

Abstract
Neurospora crassa macroconidia form germ tubes that are involved in colony establishment and conidial anastomosis tubes (CATs) that fuse to form interconnected networks of conidial germlings. Nuclear and cytoskeletal behaviors were analyzed in macroconidia, germ tubes, and CATs in strains that expressed fluorescently labeled proteins. Heterokaryons formed by CAT fusion provided a rapid method for the imaging of multiple labeled fusion proteins and minimized the potential risk of overexpression artifacts. Mitosis occurred more slowly in nongerminated macroconidia (1.0 to 1.5 h) than in germ tubes (16 to 20 min). The nucleoporin SON-1 was not released from the nuclear envelope during mitosis, which suggests that N. crassa exhibits a form of “closed mitosis.” During CAT homing, nuclei did not enter CATs, and mitosis was arrested. Benomyl treatment showed that CAT induction, homing, fusion, as well as nuclear migration through fused CATs do not require microtubules or mitosis. Three ropy mutants (ro-1, ro-3, and ro-11) defective in the dynein/dynactin microtubule motor were impaired in nuclear positioning, but nuclei still migrated through fused CATs. Latrunculin B treatment, imaging of F-actin in living cells using Lifeact-red fluorescent protein (RFP), and analysis of mutants defective in the Arp2/3 complex demonstrated that actin plays important roles in CAT fusion.
The initiation of a colony from asexual spores (conidia) of Neurospora crassa involves the production of at least two types of specialized hyphae: germ tubes and conidial anastomosis tubes (CATs) (58). Germ tubes are important for colony establishment and eventually develop into the vegetative hyphae of the mature and differentiated colony. CATs function to interconnect germlings, a process that may allow the young colony to act as a coordinated individual and regulate its overall homeostasis (50, 51).
The cytology of germ tube development in N. crassa has been analyzed in detail in living cells (4). Macroconidia and germlings undergo isotropic growth during the first 2 h following hydration in minimal growth medium at 25°C. Typically, it takes 3 h for the polarized emergence of a single germ tube from a macroconidium. Following germination (up to 9 h), the nuclei, mitochondria, and other organelles display a more-or-less uniform distribution inside growing germ tubes. After ∼10 h (i.e., when the germ tubes are >150 μm in length), organelle distribution becomes polarized, and a small exclusion zone appears at the tip. Within this exclusion zone, the phase-dark Spitzenkörper, a vesicle-dominated complex intimately involved in hyphal tip growth, forms. The development of the mature Spitzenkörper was suggested previously to represent the transition from germ tube to vegetative hypha (4).
Conidial anastomosis tubes have been shown to be morphologically and physiologically distinct from germ tubes and under separate genetic control (50, 58, 59). They are shorter and thinner than germ tubes and are chemoattracted to other CATs, which they eventually fuse with. The Spitzenkörper has not been observed in growing CATs (7, 21, 50, 51, 58, 59).
Neurospora crassa undergoes asynchronous mitotic division in its multinucleate hyphae (24, 25, 35, 48, 62). Although several studies have documented the behavior of nuclei in conidia and hyphae, none have yet provided a detailed description of mitosis in living cells of N. crassa. This is in stark contrast to studies of mitosis performed with other filamentous fungi, including Aspergillus nidulans (43, 44), Nectria haematococca (1, 2, 69), and Ashbya gossypii (26). Green fluorescent protein (GFP) labeling has greatly facilitated the observation of nuclei in living filamentous fungi (23, 24, 26, 66, 70) and has been particularly useful for time-lapse studies (22, 44). Vegetative hyphae, however, are too large (∼15 μm wide) for time-lapse imaging of nuclei in N. crassa. In addition, nuclei are extremely mobile, making the tracking of individual nuclei in mature leading hyphae difficult (24).
In this study we analyzed and compared nuclear dynamics and mitosis during the early stages of colony initiation in macroconidia, germ tubes, and CATs. Time-lapse imaging of nuclei and their associated microtubules was facilitated in all three cell types because of their small size and because nuclei in these cells were less mobile than in vegetative hyphae. Besides providing the first detailed description of mitosis in N. crassa, we also addressed (i) if N. crassa exhibits “open” or “closed” mitosis; (ii) if nuclear division is required for germ tube formation, CAT formation, and/or CAT fusion; (iii) if the cell cycle arrests during CAT induction, homing, and/or fusion; (iv) if CAT fusion is microtubule and/or actin dependent; and (v) if microtubules and associated dynein/dynactin motors are essential for nuclear migration through fused CATs.
MATERIALS AND METHODS
Strains, culture conditions, and production of conidia.
Neurospora strains are listed in Table 1. Strains were maintained and grown on solid Vogel's minimal medium with 2% (wt/vol) sucrose (12). Macroconidia were harvested from 4- to 5-day-old cultures grown at 25°C in constant light.
Table 1.
Neurospora strains used in this study
| Strain | Genotype | Protein studieda | Origin or referenceb |
|---|---|---|---|
| WT 74a | mat a | NAc | FGSC4200 |
| WT 74-OR23-1VA | mat A | NA | FGSC2489 |
| N623 | mat A his-3 | NA | FGSC6103 |
| N2281-3 | mat A his-3+::Pccg-1-hH1+-sgfp+ | Histone H1 | 24 |
| N2282 | mat A his-3+::Pccg-1-hH1+-sgfp+ | Histone H1 | 24 |
| N2283 | mat a his-3+::Pccg-1-hH1+-sgfp+ | Histone H1 | 24 |
| N2505 | ridRIP1 mat A his-3+::Pccg-1-Bml1+-sgfp+ | β-Tubulin (BML) | 24 |
| GR53 | mat A his-3+::Pccg-1-son-1+-sgfp+ | Nucleoporin SON-1 | This study |
| NMF152 | mat a his-3+::Pccg1-hH1+-sgfp+; ro-1 | Dynein heavy chain RO-1 | 49 |
| NMF153 | mat a his-3+::Pccg1-hH1+-sgfp+; ro-3 | Dynactin p150Glued RO-3 | 49 |
| FGSC11947 | mat A; NCU08566::hph+ | Dynactin subunit RO-11 | FGSC11947 |
| FGSC11946 | mat a; NCU08566::hph+ | Dynactin subunit RO-11 | FGSC11946 |
| Edi160 | mat a; NCU03911::hph+ | CAP-1 | FGSC11697 |
| Edi 161 | mat A; NCU03911::hph+ | CAP-1 | FGSC11698 |
| Edi 239 | mat A; NCU07471::hph+ | CAP-2 | FGSC11855 |
| Edi 241 | mat a; NCU07471::hph+ | CAP-2 | FGSC11854 |
| Edi502 | mat a; NCU07171::hph+ | ARP2/3 complex subunit (ARP2) | FGSC11716 |
| Edi529 | mat a; NCU09572:: hph+ | ARP2/3 complex (ARPC3) | FGSC15057 |
| Edi532 | mat a; NCU01918:: hph+ | ARP2/3 complex (20 kDa) | FGSC15991 |
| HK107 | mat a; his-3+::Pccg-1-hH1+-sgfp+; NCU08566::hph+ | Dynactin subunit RO-11 | This study |
| HK108 | mat A; his-3+::Pccg-1-hH1+-sgfp+; NCU08566::hph+ | Dynactin subunit RO-11 | This study |
| NCAL003-4 | mat a bar+::Pccg-1-Lifeact-rfp | Lifeact-RFP | This study |
CAP-1 and CAP-2 are the F-actin-capping protein alpha and beta subunit family members, respectively. Orthologues in S. cerevisiae are Tpm2p for RO-11, Cap1p for CAP-1, Cap2p for CAP-2, Arp2p for NCU07171, Arc18p for NCU09572, and Arc19p for NCU01918.
FGSC, Fungal Genetics Stock Center.
NA, not applicable.
Nuclear labeling with GFP.
Nuclei of ro-11 mutants (FGSC11946 and FGSC11947) were labeled with H1-GFP by crossing. FGSC11946 (mat a; ro-11) and FGSC11947 (mat A; ro-11) were used as recipient strains to cross with the donor strains N2282 (mat A; hH1+-sgfp+) and N2283 (mat a; hH1+-sgfp+), respectively (Table 1). Crosses were performed on solid synthetic crossing medium (12) at 25°C under constant light. Conidia collected from 5-day-old cultures were used as males for fertilization. Fourteen days after fertilization, ascospores were collected from petri dish lids, heat shocked at 60°C for 1 h, and spread onto Vogel's minimal medium supplemented with 150 μg/ml hygromycin B. Single germinated ascospores were isolated and germinated, and individual GFP-expressing colonies were selected under a Nikon SMZ1500 fluorescence stereomicroscope. Progeny with HygR and H1-GFP+ was crossed with FGSC4200 and FGSC2489 to screen for mating type. N2281-3A (mat A his-3+::Pccg-1-hH1+-sgfp+) (24) was crossed to ro-1 and ro-3 strains to obtain a homokaryotic hH1+-sgfp+ ro-1 or hH1+-sgfp+ ro-3 strain (49).
