

Abstract
In recent years, small protein oligomers have been implicated in the aetiology of a number of important amyloid diseases, such as type 2 diabetes, Parkinson’s disease and Alzheimer’s disease. As a consequence, research efforts are being directed away from traditional targets, such as amyloid plaques, and towards characterization of early oligomer states. Here we present a new analysis method, ion mobility coupled with mass spectrometry, for this challenging problem, which allows determination of in vitro oligomer distributions and the qualitative structure of each of the aggregates. We applied these methods to a number of the amyloid-β protein isoforms of Aβ40 and Aβ42 and showed that their oligomer-size distributions are very different. Our results are consistent with previous observations that Aβ40 and Aβ42 self-assemble via different pathways and provide a candidate in the Aβ42 dodecamer for the primary toxic species in Alzheimer’s disease.
Many diseases share the common trait of peptide–protein misfolding that leads to oligomerization and, eventually, formation of plaques of β-sheet structure. Prominent among these are type 2 diabetes1, Parkinson’s disease2 and Alzheimer’s disease3,4. Of these, Alzheimer’s disease is the leading cause of late-life dementia and is the focus of this paper. An increasing body of evidence links oligomerization of a ubiquitous peptide, the amyloid-β protein, to disease causation3-6. For this reason, elucidation of pathways of oligomer formation may be critical for the identification of therapeutic targets.
Many types of oligomeric amyloid-β assemblies have been described (for a review, see Lazo et al.7). Recently, Bitan et al.8-10 used photoinduced cross-linking of unmodified proteins (PICUP) to reveal that the 42-residue form of amyloid-β, Aβ42, formed (Aβ42)5 and (Aβ42)6 oligomers (‘paranuclei’) that could oligomerize to form structures of higher order. Aβ40 did not form paranuclei, but instead existed as a mixture of monomers, dimers, trimers and tetramers. Chen and Glabe11, in contrast, used fluorescence and gel electrophoresis to determine oligomer states of amyloid-β refolded from denaturing solutions. They observed only Aβ42 monomer and trimer bands, and no oligomers of Aβ40. Differences such as these may exist because of the diverse experimental systems used to monitor amyloid-β self-association. Also, it has been argued that, in addition to the intrinsic potential of amyloid-β to traverse different assembly pathways, flaws in experimental design may have misled researchers in their quest to elucidate fully the amyloid-β oligomerization process12. Hence there is significant uncertainty about amyloid-β oligomer states and their position and relevance to amyloid-β aggregation.
Results and discussion
We used a different, more direct, method to probe the amyloid-β oligomerization process: ion mobility coupled with mass spectrometry13-15. Details are given in the Methods section. Here the results for Aβ40 are given as an example. The mass spectrum of Aβ40 is shown in Fig. 1a and arrival time distributions (ATDs) of the z/n = −2 and −5/2 peaks (z = total charge, n = oligomer order) are given in Fig. 1b,c, respectively, for both high and low injection energies. High-resolution 13C isotope distribution measurements (insets in Fig. 1a) on the z/n = −3 peak and z/n = −2 peak indicate that the z/n = −3 peak is caused by monomer (i.e. n = 1, Δ(m/z) = 0.33), whereas the z/n = −2 peak results from both monomer (z = −2, n = 1, Δ(m/z) = 0.5) and dimer (z = −4, n = 2, Δ(m/z) = 0.25). The ATD of the z/n = −2 peak (Fig. 1b) is bimodal (peaks at ~520 and ~660 μs), which indicates that at least two non-interconverting species exist on the timescale of the experiment, in agreement with the mass spectrum.
Figure 1. Negative-ion mass spectrum and ATDs of Aβ40.

a, Mass spectrum from a 30 μM solution at pH 7.4 labelled with z/n values (z = charge, n = oligomer order). The insets show high-resolution 13C isotope patterns. The spacing of 0.33 for the z/n = −3 peak indicates that it is a monomer. The more complex pattern for z/n = −2 indicates that both monomer and dimer are present (dimer: z = −4, n = 2). b,c, ATDs at high and low injection energies (IE) for z/n = −2 (b) and z/n = −5/2 (c). The vertical dotted lines show the expected average experimental arrival times of the dimer and tetramer. M = monomer, D = dimer, Te = tetramer (see text).
