

Abstract
The Saccharomyces cerevisiae UGA4 gene encodes a permease capable of importing γ-aminobutyric acid (GABA) and δ-aminolevulinic acid (ALA) into the cell. GABA-dependent induction of this permease requires at least two positive-acting proteins, the specific factor Uga3 and the pleiotropic factor Uga35/Dal81. UGA4 is subjected to a very complex regulation, and its induction is affected by the presence of extracellular amino acids; this effect is mediated by the plasma membrane amino acid sensor SPS. Our results show that leucine affects UGA4 induction and that the SPS sensor and the downstream effectors Stp1 and Stp2 participate in this regulation. Moreover, we found that the Uga3 and Uga35/Dal81 transcription factors bind to the UGA4 promoter in a GABA-dependent manner and that this binding is impaired by the presence of leucine. We also found that the Leu3 transcription factor negatively regulates UGA4 transcription, although this seems to be through an indirect mechanism.
The utilization of nonpreferred nitrogen sources in the absence of preferred sources requires control at the level of transcription for the synthesis of pathway-specific catabolic enzymes and permeases. This transcriptional control requires two positive signals, the first being a global signal indicating nitrogen limitation and the second being a pathway-specific signal that involves the presence of a substrate or intermediate of a metabolic pathway (35). γ-Aminobutyric acid (GABA) can be used as a nitrogen source by the unicellular budding yeast Saccharomyces cerevisiae, being a poor source. The UGA4 gene encodes the GABA and δ-aminolevulinic acid (ALA) permease Uga4 in this organism. Its expression depends on nitrogen catabolite repression (NCR) and GABA induction (2, 5). Induction of this permease requires at least two positive-acting proteins, the specific Uga3 and the pleiotropic Uga35/Dal81 factors (3, 10). These factors act through a 19-bp CG-rich upstream activating sequence named UASGABA. The participation of both Uga3 and Uga35/Dal81 in UGA4 induction was demonstrated by genetic analysis (2, 39), and the interaction of Uga3 with the UASGABA region was shown in vitro in terms of binding using electromobility shift assays (25). The promoter region of UGA4 also contains four adjacent repeats of the heptanucleotide 5′-CGAT(A/T)AG-3′, which constitute a UASGATA element (12). This element, together with the GATA transcription factors, is responsible for the effect of NCR on UGA4.
Yeast cells assess the availability of extracellular nutrients through plasma membrane sensors. Ssy1 is a nutrient receptor that functions together with the two peripheral membrane-associated proteins Ptr3 and Ssy5 as a sensor of extracellular amino acids. Ssy1, Ptr3, and Ssy5 constitute a plasma membrane-associated complex named SPS (18). The homologous zinc finger transcription factors Stp1 and Stp2 are downstream effector components of the SPS sensor pathway. These factors are synthesized as latent cytoplasmic proteins with N-terminal regulatory domains crucial for the regulation of their activity (4, 31). In response to amino acids, Stp1 and Stp2 are activated by endoproteolytic removal of their N-terminal domains and act through specific upstream activating sequences named UASaa, present within SPS sensor-regulated promoters (14, 37). Uga35/Dal81 is required for full induction of amino acid-induced SPS sensor-dependent expression of the AGP1, PTR2, and BAP2 genes (1, 7, 26) and increases the efficiency of Stp1 binding to the AGP1 promoter (8).
Using whole-genome expression analysis of amino acid sensing (16, 17, 28), several groups reported that genes encoding amino acid and peptide transporters are induced by amino acids and that genes under NCR are repressed by amino acids and/or are strongly expressed in a ssy1Δ mutant. The UGA4 gene could be included in both groups since it encodes a transporter and it is under the control of NCR. Previously, we demonstrated that UGA4 induction diminished in the presence of extracellular amino acids (6).