The son-1 gene was labeled with GFP by integrating a 1,211-bp PCR fragment containing the son-1 coding region into XbaI- and XmaI-digested pMF272 and transforming strain N623 with the resulting plasmid, pMF361. Primary transformants were obtained by selection on minimal medium and screened as described previously (24). Primers used were SON1XAF (5′-GCCTCTAGAGGCGCGCCTAACATGGCTGGTCT-3′) and SON1XFR (5′-GCCCCCGGGCCGGCCCCTCTTCTTAACGCTCG-3′).
F-actin labeling with Lifeact-RFP.
A 1.4-kb EcoRI fragment encoding Lifeact (52), a 17-amino-acid peptide constituting the N terminus of the yeast actin-binding protein Abp140 (5, 75), and linked to the N terminus of tdTomato (Lifeact-red fluorescent protein [RFP]) was integrated into pBARGRG1 (46) to generate pAL2-Lifeact. Junctions of constructs were verified by sequencing. The expression of the Lifeact-RFP fusion protein was under the control of the inducible N. crassa ccg-1 promoter. Lifeact-RFP-expressing strains were generated as described previously (24), by transforming pAL2-Lifeact into FGSC4200. Transformants were selected on nitrogen-free Vogel's minimal medium containing Ignite (“Basta,” as described in reference 45) and screened as described above. Cells were harvested from subcultures, and the intracellular expression of Lifeact-RFP in 12 different isolates was analyzed by epifluorescence microscopy.
Heterokaryon formation for live-cell imaging.
Heterokaryons expressing multiple fusion proteins were formed by mixing suspensions of macroconidia of two individual homokaryons in a 1:1 ratio at concentrations of 105 to 106 conidia/ml. Heterokaryons were either observed directly under the microscope or incubated for 4 to 5 days to form a colony from which heterokaryotic macroconidia were harvested.
Quantification of nuclei and analysis of CAT fusion.
CAT fusion was quantified with a Nikon TE2000E inverted microscope with a 60×/1.2-numerical-aperture (NA) water immersion Plan Apo objective (Nikon, Kingston-Upon-Thames, United Kingdom). Quantification of mitosis involved the epifluorescence imaging of heterokaryons expressing the hH1-sgfp and Bml-sgfp genes (to label the nuclear histone H1 and cytoplasmic β-tubulin, respectively) under the control of the ccg-1 promoter (23). Mitotic nuclei were identified with the confocal microscope on the basis of the presence of mitotic spindles. For this, 200-μl drops of conidial suspension in liquid Vogel's minimal medium with 105 to 106 conidia per ml were placed into wells of an eight-well culture chamber slide (Nalge Nunc International, Rochester, NY) at 25°C. Nuclear size measurements were obtained from projection profiles of individual nuclei in optical sections obtained by confocal microscopy (see below for details).
Use of microtubule and actin inhibitors.
Macroconidia were incubated in the presence of 2 to 50 μg ml−1 of benomyl (Chem Service, West Chester, PA) to study the effects of the inhibition of microtubule polymerization on mitosis, germ tube formation, CAT induction, homing, fusion, and nuclear migration through fused CATs. Benomyl stocks were prepared at a concentration of 12.5 mg ml−1. Macroconidia were used at a concentration of 5.5 × 105 macroconidia ml−1 in eight-well culture chamber slides and incubated at 25°C for 6 h before the quantification of germination and CAT fusion. Two chamber slides per treatment plus controls without benomyl were tested. Time-lapse imaging was performed with images captured over the first 6 h of incubation (frame rate of 1 image/h).
To analyze the importance of actin during conidial germination, we used the actin polymerization blocker latrunculin B at a sublethal concentration of 40 μg ml−1 (diluted in dimethyl sulfoxide [DMSO] to a final concentration of <0.025%). Macroconidial germination and CAT fusion were analyzed in the same way as described above for benomyl treatments.
Controls for benomyl and latrunculin B treatments were performed in liquid Vogel's medium containing DMSO at the same concentration used with the diluted inhibitors.
Live-cell imaging.
Living conidia and conidial germlings were imaged by confocal laser scanning microscopy (CLSM), wide-field epifluorescence imaging, and deconvolution microscopy. All material was prepared for wide-field analysis by using the “inverted agar block” method (29).
For confocal laser scanning microscopy, a Bio-Rad Radiance 2100 system equipped with blue diode and argon ion lasers (Bio-Rad Microscience, Hemel Hempstead, United Kingdom) mounted onto a Nikon TE2000U Eclipse inverted microscope was used. GFP was imaged by excitation at 488 nm and fluorescence detection at 500/30 nm. The staining of cell walls was performed by the addition of 0.12 μM calcofluor white M2R (prepared from a 1.2 M stock in ethanol; Sigma, Welwyn Garden, United Kingdom) to macroconidial suspensions (105 to 106 conidia/ml) immediately after the conidia were harvested. Calcofluor white and GFP were imaged simultaneously by excitation at 405 and 488 nm, respectively, and fluorescence detection at 420/27 nm (for calcofluor white) and 500/30 nm (for GFP). A 60×/1.2-NA water immersion objective lens was used for imaging. Laser intensity and laser dwell time on individual germlings were kept to a minimum to reduce phototoxic effects. Simultaneous bright-field images were captured with a transmitted light detector. Where appropriate, Kalman filtering (n = 1) was used to improve the signal-to-noise ratio of individual images. Time-lapse imaging was performed at 1-min intervals for periods of up to 5 min or at longer time intervals for periods of up to 8 h. Images were captured with a laser scan speed of 166 lines per s and a resolution of 1,024 by 1,024 pixels by using Lasersharp 2000 software (version 5.1; Bio-Rad Microscience) and viewed with Confocal Assistant software (v. 4.02; www.nephrology.iupui.edu/imaging/software.htm). Further image processing was performed by using Paintshop Pro software (version 8.0; JASC, Reading, United Kingdom).
Wide-field epifluorescence imaging was performed with an inverted Nikon Eclipse TE 2000E microscope equipped with an Orca ER cooled charge-coupled-device (CCD) camera (Hamamatsu, Welwyn Garden City, United Kingdom), with excitation of GFP at 470/20 nm provided by a monochromator light source (Till Photonics, Graefelfing, Germany), a 500-nm dichroic filter, and a 515-nm-long-pass emission filter. Images were collected at 0.9 frames per s by using SimplePCI (v. 5.3.1) software (Compix Inc., Cranberry Township, PA).
Deconvolution microscopy was performed by using the DeltaVision RT microscope system (Applied Precision, Issaquah, WA) with a 100-W Hg light source. Excitation of GFP was at 490/20 nm, and fluorescence detection was done with a 510-nm dichroic mirror and a 528/38-nm emission filter. The excitation of RFP was performed at 555/28 nm, and fluorescence detection was done with a 570-nm dichroic mirror and a 617/73-nm emission filter. A Coolsnap HQ camera (Photometrics UK Marlow, Buckinghamshire, United Kingdom) was used to capture images. Image deconvolution and image projections were produced by using software from Applied Precision. Movies were manipulated and generated with ImagePro Plus 6.3 (Photometrics UK).
Nuclear diameter was measured with the “line profile” option of the Lasersharp software. Optical sections imaged with a 100×/1.4-NA objective and maximal profiles of individual nuclei were selected for measurement from a z stack of optical sections. For displaying the individual nuclei in Fig. 2A and B, the images were processed with Paintshop Pro software, the surrounding background was cropped, and individual selected nuclei were cloned and arranged in sequence for each time course.
Fig. 2.

Time courses of mitosis in multinucleate nongerminated conidia (A to C) and germ tubes (D and E). The interphase, prophase, metaphase, anaphase, and telophase stages of mitosis have been indicated where they can be clearly recognized. (A) Mitosis of a conidial nucleus with H1-GFP labeling. A bright fluorescent nuclear region (arrows) was often evident where a spindle pole body (SPB) should be located. The segregating H1-containing chromatin was connected by a “chromatin bridge” (*) from late anaphase to telophase. Bar, 2 μm. (B) Mitosis of a conidial nucleus labeled with β-tubulin-GFP. Microtubules were formed from a focal point presumed to be the SPB (arrows). During late prophase/early metaphase, microtubules bundled to form a compact spindle (*), which is evident from metaphase to telophase. Bar, 2 μm. (C) Mitosis in a heterokaryotic conidium in which two nuclei labeled with H1-GFP and β-tubulin-GFP were dividing asynchronously (only one nucleus divided during this time course). Astral microtubules are formed from the single SPBs (arrows) in nuclei during both interphase and early prophase. These microtubules often possessed a bright focal point where they were in contact with the plasma membrane (*). SPB duplication occurred during prophase (at 15 min). The spindle developed as a compact bundle of microtubules (15 min to 1 h). During this time, H1-GFP was associated with the spindle and by telophase had segregated to both SPBs (the chromatin bridge is masked by the BML-GFP in this image). Astral ray microtubules can be visualized at the 1-h time point. Bar, 4 μm. (D) Mitosis in a germ tube nucleus labeled with H1-GFP. In this sequence, the expression level of H1-GFP was much lower than that in C. Bar, 2 μm. (E) Germ tube nucleus labeled with SON-1-GFP to show the nuclear pores in the nuclear envelope. Telophase was easily observed because the bridge formed between the nuclei daughters. Projections of 12 deconvolved images are shown. Bar, 2 μm.