Injection-energy studies (Fig. 1b) unambiguously identified the 660 μs peak as the monomer (z = −2, n = 1) because it occurred only at high injection-energies, and thus resulted from oligomer dissociation, and because no additional peaks were observed at longer times. Only two peaks were observed in the ATD at lowest injection energy, so the peak at 520 μs can be assigned to the dimer (z = −4, n = 2). Similar injection energy studies on the z/n = −5/2 peak identified both a dimer with z = −5 and n = 2 at 100 eV injection energies and a tetramer with z = −10, n = 4 at 23 eV injection energies, although the resolution of the experiment was insufficient to observe a clean bimodal ATD. (This issue is dealt with in more detail in Supplementary Information for the z/n = −5/2 ATD of both Aβ40 and Aβ42. A peak-width analysis is given based on the diffusion equations for ions drifting in gases.) Hence, the Aβ40 solution dominantly contained monomer, dimer and tetramer, with no trimer and larger oligomers observed.
Similar experiments were carried out on the Pro19 and Met35(O) alloforms of Aβ40 and on the Met35(O) alloform of Aβ42 (details are given in Supplementary Information). These systems were chosen because PICUP experiments have shown that both substitutions are known to either reduce or eliminate fibril formation16,17 and restrict oligomer production to relatively small sizes according to Klein et al.18 and Bitan et al.19 In all cases the largest oligomer observed was the tetramer. The ATDs for the z/n = −5/2 peaks sprayed from solutions under identical conditions of all the systems considered here are summarized in Fig. 2. These data indicate that the ATD for Aβ42 is much more complex than that for the other five systems. Injection-energy studies15 indicate that the peak at the shortest times in the Aβ42 ATD is the dodecamer (Aβ42)12, and that the complex multipeak feature at longer times results from the dimer, tetramer and hexamer (see detailed discussion in Supplementary Information). We discuss this ATD shortly.
Figure 2. Arrival time distributions.

a–f, ATDs for z/n = −5/2 Aβ42 (a–c) and Aβ40 (d–f), showing the wild types (a,d), the alloforms Pro19 (b,e) and Met35(O) (c,f). The hexamer and dodecamer are only observed for Aβ42 wild type and are thus implicated in the increased toxicity for this alloform. The distributions in a and b are from Bernstein et al.15 D = dimer, Te = tetramer, H = hexamer, Do = dodecamer.
Ion mobility allows collision cross-sections (σ) of each oligomer to be measured accurately20 (see Supplementary Information for the measured cross-sections). A plot of σ/n versus n (Supplementary Information) has two important features. Firstly, for all systems, as n increases σ/n decreases. This indicates that the apparent ‘size’ of each monomer unit within an oligomer decreases as oligomerization proceeds. This result is not unexpected, but emphasizes that oligomerization induces a certain amount of structural accommodation. One cause of this accommodation could be the self-selection of favoured structures as oligomerization proceeds. For example, both experiment and molecular dynamics modelling are consistent with a reasonably diverse ensemble of Aβ42 monomer structures in solution21. Detailed analysis of the −5/2 ATD peak shapes of Aβ42 (Supplementary Fig. S4) indicates that multiple structures are also present for dimers and tetramers of this alloform. However, the experimental linewidth significantly narrows for the Aβ42 hexamer and dodecamer, which suggests a single structure for these two important species.
The second feature in the σ/n versus n graph is that Aβ42 remains relatively larger (that is, accommodates less) than all other alloforms. These data are consistent with the results of intrinsic fluorescence studies22, which showed that the structure of the centre (Leu17–Ala21) of Aβ42 was relatively stable compared with that of Aβ40. In addition, these data offer insight into why Aβ42 aggregation states and, by implication, the contribution of Aβ42 to the aetiology of Alzheimer’s disease are so different from those of other amyloid-β peptides. We obtained this insight by comparing measured cross-sections with those predicted by a simple structural model (for model details see Supplementary Information)23.