Leu3 has been described as a regulator of five genes that belong to the branched-chain amino acid synthesis pathway (LEU1, LEU2, LEU4, ILV2, and ILV5), one gene (BAP2) which belongs to a family of permeases involved primarily with the uptake of branched-chain amino acids, and one gene (GDH1) mainly responsible for the assimilation of ammonia. Leu3 activity depends on the presence of α-isopropylmalate (α-IPM), an early intermediate in leucine biosynthesis (29). Leu3 acts both as a repressor and as an activator of transcription in the absence or in the presence of α-IPM, respectively. α-IPM synthesis is highly regulated, since α-IPM synthase encoded by LEU4 is feedback inhibited by leucine and reversibly inactivated by coenzyme A (CoA) (29). On the basis of the transcriptional responses and in vivo binding of Leu3, Boer and collaborators identified three additional Leu3-regulated genes (BAT1, GAT1 and OAC1). They also reported increased UGA4 transcription, among other genes, under ammonium limitation in a leu3Δ mutant (9).
The transcription factors Leu3, Uga3, and Uga35/Dal81 are zinc binuclear cluster Zn(II)2-Cys6 proteins. They might interact with DNA as monomers, homodimers, or heterodimers (34). All these transcription regulators target very similar sequences; therefore, other factors are needed to ensure that each protein carries out its own specific regulatory task. The determinants of DNA binding specificity of the zinc binuclear cluster proteins are the nucleotides surrounding the CGG triplets, the orientation of these triplets, and the spacing between them (34).
It has been proposed that Leu3 and Uga3 recognize an everted CGG repeat spaced by 4 bp but that Leu3 does not recognize targets of Uga3 and vice versa, since additional specificity is provided by nucleotides located between the two CGG triplets (38). In addition to the CCG-N4-CGG motif, the nucleotides flanking this everted repeat are also essential for Uga3 in vitro binding and activation of transcription (25).
The target sequence of Uga35/Dal81 is controversial (34). Experiments showed that the Zn(II)2 Cys6 cluster-type DNA binding domain of Uga35/Dal81 is not required for its role in allophanate-induced transcription (10), as was described for tamA, an Aspergillus nidulans gene encoding a protein highly similar to Uga35/Dal81 (13).
The UASGABA element of the UGA4 promoter includes 19 bp, 5′-AAAAACCGCCGGCGGCAAT-3′, with the central core of this sequence being a GC-rich region that contains a perfect 10-bp palindrome, 5′-CCGCCGGCGG-3′ (39).
This work focuses on the interplay of global and specific factors and their influence on the regulation of the catabolic pathway-specific gene UGA4. In order to elucidate the molecular mechanisms of the regulation by amino acids of UGA4 transcription, we demonstrate herein, for the first time, the increased in vivo binding of the Uga3 and Uga35/Dal81 transcription factors to the UGA4 promoter in response to the inducer GABA. We also find that this binding is impaired in cells preincubated with leucine prior to GABA addition in an SPS-dependent manner. Moreover, we show that UGA4 is also strongly regulated by Leu3. Altogether, our results show the relevance of the transcription factors Uga35/Dal81, Uga3, and Leu3 as responsible for the regulation of UGA4 by amino acids.
MATERIALS AND METHODS
Strains and media.
The Saccharomyces cerevisiae strains used in this study, isogenic to the wild-type strain Σ1278b, are listed in Table 1. It was necessary for this work to use prototrophic strains to avoid the addition of amino acids during growth.
Table 1.