RESULTS
Heterokaryon formation by CAT fusion provides advantages for live-cell imaging.
Heterokaryons formed by CAT fusion provide a rapid and easy method to facilitate the imaging of multiple cellular organelles (or proteins) labeled with fluorescent markers in the same cell (Fig. 1 and 2, and see Fig. 4 and 5A). At the same time, this approach provides a convenient method to alleviate potential problems based on the presumed overexpression of fusion proteins under the control of the ccg-1 promoter (Fig. 1B and D and 2C and D) (4, 20, 24).
Fig. 1.

Timing of fluorescent protein labeling of heterokaryons formed by CAT fusion. (A and B) Fused conidial germlings (one labeled with H1-GFP and the other labeled with β-tubulin-GFP [BML-GFP]), one 9 min (A) and the other 53 min (B) after fusion. At the 9-min time point, BML-GFP has become incorporated into microtubules derived from the left-hand germling with nuclei carrying the hH1-sgfp gene, while HI-GFP has not yet labeled the nuclei in the germling on the right-hand side with nuclei carrying the bml-sgfp gene. Microtubules passed through the fused CATs (f) before nuclei did. The location of the spindle pole body (arrow) with associated microtubules is shown at 53 min. C, conidium; GT, germ tube. Bar, 10 μm. (C) Conidia from a homokaryotic H1-GFP strain. Each conidium has several nuclei. (D) Conidia from a heterokaryotic H1-GFP plus β-tubulin-GFP strain visualized by confocal microscopy. Variable levels of GFP expression were observed for different conidia. Bar, 5 μm.
Fig. 4.
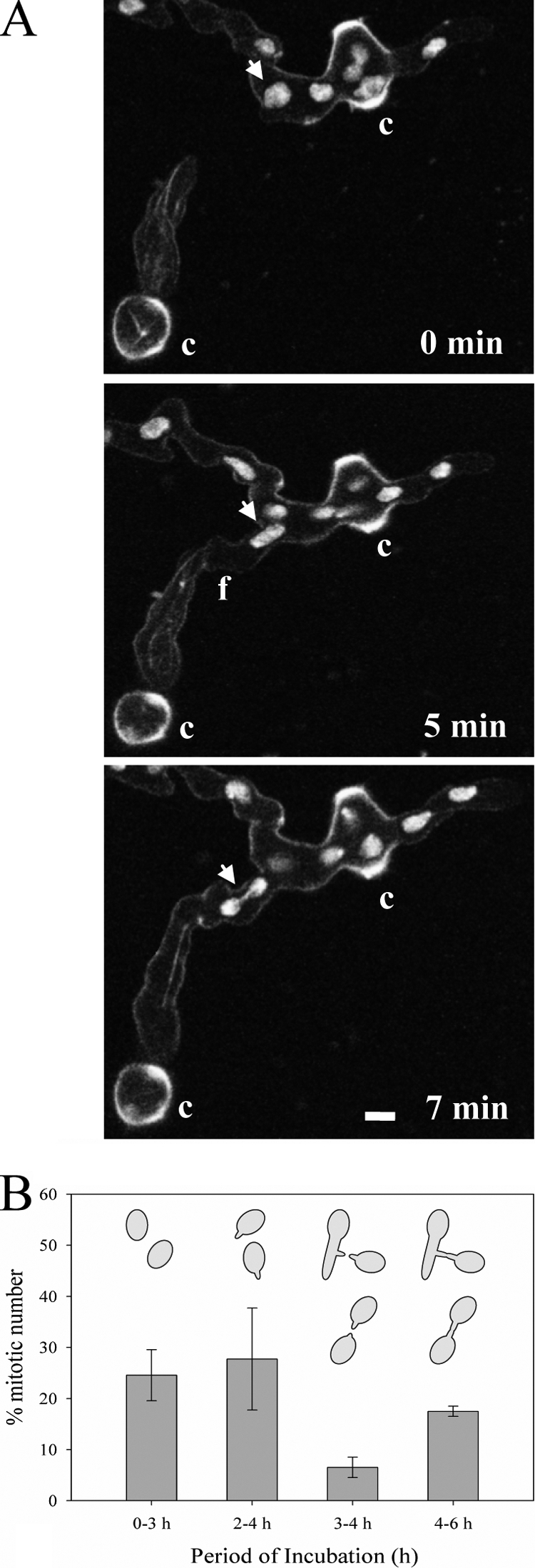
Cell cycle arrest during CAT homing. (A) CATs formed from germ tubes before (0 min) and after (5 and 7 min) fusion visualized by confocal microscopy. The upper conidial germling was labeled with H1-GFP, while the lower germling was labeled with β-tubulin-GFP. Bar, 5 μm. (B) Percentages of nuclei undergoing mitosis in ungerminated macroconidia (0 to 3 h), in macroconidia possessing only germ tubes (2 to 4 h), in macroconidia or macroconidial germlings possessing CATs (3 to 4 h), and in macroconidia or macroconidial germlings that have undergone CAT fusion (4 to 6 h). The time periods selected for the analysis of mitosis in these four different cell types were those in which the production of the individual cell types was maximal. Mitotic nuclei were identified by the presence of thick microtubule bundles in spindles labeled with BML-GFP.
Fig. 5.

Microtubular and nuclear organization and dynamics visualized by confocal and bright-field microscopy. (A) Two adjacent optical sections (0.5 μm apart) of a heterokaryotic conidium and germ tube showing five nuclei (n) after labeling with H1-GFP and β-tubulin-GFP, visualized by confocal microscopy. These two optical sections were selected from a continuous z stack of 20 optical sections captured in 0.5-μm steps. Arrows indicate the locations of spindle pole bodies with associated microtubules. Nuclei appear to be linked via microtubules. A large vacuole (v) seems to have pushed two of the nuclei to the periphery of the conidium. C, conidium; GT, germ tube. Bar, 5 μm. (B) Lack of inhibition of CAT fusion and homing by treatment with the microtubule-depolymerizing drug benomyl (40 μg ml−1). Two hours after treatment, the microtubules were reduced to punctate spots. Bar, 10 μm. (C) Nuclei labeled with H1-GFP migrate through fused CATs visualized by combined confocal and bright-field microscopy. (D) Benomyl treatment prevents the formation of long germ tubes. An H1-GFP-labeled strain was incubated with benomyl (35 μg ml−1) for 6 h visualized by combined wide-field fluorescence and differential interference contrast (DIC) microscopy. Small buds (arrow) may represent the initiation of germ tube formation, but these buds did not elongate further. Bar, 5 μm.
Homokaryotic conidia expressing hH1-sgfp (histone H1, green nuclei) were allowed to fuse with homokaryons expressing Bml-sgfp (β-tubulin, green microtubules). In the resulting heterokaryons, we noted that it took longer for H1-GFP to appear in the previously unlabeled nuclei than for BML-GFP to label previously unlabeled microtubules (Fig. 1A and B). Usually, within 2 min after successful CAT fusion (i.e., after cytoplasmic continuity between conidial germlings had been achieved), GFP-labeled microtubules extended throughout the fused CATs. Microtubules were frequently associated with a small region on the surface of each nucleus, which presumably represents the location of the spindle pole body (SPB) (Fig. 2A to C, and see Fig. 5A). Astral microtubules were formed from the SPB, which acts as a microtubule organizing center (MTOC) with a classic hub and spoke array type of organization. Many of these astral microtubules seemed to make contact with points at the plasma membrane (Fig. 2C). BML-GFP first became visibly incorporated into microtubules of the originally unlabeled homokaryon within ∼9 min of cytoplasmic continuity being achieved (Fig. 1A). In contrast, with heterokaryons initially containing labeled nuclei and nonlabeled nuclei, it took 20 to 30 min to acquire any detectable H1-GFP fluorescence; the fluorescent nuclei were best visualized >50 min following CAT fusion (Fig. 1B). This unexpected delay in GFP labeling of nuclei was consistently observed in over 30 pairs of fused germlings. We could not find convincing evidence of a spatial gradient in nuclear H1-GFP. However, H1 accumulation was not uniform among the nonexpressing nuclei, and it was not necessarily the nonexpressing nuclei that were closest to the expressing nuclei that accumulated H1 first.
The overall level of GFP labeling was lower in heterokaryotic macroconidia that contained both nuclear and microtubule GFP labels after CAT fusion. This finding suggests that the expression of both fluorescent fusion proteins under the control of the ccg-1 promoter was diluted in heterokaryons because they contained a reduced number of nuclei expressing each fusion protein. The relative fluorescences of the two fusion proteins in heterokaryons varied between macroconidia and germlings and were correlated with the ratio of nuclei that were expressing hH1-sgfp and/or Bml-sgfp or those that lacked GFP (Fig. 1C and D).
Mitosis is asynchronous and relatively slow in nongerminated conidia.
The different stages of the mitotic nuclear cycle (i.e., prophase, metaphase, anaphase, and telophase) were identified by using a combination of conventional cytogenetic criteria such as nuclear shape, nuclear size, chromatin appearance, and pairs of closely associated small nuclei (indicative of telophase) (15, 26, 55, 56).