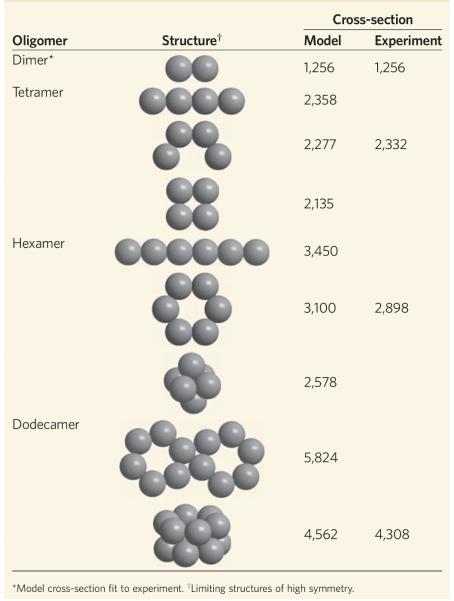
In the model, the centre-to-centre distance (diameter) of an amyloid-β dimer constructed from quasispherical monomer units was adjusted to obtain the experimental dimer cross-section. This fitting accounts for a significant fraction of the monomer accommodation as n increases. Larger structures were then assembled using these calibrated dimer centre-to-centre distances. However, as mentioned above, a number of structures contribute to the experimental dimer cross-section, which indicates that the model cross-sections will be a bit too high when compared to oligomers of higher order and hence should be upper limits because no further accommodation is included. The results for Aβ42 given in Table 1 are revealing. For example, the experimental tetramer cross-section falls between the model linear tetramer and a tetramer segment of the planar hexagon, and is much larger than the model planar square cross-section (more on this point shortly). The experimental hexamer cross-section is significantly smaller than the model linear cross-section and substantially larger than the closest-packed model cross-section, which rules these structures out. However, it is only moderately smaller than the model planar hexamer cross-section, a result consistent with the assumptions of the model and the tetramer structure, and so it supports this structure for (Aβ42)6. Similarly, the side-by-side planar hexagons can be ruled out as a structure for the dodecamer because their model cross-section is far larger than the experimental one, but the cross-section of the stacked hexagon model structure is in very good agreement with experiment. Hence, the experimental cross-sections are consistent with a quasiplanar hexagonal structure for the six-member ‘paranucleus’ and with stacked hexamers for the dodecamer.
Table 1. Experimental and model cross-sections (Å2) for Aβ42 oligomers.
|
An expanded version of the ATD for the z/n = −5/2 of Aβ42 wild type is given in Fig. 3a with the ‘structural’ assignments shown for the various peaks. There are two additional important features. Firstly, the ATD goes to the baseline where the octamer would appear. Hence, Aβ42 does not oligomerize via monomer or dimer addition past formation of the paranucleus (Aβ42)6, which suggests the mechanism in Fig. 3b occurs. Apparently, the structural stability of the paranucleus ring greatly reduces the monomer (dimer) addition rate as it accommodates homotypical growth. This critical feature indicates the potential importance of paranuclei in the aetiology of Alzheimer’s disease10.
Figure 3. Aβ42 oligomer distributions.

a, The ATDs of z/n = −5/2 with structural designations in place. b, A plausible mechanism for forming the oligomer distribution given by the ATD in a.
The second important feature in the Aβ42 ATD in Fig. 3 relates to the complete absence of any signal at arrival times prior to that of dodecamer. This is unusual because, in most instances, a sloping ATD that extends to earlier times is observed, which indicates the presence of larger oligomers that are not well resolved. The absence of ion signal at these earlier times is consistent with the absence of octadecamers (18-mers) and indicates a lack of monomer or dimer addition to the dodecamer. The absence of an 18-mer that corresponds to ((Aβ42)6)3 strongly suggests that addition of the third paranucleus is slow, and leads to accumulation of ((Aβ42)6)2. The reason aggregation of Aβ42 stops at the ((Aβ42)6)2 dodecamer is not clear. Two plausible mechanisms are discussed in the Supplementary Information24,25. Both mechanisms rationalize why the addition of a third hexamer is energetically unlikely and leads to a long-lived, but metastable, dodecamer. Apparently, this dodecamer eventually rearranges and rapidly adds monomer and forms fibrils. That Aβ42 clogged the nanospray tips used to spray the sample solution (that is, rapidly formed very large oligomers and/or fibrils), whereas each of the other five systems reported here (see Fig. 2) sprayed beautifully for a minimum of several days up to more than a week without the tip clogging supports the rearrangement hypothesis. Aggregates of higher order (Aβ42)n would rapidly move out of the mobility–mass range of the ion mass spectroscopy instrument and their dispersal would make them difficult to detect with the limited sensitivity of this instrument.