Strains used in this work
| Strain | Genotype | Parent | Primer | Source or reference |
|---|---|---|---|---|
| 23344c | matα ura3 | M. Grenson | ||
| 30995b | mataura3 ssy1Δ::KanMX2 | 7 | ||
| KW018 | matα ura3 stp1Δ | 46 | ||
| KW021 | matα ura3 stp2Δ | 46 | ||
| KW023 | matα ura3 stp1Δ stp2Δ | 46 | ||
| FA050 | matα ura3 uga35Δ::KanMX2 | 1 | ||
| SBCY01 | matα ura3 leu3Δ:: KanMX4 | 23344c | F/R-leu3 | This study |
| SBCY02 | matα ura3 LEU3-3HA-kanMX6 | 23344c | F/R- LEU3-Tag | This study |
| SBCY04 | matα ura3 leu4Δ::loxp | 23344c | F/R-leu4 | This study |
| SBCY05 | matα ura3 leu4Δ::loxp leu5Δ::KanMX4 | SBCY04 | F/R-leu5 | This study |
| SBCY08 | matα ura3 his3::KanMX leu4Δ::loxp/pSBC-LEU4fbr | SBCY04 | Plasmid M3929 | This study |
| SBCY10 | matα ura3 6HA-UGA35 | 23344c | F/R-Tag-UGA35 | This study |
| SBCY13 | matα ura3 6HA-UGA3 | 23344c | F/R-Tag-UGA3 | This study |
| SBCY17 | matα ura3 uga35Δ::natMX4 | FA050 | F/R-ME | This study |
| SBCY18 | mataura3 ssy1Δ::natMX4 | 30995b | F/R-ME | This study |
| SBCY20 | matα ura3 uga35Δ::natMX4 leu3Δ::KanMX4 | SBCY17 | F/R-leu3 | This study |
| SBCY22 | matα ura3 leu3Δ::KanMX4 6HA-UGA3 | SBCY13 | F/R-leu3 | This study |
| SBCY23 | matα ura3 leu3Δ::KanMX4 6HA-UGA35 | SBCY10 | F/R-leu3 | This study |
| SBCY24 | mataura3 ssy1Δ::natMX4 6HA-UGA35 | SBCY18 | F/R-Tag-UGA35 | This study |
| SBCY26 | mataura3 ssy1Δ::natMX4 6HA-UGA3 | SBCY18 | F/R-Tag-UGA3 | This study |
| XK14-15D | matα LEU4fbr his4 | G. B. Kohlhawa |
Gently provided by Anders Brandt (Carlsberg Laboratory, Copenhagen Valby, Denmark).
Cells were grown in minimal buffered (pH 6.1) medium (27), with 3% glucose as the carbon source and 10 mM proline as the nitrogen source.
Strain construction.
All the strains generated in this study except for the SBCY08 strain were constructed using the PCR-based gene deletion strategy described by Wach et al. (44, 45) or modified versions of it. All the parental strains are listed in Table 1, and all primers used for PCRs are listed in Table 2.
Table 2.
Primers used in this work
| Primer group and name | Sequence (5′ to 3′) |
|---|---|
| Oligonucleotides for plasmid construction | |
| F-Del1 | CGCGGAATTCGACAATTTCTTCAATCATTGAAATG |
| R-Del1 | ACATAAAACATCTCGAAATTGGTTTTTGGCGCACGA |
| F-Del2 | TCGTGCGCCAAAAACCAATTTCGAGATGTTTTATGT |
| R-Del2 | CCCCAAGCTTCATACTCATTGTTAGTAATAATAAATTATAAGACCT |
| F-LEU4fbr | CGCGGAATTCACTGCTCCTGCTTCATCG |
| R-LEU4fbr | CGCGGAATTCCGTCACTAACCGCCAAAC |
| Oligonucleotides for deletion strain construction | |
| F-leu3 | TGCAATTATGGAAGGAAGATCAGATTTTGTGGCGACTTCACACGTACGCTGCAGGTCGAC |
| R-leu3 | GGACTTTAAACCTTGGGATTGAACGCAAATTCATTCATTAAAATCGATGAATTCGAGCTC |
| F-leu4 | AAAGGATTCTCACACTAGAAGTTTACTGTAGACTTTTTCCCAGCTGAAGCTTCGTACGC |
| R-leu4 | TATAGAAATAAATAGAAGCGAATAAGTCCTGAAATACAGACATAGGCCACTAGTGGATCTG |
| F-leu5 | ACTGCTAAAATAAACACAGTTCTTAAGTATGACGCGAGATCAGCTGAAGCTTCGTACGC |
| R-leu5 | AATTAAATGCCAAAATTCCATTTCATTCTTTCATAGACGACATAGGCCACTAGTGGATCTG |
| F-ME | CGTACGCTGCAGGTCGAC |
| R-ME | ATCGATGAATTCGAGCTC |
| Oligonucleotides for tagged strain construction | |
| F-LEU3-Tag | GTTGATATTTTAATGAATGAATTTGCGTTCAATCCCAAGGTTCGGATCCCCGGGTTAATTAA |
| R-LEU3-Tag | ACGTATATAGAAAATCATTTACCTCTCCTGTAGCACCGCAGTATCGATGAATTCGAGCTC |