In this study, mitosis was monitored by time-lapse imaging of nongerminated macroconidia and germ tubes in (i) homokaryons labeled with H1-GFP, (ii) homokaryons labeled with BML-GFP, and (iii) heterokaryons containing both fusion proteins. Confocal images for each time course were optimally collected at 1- to 3-min time intervals, and selected images are shown in Fig. 2A to D. As expected, nuclei in both nongerminated macroconidia and germ tubes exhibited asynchronous mitotic divisions (Fig. 2C, and see Movie S1 in the supplemental material). Nongerminated macroconidia observed immediately after harvesting were typically multinucleate. In the H1-GFP strain (N2283), the number of nuclei per macroconidium varied between 1 and 4 (mean, 1.97; n = 600). The nuclei in nongerminated macroconidia and germ tubes varied from being spherical to being pear shaped, as previously reported for nuclei in vegetative hyphae (23, 24). Spherical nuclei varied between 2.3 and 4.3 μm in diameter. These nuclei should be in interphase or early prophase because of the lack of a spindle-induced change in nuclear morphology (see below). It was predicted that the larger nuclei would be in the G2 or M phase in the nuclear cycle following DNA duplication (S phase). This was confirmed for nuclei identified as being in early prophase (see below), which were always at the larger end of the nuclear size spectrum. Mitosis was initiated 30 to 60 min after hydration and typically took 1.0 to 1.5 h to complete for nongerminated macroconidia at 25°C (Fig. 3). Approximately 25% ± 5.0% (n = 500) of the nuclei were found to be undergoing mitosis 1 h after hydration.
Fig. 3.

Rates of conidial germination and CAT fusion and the number of nuclei in conidia/conidial germlings during the first 5 h of incubation. The nuclear number significantly increased only after 4 h, when germination was near maximal and over 50% of the conidial germlings had undergone CAT fusion.
During the first 30 min following macroconidial hydration, nuclei varied between being spherical and being pear shaped in interphase until prophase/metaphase (Fig. 2A to D). Spherical nuclei exhibited rotating and tumbling or saltatory movements. Motile nuclei were typically pear shaped, and the apices of these nuclei were usually oriented in the direction of movement and often moved along the inner surface of the plasma membrane. At this stage, bright spots of BML-GFP labeling appeared in close proximity to the plasma membrane. These fluorescent spots were smaller (0.5 ± 0.3 μm in diameter) than putative SPBs (1.2 ± 0.4 μm in diameter) (Fig. 2C, and see Movie S2 in the supplemental material). These spots may represent cytoplasmic MTOCs (74) and were always present whether mitosis was occurring or not. The majority of cytoplasmic astral microtubules extended to the plasma membrane and curved around the nucleus. They exhibited dynamic instability growing and shrinking to and from the focal point of high H1-GFP fluorescence, which probably represents the SPB (Fig. 2B and Movie S3).
We monitored mitosis in heterokaryotic conidia in which nuclei were labeled with H1-GFP and BML-GFP (Fig. 2C). In Fig. 2C, only one of the nuclei is undergoing mitosis. Microtubules are formed from the single SPBs of both nuclei during interphase and early prophase. SPB duplication during prophase is visible at the 15-min time point. Exactly when SPB duplication starts and how long it takes are as yet unclear. The precise length of prophase was difficult to determine by using H1-GFP imaging alone. Here, prophase was defined as the stage during which the nuclei increased further in size compared with interphase nuclei and during which the H1-GFP-labeled chromatin started to become distributed in a nonuniform fashion.
During late prophase and early metaphase, microtubules bundled to form a compact spindle that was ∼0.5 μm wide and visible within mitotic nuclei within 15 min following interphase and <1 h until the end of telophase. The H1-labeled chromatin became more obviously heterogeneously distributed, and the nuclear envelope became highly invaginated during metaphase (Fig. 2A and C). Astral microtubules were not evident during metaphase. At the start of metaphase, SPB division was completed, and the two daughter SPBs migrated away from each other in association with the nuclear envelope. We were not able to achieve sufficient resolution to observe the SPB position (i.e., if they were on the outer or inner surface of the nuclear envelope or embedded within it). No distinct metaphase plate (i.e., the plane of the equator of the spindle along which chromosomes are positioned during metaphase) could be distinguished.
During anaphase, the region labeled by H1-GFP became stretched out and gradually polarized toward the spindle poles. H1-GFP was clearly associated with the spindle for the same length of time. After 1 h, the distribution of H1-GFP (Fig. 2A) was completely masked by the spindle shown in Fig. 2C because the latter was imaged in a heterokaryon also containing BML-GFP. This “chromatin bridge” (when imaging H1-GFP) or “telophase bundle” (when observed with BML-GFP) was maintained from late anaphase until telophase (see Movie S1 in the supplemental material). Astral microtubules started to reappear during anaphase.
Metaphase and anaphase each took ∼20 to 30 min and telophase took ∼30 to 40 min to complete. During telophase, the concentrated H1-GFP rounded up at each spindle pole where the two daughter nuclei became defined. The telophase bundle (or chromatin bridge), which is characterized as a dense line of material at the midline of the cell during telophase (16), was less prominent (<5.0 μm long) in macroconidia than in germ tubes (<6 μm long); in both cells, it was 1.0 ± 0.5 μm wide. The remains of the telophase bundle joined the two daughter nuclei (Fig. 2D). Telophase ended when the telophase bundle disappeared, the SPB was reestablished, and cytoplasmic microtubules formed from the SPB exhibited normal dynamic instability again.
Mitosis occurs faster in germ tubes than in nongerminated macroconidia.
After ∼2.5 h of incubation at 25°C, germ tubes started to form. Mitosis was approximately three to four times faster (it took 16 to 20 min to complete) in germ tubes than in nongerminated macroconidia. After a ∼2-h lag period in nongerminated macroconidia, the number of nuclei participating in mitosis was fairly constant, only increasing after ∼4 h of incubation, when germination was maximal (Fig. 3). Nuclei were either rounded or pear shaped during this period. Metaphase and anaphase each took ∼4 min to complete. Telophase took ∼3 min, at which time the H1-GFP-labeled chromatin bridge was observed (Fig. 2D). Other features of mitosis in germ tubes were similar to those observed for macroconidia, except that telophase nuclei linked by a chromatin bridge could be further apart (<6 μm).
A form of “closed mitosis” occurs in N. crassa.
In order to visualize the nuclear envelope during mitosis in germinating conidia, we imaged a strain that expressed the nuclear pore complex marker SON-1 tagged with GFP (see Movie S4 in the supplemental material). Time courses of mitosis for 10 germlings were monitored by deconvolution microscopy with image frame rates of <1 Hz. SON-1-GFP localized in a discontinuous fashion along the nuclear envelope, and nuclear pores decorated with SON-1-GFP were visible throughout mitosis (Fig. 2E).
Mitosis is arrested during CAT homing.
CATs started to form at the same time as germ tubes (i.e., after ∼2.5 h). Few microtubules were observed in growing CATs, but those that were present exhibited dynamic instability similar to that of the microtubules in germ tubes. No unusual changes in the behavior of either nuclei or microtubules were observed at the sites of CAT induction in conidia or germ tubes. However, nuclear behavior changed when CATs homed toward each other. First, homing CATs rarely contained a nucleus; <0.5% (n = 300) of homing CATs and 7.6% ± 1.0% (n = 300) of nonhoming CATs contained nuclei. Second, a significantly lower proportion of nuclei (6.5% ± 4.3%; n = 300) were found to undergo mitosis in macroconidia with homing CATs than in other cell types (24.6% ± 7.3% [n = 300] of nongerminated macroconidia, 27.8% ± 10.3% [n = 300] of macroconidia with germ tubes only, and 17.5% ± 2.3% [n = 300] of macroconidial germlings that had undergone CAT fusion contained mitotic nuclei) (Fig. 4A). Third, nuclei in mitosis after early prophase were never observed in macroconidia with homing CATs (n = 20), which suggests that mitosis may be arrested (Fig. 4B). Furthermore, nuclei did not enter mitosis within ∼1 h following fusion (observed for 20 pairs of fused germlings). Figure 4 at 7 min shows a migrating dividing nucleus in telophase that will have entered mitosis before a macroconidium with CAT homing had occurred.
Adjacent nuclei appear to be linked via microtubules.
Microtubules continued to exhibit dynamic instability and transiently extended to the apical plasma membrane of growing germ tube tips as shown previously (24). Adjacent nuclei in germ tubes commonly appeared to be attached to the same microtubule bundles. This was best visualized by tracking individual microtubules in adjacent optical sections (Fig. 5A). These points of possible attachment were often found to be in close association with highly fluorescent foci of H1-GFP (the presumed location of SPBs) (Fig. 5A). The putatively attached nuclei were organized more or less linearly in single rows and moved mostly in the same direction and at the same rate as the extending germ tube tip. During germ tube branching, an occasional nucleus would move in a retrograde fashion.
Nuclear division and microtubules are not required for CAT induction, homing, and fusion.
During CAT homing prior to fusion, microtubules in the CAT tips exhibited marked dynamic instability. After fusion, the microtubules of the parent germlings extended through the fused CATs and became intermingled with each other, as reported previously (58). Microtubule bundles seemed to link nuclei within the fused germlings (Fig. 5A).