To test this aggregation hypothesis we looked at the Aβ42 positive-ion mass spectrum on a Q-TOF instrument specially built to observe high-mass aggregates under conditions in which Aβ42 aggregation is maximized (see Supplementary Fig. S6)26,27. A mound of ion intensity was observed that stretches from less than m/z 1,000 to m/z 8,000 and reflects a nearly continuous array of highly charged oligomers. Two sharp features at low m/z were identified as +4 and +3 residual monomers. This assignment was confirmed by tandem mass spectroscopy (MS/MS) (Supplementary Fig. S6 inset). The MS/MS result indicated that monomer loss dominated the dissociation process, which is common for collisional dissociation of large aggregates28. When Pro19Aβ42 was sprayed under identical conditions, a very clean monomer-dominated spectrum was observed (Supplementary Fig. S6), consistent with the fact that this alloform does not form fibrils17.
A clue as to why Aβ42, but none of the other five systems, forms paranuclei is found in the relative cross-sections of the tetramers. This effect can be quantified using the model developed here. Each of the systems studied terminates at the tetramer, except Aβ42. A normalized plot of the size of each tetramer experimental cross-section relative to several possible tetramer model structures is given in Fig. 4a. The model cross-section for the linear tetramer was taken as the benchmark because it is the largest possible structure and all system sizes were normalized to that value. The results indicate that the Aβ42 tetramer is much more ‘open’ than Aβ40 or the other three alloforms shown. If the experimental cross-sections are fitted to a dimer–dimer angle the structures in Fig. 4b are obtained.
Figure 4. The normalized cross-sections for the tetramers.

a, Relative sizes of all tetramers reported herein (shown to right of vertical scale). The vertical scale is the relative cross-section of the model tetramers normalized to the linear structure. The broad arrows on the vertical axis show the spread of values for the modelled structures. Aβ42 is a much more open tetramer than the other alloforms. b, The structures of the Aβ42 and Aβ40 tetramers taken from their location on the relative cross-section scale given in a, assuming the angle between dimer components is the determining parameter.
Another dimer could easily add to the ‘open’ Aβ42 tetramer structure (Fig. 4b) to form the quasiplanar hexamer paranucleus (or a monomer could add to form a pentamer, which explains the stacked dipentamer shown in Fig. 3). However, the ‘closed’ Aβ40 tetramers in Fig. 4b (and other alloforms) are not structurally disposed to the addition of either a monomer or a dimer and hence they form stable, long-lived tetramers. Our experimental data show the structure of the tetramer to be crucial in determining whether an amyloid-β alloform goes on to form paranuclei and dodecamers and subsequently contributes to the aetiology of Alzheimer’s disease. Hence the (perhaps small) folding differences in the various monomers are amplified by aggregation, and at the tetramer dramatic differences occur. From a different perspective, the ‘dimer–dimer’ interface is different from the ‘monomer–monomer’ interface, and perhaps the Aβ42 dimer is uniquely suited to form an open tetramer and the other systems are not. The tetramer is small enough to hope that serious modelling can be done to extract the factors at the molecular level in these systems that drive such crucial structural differences. These studies are underway.
A global scheme of amyloid-β oligomerization can be deduced by combining the data presented here with previous published data9,10 (Fig. 5). For Aβ42 a receptive structure in the tetramer allows further dimer addition and paranucleus formation with a planar hexagonal structure. Two paranuclei combine to form a stacked paranuclear dimer, which resists further transformation. Eventually, ((Aβ42)6)2 rearranges to form a new structure to which monomer rapidly adds to form large β-sheet oligomer assemblies. Aβ40, however, forms a more compact tetramer that firmly resists further addition and hence forms no paranuclei. A similar fate to that of Aβ40 befalls the Pro19 and Met35(O) alloforms of both Aβ42 and Aβ40. Although some of these systems eventually do form fibrils, they appear to do so by a mechanism that bypasses paranucleus formation10.
Figure 5. Mechanism of oligomerization and eventual fibril formation for Aβ42 and for Aβ40.

For Aβ40 the key structure is the tetramer that resists further monomer or dimer addition. In Aβ42 an ‘open’ tetramer promotes the formation of the planar hexamer (paranucleus) and the stacked dodecamer, which resists further reaction. For Aβ40 the tetramer apparently eventually forms fibrils (observed by others), but these were not observed in our experiments. For Aβ42 a rate-limiting slow α- to β-sheet transformation may occur for the dodecamer, but this was not explicitly observed in our experiments. Fibril formation was indirectly observed through macroscopic clogging of the spray tips used for Aβ42.