| F-Tag-UGA3 | CATGTATGGATGCCAAGAAAACAAAGTTTTTTAAAGTGAGGTATGTGCAGGTCGACAACCCTTAAT |
| R-Tag-UGA3 | CATGCTTCGAATATTTCAATTTCAGCTTCTCCACGCCATAATTGCGGCCGCATAGGCCACT |
| F-Tag-UGA35 | TGTTTAGACGAGCGGCAGAACGACAGGCAGCCATACTATCAAATGTGCAGGTCGACAACCCTTAAT |
| R-Tag-UGA35 | CTTCGTAGGCGATGCGGCATTATCAGCTGGTGATTGGTGAGGGTCGCGGCCGCATAGGCCACT |
| Oligonucleotides fos qChIPa | |
| F-UGA4qPCR | AATCGCTTATCGCTTATCGTG |
| R-UGA4qPCR | GGAACTGATTACTGTGCCAAG |
| F-LEU2qPCR | TCGCCTGACGCATATACC |
| R-LEU2qPCR | ACGATTGCTAACCACCTATTG |
| F-UGA4 UCqPCR | AGTCCAATACCTCTGTCCTC |
| R-UGA4 UCqPCR | AGCCGCAACTTCATTCTG |
qChIP, quantitative ChIP.
The leu4Δ deletion was generated using the pUG6 plasmid (23) to amplify the loxP-KanMX-loxP cassette. After the strain was generated, the KanMX cassette was excised by recombination mediated by Cre recombinase (pSH47 plasmid).
The leu3Δ and leu5Δ strains were constructed using the pFA6a-KanMX4 plasmid as a template for PCR (45).
Strains with a C-terminal tag were generated using the pFA6a-3HA-KanMX6 plasmid (32).
Strains that express N-terminal tagged proteins under the control of its natural promoter were generated using the pOM10 plasmid as a template for PCR (20), with posterior Cre-mediated excision of the KanMX cassette.
The uga35Δ::natMX and ssy1Δ::natMX deletions were generated by replacing the KanMX2 cassette of the FA050 and 30995c strains, respectively, with a natMX cassette. The natMX cassette was amplified from the pAG25 plasmid (22).
For the construction of the leu4Δ his3Δ strain, the SBCY04 strain was transformed with a BamHI-digested M3929 plasmid (43).
All yeast transformations were carried out using the lithium method (21). Transformants were selected on rich medium containing 200 μg/ml G418 or 100 μg/ml nourseothricin (ClonNat; Werner BioAgents).
Plasmids.
The plasmids used to analyze promoter activities were derived from the YEp357 plasmid (36). The UGA4-lacZ fusion gene carries the 5′ regulatory region and part of the coding region of UGA4 (positions −583 to +15 with respect to the ATG initiation codon). Two nested 5′ deletions of UGA4, called UASGATAΔ (i.e., without the UASGATA sequence, positions −406 to +15) and UASΔ (i.e., without the UASGATA and UASGABA sequences, positions −385 to +15), fused to the lacZ reporter gene were also used. The UASGABAmut-lacZ fusion gene contains the UGA4 sequence, positions −583 to +15 with respect to the ATG initiation codon, with an altered UASGABA element, where the core sequence GCCGGCGGC was replaced by ATTAGTAAT (the changed positions are underlined). All these constructions were previously described by Luzzani et al. (33). The UASGABAdel-lacZ fusion gene generated using the strategy described by Strachan and Read (38a) contains the UGA4 sequence, positions −583 to +15 with respect to the ATG initiation codon, with the sequence GCCGGCGGC deleted from the UASGABA element. The UGA3-lacZ and UGA35-lacZ fusion genes contain the UGA3 and UGA35 sequences, positions −795 to +24 and −788 to +30 with respect to the ATG initiation codon, respectively. The primers used to construct UASGABAdel-lacZ (F/R-Del1 and F/R-Del2), UGA3-lacZ (F/R-UGA3), and UGA35-lacZ (F/R-UGA35) are listed in Table 2. All constructions were verified by DNA sequence analysis.