The microtubule-depolymerizing drug benomyl was used to determine the importance of microtubules and nuclear division for CAT induction, homing, and fusion. When used at a concentration of 40 μg ml−1, benomyl caused microtubules to be reduced into small punctate spots or occasionally very short microtubules <3 μm long (Fig. 5B). Benomyl treatment also prevented nuclei from forming spindles and thus blocked mitosis in nongerminated macroconidia and in macroconidial germlings. After the addition of benomyl, <0.1% of macroconidia went on to form germ tubes longer than 10 μm, although some formed bud-like structures, which may represent the very early stages of germ tube formation (Fig. 5D). Quantification of the effects of benomyl treatment on conidial germination (emergence of a germ tube or a CAT) and CAT fusion at 5 h showed that 92% ± 2.5% (n = 300) of the benomyl-treated macroconidia underwent germination (untreated control, 91.5% ± 6.36%; n = 300) and that 89% ± 0.5% (n = 300) underwent CAT fusion (untreated control, 66% ± 5.67%; n = 300) (Fig. 6).
Fig. 6.

Quantification of effects of the antimicrotubule drug benomyl (40 μg ml−1) and the antiactin drug latrunculin B (40 μg ml−1) on macroconidial germination and CAT fusion. Conidial germination represents the percentage of macroconidia possessing germ tubes and/or CATs. CAT fusion was reduced greatly with the sublethal latrunculin B treatment.
The time for complete microtubule turnover (i.e., the time for microtubules to be reduced to punctate spots) was measured in conidial germlings that had been incubated for 4 h. The cytoplasmic microtubule turnover time was 40 ± 8.154 s (n = 6), and the spindle microtubule turnover time was 10 ± 3.03 min (n = 4). CAT homing continued to occur in the absence of microtubules after benomyl treatment (Fig. 5B).
Nuclei exhibit oscillatory motility when migrating through fused CATs.
Nuclei migrated through fused CATs, although this did not occur immediately after fusion. The nuclei of fused germlings exhibited oscillatory motility, moving back and forth from the fusion pore before eventually migrating through it. During this period, transiting nuclei were elongated and commonly squeezed though the fusion pore when it was still small (∼1 μm in diameter). When CAT fusion occurred between CATs that had emerged directly from macroconidia (58), the first nucleus passed through the fusion pore ∼1 h after cytoplasmic continuity had been established. In cases where CAT fusion occurred between CATs derived from germ tubes (58), the first nucleus passed through much earlier (after ∼30 min). Mitotic nuclei were sometimes observed migrating through fused CATs (Fig. 4A). In the presence of benomyl or latrunculin B, the velocity of nuclear migration was reduced, but nuclear migration was not prevented (Fig. 5C). When CATs formed directly from macroconidia fused, this was usually followed by the emergence of a germ tube from one or both of the macroconidia or from the fused CATs themselves (58). Once a new germ tube had appeared, the movement of the nuclei in the fused germlings was directed into the new germ tube (see Movie S5 in the supplemental material).
The speed of nuclear migration through fused CATs depended on the type of CAT fusion. Nuclear migration between fused CATs formed from germ tubes occurred quickly (∼3 min), migration between a fused macroconidium and germ tube was slower (∼20 min), and migration between fused macroconidia was slowest (20 to 60 min).
Dynein/dynactin is not essential for nuclear migration through fused CATs.
Given that nuclear movement in filamentous fungi was reported to be dependent on dynein and its activator, dynactin (e.g., see references 9, 38, 41, and 54), we analyzed a possible role for dynein/dynactin during nuclear migration through fused CATs by comparing the wild type with ro-1 (dynein heavy chain), Δro-3 (dynactin subunit), and Δro-11 (dynactin subunit) mutants. After incubation for 6 h, the Δro mutants had undergone 65 to 89% germination, compared with ∼82% in the wild type. At this time point, CAT fusion was absent (<1%) in the Δro-1 mutant, was 22% ± 5% in the Δro-3 mutant, and was 40% ± 3% in the Δro-11 mutant, compared with 68% ± 2.5% in the wild type. Only after 8 h did CAT fusion between germlings start to occur in the Δro-1 mutant. The dynein/dynactin mutants did not show any aberrant morphological defects with regard to CAT induction or homing. As previously reported (39), nuclear positioning was clearly impaired in the germ tubes of these three mutants, and their nuclei usually remained within the spore body (Fig. 7B and C). Both types of nuclear movement (i.e., saltatory movement in nongerminated conidia and migratory movement in germ tubes) were inhibited in the mutants, but nuclear migration still occurred through fused CATs (Fig. 7C, and see Movie S6 in the supplemental material).
Fig. 7.

Nuclear distribution and behavior in the wild type and ropy (dynein/dynactin) mutants visualized by combined wide-field, bright-field, and fluorescence microscopy. Nuclei were labeled with H1-GFP. (A) Network of fused wild-type germlings showing a more-or-less random distribution of nuclei. (B) ro-1 (dynein heavy chain) mutant showing CATs derived from germ tubes homing toward each other (arrow). The nuclei are concentrated in or close to the conidia. (C) Δro-11 (dynactin subunit) mutant showing nuclear migration (arrow) through fused CATs (f) after incubation for 7 h. f, points of CAT fusion. Bars, 15 μm.
Actin is required for CAT induction, homing, and fusion.
We used the actin polymerization inhibitor latrunculin B to assess the importance of actin during CAT induction and fusion and its impact on nuclear migration through fused CATs (Fig. 7A and B). The amount of CAT fusion was significantly reduced after treatment with a sublethal concentration of latrunculin B (40 μg ml−1) that did not significantly inhibit germination. At higher latrunculin B concentrations, CAT fusion was completely inhibited, and macroconidial germination became inhibited as well (data not shown). As indicated above, nuclei still moved and were able to migrate through fused CATs in the presence of latrunculin B, but these processes were delayed.
The Arp2/3 complex is a seven-subunit protein complex that promotes actin assembly, dynamics, and cross-linking (36, 57). Homokaryons of several deletion mutants encoding protein subunits within the Arp2/3 complex have been reported to be ascospore lethal (http://www.fgsc.net). We analyzed heterokaryons in which some nuclei carried the deletion alleles NCU07171, NCU09572, and NCU01918, respectively (Table 1) (these strains have not been described previously; they were generated by using a method described in reference 11). After incubation for 6 h, we found that the percentage of macroconidial germination was similar to that of the wild type in FGSC15057 (NCU09572) but significantly lower in FGSC11716 (NCU07171) (P < 0.05) and FGSC15991 (NCU01918) (P < 0.01). However, compared to the wild type, CAT fusion was much more reduced (Fig. 8) than germination in all three mutants. This further supports an important role for actin during the process of CAT fusion.
Fig. 8.

Greater inhibition of CAT fusion than conidial germination in deletion mutants defective in different protein subunits within ARP2/3 protein complex.
Lifeact-RFP reveals accumulation of F-actin cables and patches in CAT tips during homing and fusion.
To investigate F-actin dynamics during the process of CAT fusion, we constructed a Lifeact-RFP expression vector for N. crassa and randomly integrated it into the genome of a wild-type strain. The overexpression of the fusion protein by using the ccg-1 promoter had no obvious effects on cell morphology or general development.
Lifeact-RFP labeled F-actin cables and patches in living cells of N. crassa (Fig. 9). CAT formation coincided with a significant increase in F-actin fluorescence in the CAT tip, intensified during homing, and peaked while CATs attached to each other (Fig. 9A). Upon fusion pore formation, fluorescence at the fusion site began to decrease, leaving a few patches behind, which became evenly dispersed around this region (Fig. 9B). After benomyl treatment, the overall localization and abundance of actin cables and patches remained essentially unchanged, and CAT-mediated cell fusion progressed normally (Fig. 9C and D).
Fig. 9.

F-actin dynamics during CAT-mediated cell fusion are unaffected by benomyl treatment. Shown are pairs of Lifeact-RFP-expressing conidia of N. crassa during pre- and postfusion stages. (A) In the untreated control, fluorescence intensities of actin cables and patches are concentrated at the CAT tips during homing and peak as cell-to-cell contact is established. (B) After cytoplasmic continuity has been achieved by fusion pore formation, the fluorescence intensity has decreased, and the remaining patches are dispersed. (C and D) In the presence of 3 μg/ml benomyl, germ tubes are unable to become elongated, and the macroconidia show extended isotropic growth and become swollen. The localization and abundance of actin cables and patches remained essentially unchanged after benomyl treatment. Arrows mark sites of CAT contact/fusion. Scale bars, 5 μm.
DISCUSSION
In this paper we provide the first detailed live-cell analysis of nuclear behavior and mitosis during colony initiation in N. crassa (summarized in Fig. 10). Novel aspects that have been revealed in this study are as follows: (i) N. crassa exhibits a form of “closed mitosis”; (ii) mitosis is inhibited during CAT homing; (iii) microtubules are not required for CAT induction, homing, or fusion, nor are they required for nuclear migration through fused CATs; and (iv) actin is the cytoskeletal element of primary importance at different stages during CAT fusion.
Fig. 10.