In related work Barghorn et al.29 isolated a species they called ‘globulomer’ from Aβ42 using a quite complex protocol. This species is soluble and has a molecular weight between 60 and 100 kDa. It is stable against dissociation for several days and only slowly self-aggregates to form larger oligomers in the absence of Aβ42 monomer. Application of proteases truncated the N-terminus of the globulomer Aβ42 monomer constituents up to about residue F20, which indicates that the C-terminal hydrophobic residues are in the centre of the globulomer. This result is consistent with our view of both the hexamer and the dodecamer complexes, in which the hydrophobic tails are envisioned as occupying the centre of the structures (see Supplementary Fig. S4 and the cartoon in Baumketner et al.21). Barghorn et al. suggest the globulomer is made up of 12 Aβ42 molecules, but their evidence for this is not conclusive29.They also assume that the globulomer is formed by an independent mechanism than through Aβ42 fibrils, a suggestion at odds with the mechanism we give in Fig. 5 and one that would be difficult to rationalize with a kinetic model (which they did not present).
One additional important point is worth making. Lesné et al.6 used amyloid precursor protein transgenic Tg2576 mice to demonstrate that memory defects occur in ‘middle-aged’ mice (6–14 months) without neuronal loss, but in older mice abundant neuritic plaques that contain Aβ42 were observed. What they observed in these middle-aged mice was an extracellular accumulation of a ~56 kDa soluble amyloid assembly, Aβ*56. These authors proposed that this 56 kDa assembly impairs memory by an unknown mechanism independent of either plaque formation or neuronal loss. Their observations are supported by recent results of Cheng et al.5, who also used transgenic mice and the ‘Arctic’ (E22G) mutant alloform of Aβ42, which implicate Aβ*56 rather than fibrils as a determinant of Alzheimer’s disease. Here we show that Aβ42 forms in vitro a dodecamer composed of stacked hexamer paranuclei. This dodecamer is the terminal species observed in our experiments, and has a mass of ~55.2 kDa, which suggests it is the soluble assembly that Lesné et al. observed6. This assembly has not yet rearranged into a β-sheet structure and may possibly be the key neurotoxic assembly in Alzheimer’s disease.
Methods
Materials
All peptides were synthesized on an automated peptide synthesizer (Applied Biosystems model 430A) using methods based on 9-fluorenylmethoxycarbonyl (FMOC) essentially as described by Lomakin et al.30 For Aβ42 an FMOC-l-Ala-2-chlorotrityl resin was used (0.51 mmol g−1, Biosearch Technologies). Lyophilized peptide was dissolved at a concentration of 4 mg ml−1 in deionized water. To this solution, 0.006 times the volume of 1 N NaOH was added, followed by filtered 50 mM ammonium acetate solution (pH 7.4) to reduce the peptide concentration to about 2 mg ml−1. The solution was sonicated for one minute and then 100 μl was filtered through a 10,000 amu Gel Filtration G-10 Macro Spin Column purchased from Nest Group. The filters were prehydrated and washed in 25 mM ammonium acetate pH 7.4 according to the manufacturer’s suggestions for desalting and buffer exchange to ensure the pH of the peptide solutions was maintained at 7.4, and cleaned of interfering salts for mass-spectrometry purposes. The sample was centrifuged for four minutes at 2,000g and the filtrate was collected and used immediately. The final concentration of peptide ranged from 30 to 40 μM (determined by amino-acid analysis) in 25 mM ammonium acetate and adjusted to pH 7.4.
For Aβ42 it was necessary to carry out the experiments as quickly as possible. Although the procedure outlined here led to stable solutions, the signal still rapidly diminished over a period of about a day because of aggregation of the peptide (eventual clogging of the spray tips confirmed this). No new features were observed in either the mass spectrum or ATD for Aβ42 for as long as observations were possible.
The other peptides were much better behaved with solutions stable up to a week (several weeks for Pro19Aβ40 and Pro19Aβ42). The mass spectra and ATDs did not change over this time period for any of these peptides, which indicates that hexamers and dodecamers were not formed in measurable quantities. Hexamers and dodecamers were exclusively formed in Aβ42 wild type. All experiments were completely reproducible over many trials over a period of more than a year. Circular dichroism spectra were taken of all samples to ensure consistency from batch to batch. In all cases the spectra were identical for a given alloform over the complete timescale of the project, which indicates excellent sample consistency.
