSH, Et2OBerkowitz DB, Choi S, Maeng J-H. J. Org Chem. 2000;65:847–860. doi: 10.1021/jo991582+. SEM deprotection from phenol (j) (TBAF, HMPA) Saimoto H, Hiyama T. Tetrahedron Lett. 1986;27:597–600. P2I4 Saimoto H, Kusano Y, Hiyama T. Tetrahedron Lett. 1986;27:1607–1610. H2SO4 Shih TL, Wyvratt MJ, Mrozik H. J. Org Chem. 1987;52:2029–2033. TBAF, MS4Å, DMPUKoreeda M, Dixon LA, His JD. Synlett. 1993:555–556. HCl Graybill TL, Casillas EG, Pal K, Townsend CA. J. Am. Chem. Soc. 1999;121:7729–7746. "Anhydrous TBAF"Sugimura T, Paquette LA. J. Am. Chem. Soc. 1987;109:3017–3024. HF, CH3CN White JD, Kawasaki M. J. Am. Chem. Soc. 1990;112:4991–4993. I2, hv Karim S, Parmee ER, Thomas EJ. Tetrahedron Lett. 1991;32:2269–2272.
- 37.Preparation of trialkylsilyloxymethyl chlorides: Pitsch S, Weiss PA, Wu X, Ackermann D, Honegger T. Helv. Chim. Acta. 1999;82:1753–1761. Pitsch S, Weiss PA, Jenny L, Stutz A, Wu X. Helv. Chim. Acta. 2001;84:3773–3795.
- 38.Gioeli C, Balgobin N, Joesephson S, Chattopadhyaya JB. Tetrahedron Lett. 1981;22:969–972. [Google Scholar]
- 39.See the Supplementary Data for the preparation of the buffered HF solutions.
- 40.Brigl PZ. Physiol. Chem. 1922;122:245. [Google Scholar]
- 41.Liu G, Wurst JM, Tan DS. Org. Lett. 2009;11:3670–3673. doi: 10.1021/ol901437f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.