Summary of the main features of the different stages of mitosis in a nongerminated macroconidium of N. crassa. During interphase, nuclei (dark green) are very mobile, rotate, and are rounded or pear shaped. When rounded, they are ∼2 μm in diameter. Astral ray microtubules exhibiting pronounced dynamic instability (red lines) form from a single spindle pole body (SPB) (yellow spot) on the surface of the nucleus. During prophase, nuclei are very mobile, rotate, and are rounded or pear shaped. When rounded, they are <4 μm in diameter. The SPB duplicates, and the two daughter SPBs start to move apart. Astral ray microtubules continue to exhibit pronounced dynamic instability. Condensed chromatin starts to appear toward the end of prophase. During metaphase, nuclei are less mobile, rotate less, and have an irregular shape. Astral microtubules seem to disappear. Condensed chromatin (darker green regions within the nucleoplasm) becomes prominent. A spindle composed of a bundle of microtubules (thick red line) grows in length between the SPBs that continue to move apart. During anaphase, future daughter nuclei start to move apart and start to become mobile and rotate again. Short astral microtubules start to form again. The SPBs are located at the two poles of the dividing nuclei with a long spindle bundle between them. During telophase, constriction between future daughter nuclei becomes prominent. Nuclear mobility and rotations are similar to those in anaphase. A long spindle composed of a microtubule bundle (termed “telophase bundle”) has chromatin (the so-called “chromatin bridge”) associated with it. This is visible until the daughter nuclei separate during nucleokinesis. During interphase, daughter nuclei with variable morphologies have been formed (see above).
Neurospora exhibits features of typical “closed mitosis” and forms chromatin bridges and telophase bundles.
Until recently, ascomycete fungi have been described as exhibiting “closed mitosis,” in which the nuclear envelope remains intact and chromosome separation to the poles of the spindle occurs inside the nucleus (25). This has been best characterized for Saccharomyces cerevisiae (67). However, recent reports showed that Aspergillus nidulans and the basidiomycete Ustilago maydis undergo a form of “open mitosis.” In A. nidulans, the nuclear pore complex breaks down partially to allow the influx of regulatory molecules (13–15). In U. maydis, the nuclear envelope breaks down after chromosomes have migrated from the old nuclear envelope to the new envelope (69). Observations of mitosis in cells expressing GFP-labeled nucleoporin SON-1 strongly suggest that N. crassa, like yeasts, exhibits a form of closed mitosis, as SON-1 remains visible in the nuclear pore complex throughout the nuclear cycle. This is in contrast to the situation for A. nidulans, in which the nucleoporin SONA (the homologue of N. crassa SON-1) transiently disappears during metaphase (15).
Our observations support the idea that the influx of proteins before and during mitosis in N. crassa is more highly regulated than that in A. nidulans and may thus be more reminiscent of the situation in budding yeast (reviewed in reference 13). The more highly regulated protein influx in N. crassa may provide a convenient way to regulate asynchronous division in Neurospora, which contrasts with the synchronous or semisynchronous division that occurs in A. nidulans (28, 56).
A closed mitosis may explain the presence of distinct, bright telophase bundles (when observing β-tubulin-GFP) and chromatin bridges (when observing H1-GFP). This feature is not evident during mitosis in A. nidulans. Nevertheless, organisms in which the nuclear envelope breaks down completely, e.g., mammalian HeLa cells or the marine diatom Lithodesmium undulatum (10, 37), also have telophase bundles.
Mitosis takes longer in ungerminated N. crassa macroconidia than it does in germ tubes (Fig. 3). Previously, it was shown that most of the nuclei were arrested in the G1 or G2 phase during germination in N. crassa (34). At this time, most nuclei are thought to transcribe genes required for germination (32). This may explain why mitosis takes longer in ungerminated macroconidia than in germ tubes.
In N. crassa, the nuclear envelope becomes invaginated at the end of metaphase, which may be important for the segregation of the nucleolus at the end of metaphase, as was recently shown for A. nidulans (71). In A. nidulans, astral microtubules disassemble during mitosis (30), as is the case for N. crassa (this study). This contrasts with budding yeast and Ustilago maydis, in which astral microtubules do not disassemble during mitosis (63, 68). In N. crassa, the presence of the nonmitotic nuclei together in the same cytoplasm with mitotic nuclei (asynchronous mitosis) explains why microtubules are still observed during mitosis.
We observed that following heterokaryon formation after CAT fusion between homokaryons expressing GFP-labeled tubulin and H1-GFP, respectively, H1-GFP took much longer (>30 min) to become incorporated into unlabeled nuclei compared with the rapid incorporation of GFP-labeled tubulin into unlabeled microtubules (<8 min). One possible reason for this delay in nuclear H1-GFP labeling may be a relatively long turnover time for the H1 histone in nuclei. Differences in the timing of the nonfluorescent nuclei becoming fluorescent may relate to the nuclei being in different stages of the cell cycle.
Mitosis is inhibited during CAT homing.
During CAT chemoattraction, nuclei did not enter CATs, and mitosis was arrested throughout the homing germlings. The significance of this is unclear. During nonself signaling in budding yeast, peptide sex pheromones produced by mating yeast cells initiate cell cycle arrest in G1 phase, promote actin nucleation and polymerization leading to the polarized outgrowth of mating projections, and induce the expression of mating-related proteins that are involved in the formation of mating projections and cell-cell fusion (17). The self-signaling ligand involved in CAT chemoattraction is unknown (21, 50), but we hypothesize that it may be the primary signal that initiates cell cycle arrest in preparation for CAT fusion.
Microtubules and dynein/dynactin are not required for CAT fusion or nuclear migration through fused CATs.
Microtubules play important roles in tip growth and organelle transport within hyphae. However, these roles are not well understood and may differ in different types of hyphae and in different species. The available evidence suggests that microtubules are required for maintaining the fast growth rate, the shape of the tip, and the directionality of mature vegetative hyphae (30, 41, 53). Horio and Oakley (30) reported previously that cytoplasmic microtubules in A. nidulans disappear altogether during the synchronized mitoses that occur within germ tubes but that germ tubes continued to grow during this period. In the present study, we found that cytoplasmic microtubules persist during the asynchronous mitoses that occur within the multinucleate macroconidia and germ tubes of N. crassa. The complete depolymerization of microtubules with benomyl prevented the formation of long germ tubes altogether, although small buds, suggestive of initiated germ tubes, were formed by benomyl-treated macroconidia. However, the benomyl treatment did not significantly inhibit the processes of CAT induction, homing, or fusion, nor did it prevent nuclear migration through fused CATs. Furthermore, we showed that nuclear division is not essential for progression through the process of CAT induction, homing, or fusion because the benomyl treatment blocks mitosis at metaphase by inhibiting spindle formation. The turnover time for cytoplasmic microtubules was shown to be ∼40 s, and for spindle microtubules, it was ∼10 min.
The migration and positioning of nuclei within fungal hyphae have been reported to be regulated by the microtubule-associated motor protein dynein and its activator, dynactin (9, 38, 41, 49). Our analysis of three ropy mutants that were defective in different components of the cytoplasmic dynein/dynactin complex confirmed its role in nuclear positioning because the nuclei of these mutants had a strong tendency to remain in the conidium after germination. However, we found that nuclear migration still occurred between fused germlings through fused CATs, indicating that this process does not require dynein/dynactin. Recently, it was shown that the movement and distribution of nuclei in mature vegetative hyphae appear to be determined by a combination of forces, with cytoplasmic bulk flow being a major determinant (49). Whether bulk flow plays a role in nuclear migration through fused CATs is not known.
Actin plays important roles in the whole process of CAT fusion.
F-actin has been demonstrated to play important roles in cell polarity regulation, septation, exocytosis, endocytosis, and organelle movement in fungi (6, 7, 27, 40, 42, 64, 65, 73). Three F-actin-containing structures are found in fungi: actin patches that are associated with endocytic vesicles (3, 7, 31, 72); actin cables that are assembled at sites of growth, where they form tracks for secretion and organelle transport (7, 8, 18, 19, 31, 47, 60, 61); and contractile actomyosin rings that are involved in septum formation (7, 27, 33, 40).
Recently, we used the Lifeact probe fused to either GFP or the red fluorescent protein TagRFP to image the F-actin cytoskeleton during colony initiation and growth of mature hyphae in living cells of N. crassa (7). During colony establishment, F-actin cables and patches localized to sites of active growth during the establishment and maintenance of the polarized growth of germ tubes and CATs. Upon CATs making contact and their polarized growth ceasing, actin cables gradually disappeared from the fusion site. Once cytoplasmic continuity was fully established, actin cables completely disappeared from the site of fusion, whereas cortical actin patches remained, suggesting that endocytic recycling may be required after fusion has taken place. The results obtained in the present study using Lifeact fused to a different fluorescent protein (tdTomato) were consistent with results obtained in a parallel study (7). However, due to the larger size of tdTomato and its tendency to dimerize, the utilization of the monomeric TagRFP as a red fluorescent marker is preferable.