Mass spectrometry
Standard mass spectra and ion mobility experiments were accomplished on an instrument built in-house and composed of a nano-electrospray ionization (N-ESI) source, an ion funnel, a temperature-controlled drift cell and a quadrupole mass filter followed by an electron multiplier for ion detection31. The high-resolution 13C isotope distributions for each peak in the mass spectra were obtained on a Q-TOF mass spectrometer (Micromass UK) equipped with a N-ESI source.
In the ion-mobility measurements, the ions were stored at the end of the ion funnel and then pulsed into the drift cell filled with 5 torr of helium gas and drawn through the cell under the influence of a weak electric field. The ions’ injection energy into the drift cell was varied from 0 to 150 eV. At low injection voltages, the ions were gently pulsed into the mobility cell and only needed a few ‘cooling’ collisions to reach thermal equilibrium with the buffer gas. At high injection voltages, the larger collision energy led to internal excitation of the ions before cooling and equilibrium occurred. This transient internal excitation can lead to annealing, that is partial or complete isomerization, to give the most stable conformers or dissociation of dimers and oligomers of higher order15. The ions exited the drift cell, passed through a quadrupole mass filter and were detected as a function of time to produce an ATD. The arrival time is simply related to the mobility, which in turn is inversely proportional to the collision cross-section32. All factors in these equations20 are either universal constants or parameters that were accurately measured in the experiment. Hence accurate (±1%) collision cross-sections were obtained.
Supplementary Material
Acknowledgements
M.T.B., G.B. and D.B.T. thank the National Institutes of Health, J.E.S. thanks the National Science Foundation, the Alfred P. Sloan Foundation and the David and Lucile Packard Foundation, and C.V.R. thanks the Biotechnology and Biological Sciences Research Council for support of this work. We gratefully acknowledge C. Carpenter for her help in producing the manuscript and figures.
Footnotes
Additional information
Supplementary information accompanies this paper at www.nature.com/naturechemistry.
References
- 1.Meier JJ, et al. Inhibition of human IAPP fibril formation does not prevent β-cell death: evidence for distinct actions of oligomers and fibrils of human IAPP. Am. J. Physiol. Endocrinol. Metab. 2006;291:E1317–E1324. doi: 10.1152/ajpendo.00082.2006. [DOI] [PubMed] [Google Scholar]
- 2.Conway KA, et al. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc. Natl Acad. Sci. USA. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klein WL, Krafft GA, Finch CE. Targeting small Aβ oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 4.Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J. Neurosci. Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 5.Cheng IH, et al. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- 6.Lesné S, et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 7.Lazo ND, Maji SK, Fradinger EA, Bitan G, Teplow DB. In: Amyloid Proteins: The Beta Sheet Conformation and Disease. Sipe JD, editor. Wiley; 2005. pp. 385–491. [Google Scholar]
- 8.Bitan G, Lomakin A, Teplow DB. Amyloid β-protein oligomerization. Prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J. Biol. Chem. 2001;276:35176–35184. doi: 10.1074/jbc.M102223200. [DOI] [PubMed] [Google Scholar]
- 9.Bitan G, Vollers SS, Teplow DB. Elucidation of primary structure elements controlling early amyloid β-protein oligomerization. J. Biol. Chem. 2003;278:34882–34889. doi: 10.1074/jbc.M300825200. [DOI] [PubMed] [Google Scholar]
- 10.Bitan G, et al. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl Acad. Sci. USA. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y-R, Glabe CG. Distinct early folding and aggregation properties of Alzheimer amyloid-β peptides Aβ40 and Aβ42: stable trimer or tetramer formation by Aβ42. J. Biol. Chem. 2006;281:24414–24422. doi: 10.1074/jbc.M602363200. [DOI] [PubMed] [Google Scholar]
- 12.Bitan G, Fradinger EA, Spring SM, Teplow DB. Neurotoxic protein oligomers – what you see is not always what you get. Amyloid. 2005;12:88–95. doi: 10.1080/13506120500106958. [DOI] [PubMed] [Google Scholar]
- 13.von Helden G, Hsu M-T, Kemper PR, Bowers MT. Structures of carbon cluster ions from 3 to 60 atoms: linears to rings to fullerenes. J. Chem. Phys. 1991;95:3835–3837. [Google Scholar]
- 14.Wyttenbach T, Bowers MT. Gas-phase conformations: the ion mobility/ion chromatography method. Top. Curr. Chem. 2003;225:207–232. [Google Scholar]
- 15.Bernstein SL, et al. Amyloid β-protein: monomer structure and early aggregation states of Aβ42 and its Pro19 alloform. J. Am. Chem. Soc. 2005;127:2075–2084. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]
- 16.Hou L, Kang I, Marchant RE, Zagorski MG. Methionine 35 oxidation reduces fibril assembly of the amyloid Aβ-(1-42) peptide of Alzheimer’s disease. J. Biol. Chem. 2002;277:40173–40176. doi: 10.1074/jbc.C200338200. [DOI] [PubMed] [Google Scholar]
- 17.Wood SJ, Wetzel R, Martin JD, Hurle MR. Prolines and amyloidogenicity in fragments of the Alzheimer’s peptide β/A4. Biochemistry. 1995;34:724–730. doi: 10.1021/bi00003a003. [DOI] [PubMed] [Google Scholar]
- 18.Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol. Aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Bitan G, et al. A molecular switch in amyloid assembly: Met35 and amyloid β-protein oligomerization. J. Am. Chem. Soc. 2003;125:15359–15365. doi: 10.1021/ja0349296. [DOI] [PubMed] [Google Scholar]
- 20.Gidden J, Ferzoco A, Baker ES, Bowers MT. Duplex formation and the onset of helicity in poly d(CG)n oligonucleotides in a solvent-free environment. J. Am. Chem. Soc. 2004;126:15132–15140. doi: 10.1021/ja046433+. [DOI] [PubMed] [Google Scholar]
- 21.Baumketner A, et al. Amyloid β-protein monomer structure: a computational and experimental study. Protein Sci. 2006;15:420–428. doi: 10.1110/ps.051762406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maji SK, Amsden JJ, Rothschild KJ, Condron MM, Teplow DB. Conformational dynamics of amyloid β-protein assembly probed using intrinsic fluorescence. Biochemistry. 2005;44:13365–13376. doi: 10.1021/bi0508284. [DOI] [PubMed] [Google Scholar]
- 23.Wyttenbach T, von Helden G, Batka JJ, Carlat D, Bowers MT. Effect of the long-range potential on ion mobility measurements. J. Am. Soc. Mass Spectrom. 1997;8:275–282. [Google Scholar]
- 24.Oosawa F, Kasai M. Theory of linear and helical aggregations of macromolecules. J. Mol. Biol. 1962;4:10–21. doi: 10.1016/s0022-2836(62)80112-0. [DOI] [PubMed] [Google Scholar]
- 25.Tobacman LS, Korn ED. The kinetics of actin nucleation and polymerization. J. Biol. Chem. 1983;258:3207–3214. [PubMed] [Google Scholar]
- 26.Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal. Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 27.Ruotolo BT, et al. Evidence for macromolecular protein rings in the absence of bulk water. Science. 2005;310:1658–1661. doi: 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- 28.McCammon MG, Hernandez H, Sobott F, Robinson CV. Tandem mass spectrometry defines the stoichiometry and quaternary structural arrangement of tryptophan molecules in the multiprotein complex TRAP. J. Am. Chem. Soc. 2004;126:5950–5951. doi: 10.1021/ja0317170. [DOI] [PubMed] [Google Scholar]
- 29.Barghorn S, et al. Globular amyloid β-peptide1-42 oligomer – a homogenous and stable neuropathological protein in Alzheimer’s disease. J. Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- 30.Lomakin A, Chung DS, Benedek GB, Kirschner DA, Teplow DB. On the nucleation and growth of amyloid β-protein fibrils: detection of nuclei and quantitation of rate constants. Proc. Natl Acad. Sci. USA. 1996;93:1125–1129. doi: 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyttenbach T, Kemper PR, Bowers MT. Design of a new electrospray ion mobility mass spectrometer. Int. J. Mass Spectrom. 2001;212:13–23. [Google Scholar]
- 32.Mason EA, McDaniel EW. Transport Properties of Ions in Gases. Wiley; 1988. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.