Latrunculin A treatment was previously found to cause the rapid dissolution of the actin cytoskeleton in conidial germlings (7). In the present study, both conidial germination (germ tube and CAT formation) and CAT fusion were very sensitive to the less potent actin-depolymerizing agent latrunculin B, which is consistent with F-actin having important roles in these processes. The fact that benomyl treatment did not significantly inhibit the process of CAT induction, homing, or fusion did not prevent nuclear migration through fused CATs, or disrupt the actin cytoskeleton, strongly suggests that F-actin may be the primary cytoskeletal element involved in these activities. Further evidence supporting a key role for actin in CAT fusion came from an analysis of heterokaryotic strains that carried deletion alleles encoding components of the actin-nucleating Arp2/3 complex.
Overall, our results indicate that actin and microtubules have different roles during colony initiation compared with the growth of vegetative hyphae in the mature colony. CATs share important features in common with budding yeast. In particular, neither CATs nor yeast cells have a requirement for microtubule-mediated polarized growth or long-distance organelle transport (40). These features may be related to their small, determinate size, in contrast to mature vegetative hyphae, which tend to be much larger and exhibit indeterminate and faster growth.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lisete Chamma Davide and Blythe Nilson for suggestions on the manuscript. We also thank Jens Tilsner from the University of Edinburgh for providing the plasmid from which Lifeact-tdTomato was subcloned.
Funds for this research were provided by the BBSRC (grant number BB/E010741/1) to N.D.R., the Welcome Trust (VIP award) to M.G.R., and the OHSU Medical Research Foundation and American Cancer Society (grant number RSG-08-030-01-CCG) to M.F.
Footnotes
Supplemental material for this article may be found at http://ec.asm.org/.
Published ahead of print on 5 March 2010.
REFERENCES
- 1.Aist J. R. 2002. Mitosis and motor proteins in the filamentous ascomycete, Nectria haematococca, and some related fungi. Int. Rev. Cytol. 212:239–263 [DOI] [PubMed] [Google Scholar]
- 2.Aist J. R., Morris N. R. 1999. Mitosis in filamentous fungi: how we got where we are. Fungal Genet. Biol. 27:1–25 [DOI] [PubMed] [Google Scholar]
- 3.Araujo-Bazán L., Peñalva M. A., Espeso E. A. 2008. Preferential localization of the endocytic internalization machinery to hyphal tips underlies polarization of the actin cytoskeleton in Aspergillus nidulans. Mol. Microbiol. 67:891–905 [DOI] [PubMed] [Google Scholar]
- 4.Araujo-Palomares C. L., Castro-Longoria E., Riquelme M. 2007. Ontogeny of the Spitzenkörper in germlings of Neurospora crassa. Fungal Genet. Biol. 44:492–503 [DOI] [PubMed] [Google Scholar]
- 5.Asakura T., Sasaki T., Nagano F., Satoh A., Obaishi H., Nishioka H., Imamura H., Hotta K., Tanaka K., Nakanishi H., Takai Y. 1998. Isolation and characterization of a novel actin filament-binding protein from Saccharomyces cerevisiae. Oncogene 16:121–130 [DOI] [PubMed] [Google Scholar]
- 6.Barja F., Chappuis M.-L., Turian G. 1993. Differential effects of anticytoskeletal compounds on the localization and chemical patterns of actin in germinating conidia of Neurospora crassa. FEMS Microbiol. Lett. 107:261–266 [DOI] [PubMed] [Google Scholar]
- 7.Berepiki A., Lichius A., Shoji J.-Y., Tilsner J., Read N. D. 2010. F-actin dynamics in Neurospora crassa. Eukaryot. Cell 9:547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bretscher A. 2003. Polarized growth and organelle segregation in yeast: the tracks, motors and receptors. J. Cell Biol. 160:811–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruno K. S., Tinsley J. H., Minke P. F., Plamann M. 1996. Genetic interactions among cytoplasmic dynein, dynactin, and nuclear distribution mutants of Neurospora crassa. Proc. Natl. Acad. Sci. U. S. A. 93:4775–4780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Byers B., Abramson D. H. 1968. Cytokinesis in HeLa: post-telophase delay and microtubule-associated motility. Protoplasma 66:413–435 [DOI] [PubMed] [Google Scholar]
- 11.Colot H. V., Park G., Turner G. E., Ringelberg C., Crew C. M., Litvinkova L., Weiss R. L., Borkovich K. A., Dunlap J. C. 2006. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. U. S. A. 103:10352–10357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis R. 2000. Neurospora: contributions of a model organism. Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 13.De Souza C. P., Horn K. P., Master K., Osmani S. A. 2003. The SONB(NUP98) nucleoporin interacts with the NIMA kinase in Aspergillus nidulans. Genetics 165:1071–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Souza C. P., Osmani S. A. 2007. Mitosis, not just open or closed. Eukaryot. Cell 6:1521–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Souza C. P., Osmani A. H., Hashmi S. B., Osmani S. A. 2004. Partial nuclear pore complex disassembly during closed mitosis in Aspergillus nidulans. Curr. Biol. 14:1973–1984 [DOI] [PubMed] [Google Scholar]
- 16.Dustin P. 1978. Microtubules. Springer-Verlag, Berlin, Germany [Google Scholar]
- 17.Elion E. A. 2000. Pheromone response, mating and cell biology. Curr. Opin. Microbiol. 3:573–581 [DOI] [PubMed] [Google Scholar]
- 18.Evangelista M., Pruyne D., Amberg D. C., Boone C., Bretscher A. 2002. Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat. Cell Biol. 4:32–34 [DOI] [PubMed] [Google Scholar]
- 19.Fehrenbacher K. L., Yang H. C., Gay A. C., Huckaba T. M., Pon L. A. 2004. Live-cell imaging of mitochondrial movement along actin cables in budding yeast. Curr. Biol. 14:1996–2004 [DOI] [PubMed] [Google Scholar]
- 20.Fleißner A., Sarkar S., Jacobson D. J., Roca M. G., Read N. D., Glass N. L. 2005. The so locus is required for vegetative cell fusion and postfertilization events in Neurospora crassa. Eukaryot. Cell 4:920–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fleißner A., Leeder S., Roca M. G., Read N. D., Glass N. L. Oscillatory recruitment of signaling proteins to cell tips promotes coordinated behavior during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 106:19387–19392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fox H., Hickey P. C., Fernandez-Abalos J. M., Lunness P., Read N. D., Doonan J. H. 2002. Dynamic distribution of BIMG(PP1) in living hyphae of Aspergillus indicates a novel role in septum formation. Mol. Microbiol. 45:1219–1230 [DOI] [PubMed] [Google Scholar]
- 23.Freitag M., Hickey P. C., Khlafallah T. K., Read N. D., Selker E. U. 2004. HP1 is essential for DNA methylation in Neurospora. Mol. Cell 13:427–434 [DOI] [PubMed] [Google Scholar]
- 24.Freitag M., Hickey P. C., Raju N. B., Selker E. U., Read N. D. 2004. GFP as a tool to analyze the organization, dynamics and function of nuclei and microtubules in Neurospora crassa. Fungal Genet. Biol. 41:897–910 [DOI] [PubMed] [Google Scholar]
- 25.Gant T. M., Wilson K. L. 1997. Nuclear assembly. Annu. Rev. Cell Dev. Biol. 13:669–695 [DOI] [PubMed] [Google Scholar]
- 26.Gladfelter A. S., Hungerbuehler A. K., Philippsen P. 2006. Asynchronous nuclear division cycles in multinucleated cells. J. Cell Biol. 172:347–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris S. D., Morrell J. L., Hamer J. E. 1994. Identification and characterization of Aspergillus nidulans mutants defective in cytokinesis. Genetics 136:517–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hickey P., Read N. D. 2009. Imaging living cells of Aspergillus in vitro. Med. Mycol. 37:1460–2709 [DOI] [PubMed] [Google Scholar]
- 29.Hickey P. C., Swift S. R., Roca M. G., Read N. D. 2005. Live-cell imaging of filamentous fungi using vital fluorescent dyes, p. 63–87InSavidge T., Pothoulakis C. (ed.), Microbial imaging, Elsevier, Amsterdam, Netherlands: . [Google Scholar]
- 30.Horio T., Oakley B. R. 2005. The role of microtubules in rapid hyphal tip growth of Aspergillus nidulans. Mol. Biol. Cell 16:918–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huckaba T. M., Gay A. C., Pantalena L. F., Yang H. C., Pon L. A. 2004. Live-cell imaging of the assembly, disassembly, and actin cable-dependent movement of endosomes and actin patches in the budding yeast, Saccharomyces cerevisiae. J. Cell Biol. 167:519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kasuga T., Townsend J. P., Chaoguang T., Gilbert L. B., Mannhaupt G., Taylor J. W., Glass N. L. 2005. Long-oligomer profiling in Neurospora crassa reveals the transcriptional program underlying biochemical and physiological events of conidial germination. Nucleic Acid Res. 33:6469–6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knechtle P. F., Dietrich F., Philippsen P. 2003. Maximal polar growth potential depends on the polarisome component AgSpa2 in the filamentous fungus Ashbya gossypii. Mol. Biol. Cell 14:4140–4154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loo M. 1976. Some required events in conidial germination of Neurospora crassa. Dev. Biol. 54:201–213 [DOI] [PubMed] [Google Scholar]
- 35.Lu B. C. 2006. Karyotyping of Neurospora crassa using synaptonemal complex spreads of translocation quadrivalents. Genome 49:612–618 [DOI] [PubMed] [Google Scholar]
- 36.Machesky L., Gould K. L. 1999. The Arp2/3 complex: a multifunctional actin organizer. Curr. Opin. Cell Biol. 11:117–121 [DOI] [PubMed] [Google Scholar]
- 37.Manton I., Kowallik K., Von Stosch H. A. 1970. Observations on the fine structure and development of the spindle at mitosis and meiosis in a marine centric diatom (Lithodesmium undulatum). J. Cell Sci. 6:131–157 [DOI] [PubMed] [Google Scholar]
- 38.Minke P. F., Lee I. H., Plamann M. 1999. Microscopic analysis of Neurospora ropy mutants defective in nuclear distribution. Fungal Genet. Biol. 28:55–67 [DOI] [PubMed] [Google Scholar]
- 39.Minke P. F., Lee I. H., Tinsley J. H., Bruno K. S., Plamann M. 1999. Neurospora crassa ro-10 and ro-11 genes encode novel proteins required for nuclear distribution. Mol. Microbiol. 32:1065–1076 [DOI] [PubMed] [Google Scholar]
- 40.Moseley J. B., Goode B. L. 2006. The yeast actin cytoskeleton: from cellular function to biochemical mechanism. Microbiol. Mol. Biol. Rev. 70:605–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mouriño-Pérez R. R., Roberson R. W., Bartnicki-Garcia S. 2006. Microtubule dynamics and organization during hyphal growth and branching in Neurospora crassa. Fungal Genet. Biol. 43:389–400 [DOI] [PubMed] [Google Scholar]
- 42.Novick P., Botstein D. 1985. Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell 40:405–416 [DOI] [PubMed] [Google Scholar]
- 43.Osmani A. H., Davies J., Liu H. L., Nile A., Osmani S. A. 2006. Systematic deletion and mitotic localization of the nuclear pore complex proteins of Aspergillus nidulans. Mol. Biol. Cell 17:4946–4961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osmani A. H., Davies J., Oakley C. E., Oakley B. R., Osmani S. A. 2003. TINA interacts with the NIMA kinase in Aspergillus nidulans and negatively regulates astral microtubules during metaphase arrest. Mol. Biol. Cell 14:3169–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pall M. L. 1993. The use of Ignite (Basta;glufosinate;phosphinothricin) to select transformants of bar-containing plasmids in Neurospora crassa. Fungal Genet. Newsl. 40:58 [Google Scholar]
- 46.Pall M. L., Brunelli J. P. 1994. New plasmid and plasmid hybrid vectors and a Neurospora crassa genomic library containing the bar selectable marker and the Cre/lox site-specific recombination system for use in filamentous fungi. Fungal Genet. Newsl. 41:63–65 [Google Scholar]
- 47.Pruyne D. W., Schott D. H., Bretscher A. 1998. Tropomyosin-containing actin cables direct the Myo2p-dependent polarized delivery of secretory vesicles in budding yeast. J. Cell Biol. 143:1931–1945 [DOI] [PubMed] [Google Scholar]
- 48.Raju N. B. 1984. Use of enlarged cells and nuclei for studying mitosis in Neurospora. Protoplasma 121:87–98 [Google Scholar]
- 49.Ramos-García S. L., Roberson R. W., Freitag M., Bartnicki-Garcia S., Mouriño-Pérez R. R. 2009. Cytoplasmic bulk flow propels nuclei in mature hyphae of Neurospora crassa. Eukaryot. Cell 8:1880–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Read N. D., Fleißner A., Roca M. G., Glass N. L. 2010. Hyphal fusion, p. 260–273InBorkovich K. A., Ebbole D. (ed.), Cellular and molecular biology of filamentous fungi. ASM Press, Washington, DC [Google Scholar]
- 51.Read N. D., Lichius A., Shoji J., Goryachev A. 2009. Self-signalling and self-fusion in filamentous fungi. Curr. Opin. Microbiol. 12:608–615 [DOI] [PubMed] [Google Scholar]
- 52.Riedl J., Crevenna A. H., Kessenbrock K., Yu J. H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T. A., Werb Z., Sixt M., Wedlich-Soldner R. 2008. Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5:605–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riquelme M., Reynaga-Peña C. G., Gierz G., Bartnicki-García S. 1998. What determines growth direction in fungal hyphae? Fungal Genet. Biol. 24:101–109 [DOI] [PubMed] [Google Scholar]
- 54.Riquelme M., Roberson R. W., McDaniel D. P., Bartnicki-Garcia S. 2002. The effects of ropy-1 mutation on cytoplasmic organization and intracellular motility in mature hyphae of Neurospora crassa. Fungal Genet. Biol. 37:171–179 [DOI] [PubMed] [Google Scholar]
- 55.Robinow C. F. 1963. Observations on cell growth, mitosis, and division in the fungus Basidiobolus ranarum. J. Cell Biol. 17:123–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinow C. F., Caten C. E. 1969. Mitosis in Aspergillus nidulans. J. Cell Sci. 5:403–431 [DOI] [PubMed] [Google Scholar]
- 57.Robinson R. C., Turbedsky K., Kaiser D. A., Marchand J.-B., Higgs H. N., Choe S., Thomasa D., Pollard D. 2001. Crystal structure of Arp2/3 complex. Science 294:1679–1684 [DOI] [PubMed] [Google Scholar]
- 58.Roca M. G., Arlt J., Jeffree C. E., Read N. D. 2005. Cell biology of conidial anastomosis tubes in Neurospora crassa. Eukaryot. Cell 4:911–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roca M. G., Read N. D., Wheals A. E. 2005. Conidial anastomosis tubes in filamentous fungi. FEMS Microbiol. Lett. 249:191–198 [DOI] [PubMed] [Google Scholar]
- 60.Sagot I., Klee S. K., Pellman D. 2002. Yeast formins regulate cell polarity by controlling the assembly of actin cables. Nat. Cell Biol. 4:42–50 [DOI] [PubMed] [Google Scholar]
- 61.Schott D. H., Collins R. N., Bretscher A. 2002. Secretory vesicle transport velocity in living cells depends on the myosin-V lever arm length. J. Cell Biol. 156:35–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Serna L., Stadler D. 1978. Nuclear division cycle in germinating conidia of Neurospora crassa. J. Bacteriol. 136:341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shaw S. L., Yeh E., Maddox P., Salmon E. D., Bloom K. 1997. Astral microtubule dynamics in yeast: a microtubule-based searching mechanism for spindle orientation and nuclear migration into the bud. J. Cell Biol. 139:985–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shortle D., Haber J. E., Botstein D. 1982. Lethal disruption of the yeast actin gene by integrative DNA transformation. Science 217:371–373 [DOI] [PubMed] [Google Scholar]
- 65.Shortle D., Novick P., Botstein D. 1984. Construction and genetic characterization of temperature-sensitive alleles of the yeast actin gene. Proc. Natl. Acad. Sci. U. S. A. 81:4889–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Siervers N., Bertsch E., Fischer R. 1999. Isolation of nuclear migration mutants of Aspergillus nidulans using GFP expressing strains. Mycol. Res. 103:961–966 [Google Scholar]
- 67.Simos G. 2002. Structure, function and biogenesis of the nuclear envelope in the yeast Saccharomyces cerevisiae, p. 87–101InCollas P. (ed.), Nuclear envelope dynamics in embryos and somatic cells. Landes Bioscience, Georgetown, TX [Google Scholar]
- 68.Steinberg G., Wedlich-Soldner R., Brill M., Schulz I. 2001. Microtubules in the fungal pathogen Ustilago maydis are highly dynamic and determine cell polarity. J. Cell Sci. 114:609–622 [DOI] [PubMed] [Google Scholar]
- 69.Straube A., Weber I., Steinberg G. 2005. A novel mechanism of nuclear envelope break-down in a fungus: nuclear migration strips off the envelope. EMBO J. 24:1674–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suelmann R., Sievers N., Fischer R. 1997. Nuclear traffic in fungal hyphae: in vivo study of nuclear migration and positioning in Aspergillus nidulans. Mol. Microbiol. 25:757–769 [DOI] [PubMed] [Google Scholar]
- 71.Ukil L., De Souza C. P., Liu H.-L., Osmani S. 2009. Nucleolar separation from chromosome during Aspergillus nidulans mitosis can occur without spindle forces. Mol. Biol. Cell 20:2132–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Upadhyay S., Shaw B. D. 2008. The role of actin, fimbrin and endocytosis in growth of hyphae in Aspergillus nidulans. Mol. Microbiol. 68:690–705 [DOI] [PubMed] [Google Scholar]
- 73.Virag A., Griffiths A. J. 2004. A mutation in the Neurospora crassa actin gene results in multiple defects in tip growth and branching. Fungal Genet. Biol. 41:213–225 [DOI] [PubMed] [Google Scholar]
- 74.Xiang X., Fischer R. 2004. Nuclear migration and positioning in filamentous fungi. Fungal Genet. Biol. 41:411–419 [DOI] [PubMed] [Google Scholar]
- 75.Yang H. C., Pon L. A. 2002. Actin cable dynamics in budding yeast. Proc. Natl. Acad. Sci. U. S. A. 99:751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.