The pSBC-LEU4fbr plasmid was constructed cloning a fragment containing the promoter, coding region, and 3′ noncoding region (positions −996 to +2016) of the LEU4fbr gene into the pRS413 plasmid (11). The LEU4fbr gene was amplified from genomic DNA of the XK14-15D strain (gently provided by Anders Brandt [Carlsberg Laboratory, Copenhagen Valby, Denmark]) and was sequenced (GenBank accession no. GU598519).
β-Galactosidase assays.
Cells grown on the minimal buffered medium up to an absorbance at 570 nm of 0.5 to 0.9 were harvested and transferred to fresh medium with or without 1.3 mM leucine. After a 30-min incubation at 30°C, 0.1 mM GABA was added. At the indicated time points, an aliquot (10 ml) of each culture was collected by centrifugation and resuspended in 2 ml buffer Z (Miller, 1972). β-Galactosidase activity measured according to Miller (35a) was expressed as Miller units. The results shown are the means for duplicates within a representative assay. At least duplicate assays for each of two independent transformants were performed. The deviation of these values from the mean was less than 15%.
Chromatin immunoprecipitation (ChIP) assays.
Cells (a 100-ml culture) were grown to an optical density at 600 nm (OD600) of 0.8 and after different treatments were fixed for 20 min at room temperature in the presence of 1% formaldehyde. Glycine was then added to give a final concentration of 125 mM and incubated for 5 min. Cells were harvested, washed with ice-cold 125 mM Tris-buffered saline (TBS)–glycine and ice-cold TBS and resuspended in 0.4 ml of FA lysis buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, and 2 mM phenylmethylsulfonyl fluoride). An equal volume of glass beads (0.5 mm in diameter; Sigma) was added, and the cells were disrupted by vortexing them for 40 min at 4°C (4 × 10 min with intervals on ice). The lysates were separated from the glass beads, and the chromatin was then pelleted by centrifugation (17,000 × g for 30 min) and resuspended in 0.4 ml of fresh FA lysis buffer. Samples were sonicated to obtain DNA fragments with an average size of 500 bp (Branson Sonifier; 3 × 10 s at 15% amplitude) and clarified by centrifugation at 17,000 × g for 30 min. Protein content was measured using the Bradford assay, and 1 mg of protein was used for each immunoprecipitation. Samples were stored at −80°C. Normal mouse IgG (Santa Cruz) or monoclonal antihemagglutinin (anti-HA) antibody (12CA5 Roche) were added to 25 μl of preblocked (1 mg/ml salmon sperm DNA and 1 mg/ml bovine serum albumin) magnetic beads coupled to protein G (Dynal). After a 5-hour incubation, beads were added to each lysate and were incubated overnight at 4°C in a rotator. Immune complexes were sequentially washed five times with FA lysis buffer, four times with FA lysis buffer containing 500 mM NaCl, five times with wash buffer (10 mM Tris-Cl, pH 8, 0.25 mM LiCl, 1 mM EDTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate), and two times with Tris-EDTA (TE) buffer. Bound proteins were eluted from the beads by adding 150 μl elution buffer (50 mM Tris-Cl, pH 8, 10 mM EDTA, 1% SDS) and incubating for 15 min at 65°C. Cross-linking was reversed by an overnight incubation at 65°C in the presence of proteinase K (0.25 mg/ml). DNA was purified using a QIAquick PCR purification kit (Qiagen). Real-time quantitative PCR (qPCR) was carried out with an Opticon Monitor 3 (Bio-Rad) with primers that amplified promoter regions of the UGA4 (F/R-UGA4qPCR) and LEU2 (F/R-LEU2qPCR) genes (Table 2). A pair of primers that amplify a region located 2.5 kb downstream of the UGA4 promoter was used as an unbound control (F/R-UGA4 UCqPCR).
ChIP DNA was normalized to input DNA and calculated as a signal-to-noise ratio over an IgG control ChIP. The ΔΔCT method was used to calculate the fold change of binding to the promoter of interest (30). Propagation of error was handled using standard root mean square methods.
Nucleotide sequence accession number.
The sequence determined for the present study has been deposited in GenBank under accession no. GU598519.
RESULTS
Previous studies have shown that the induction of UGA4 was inhibited by the addition of a mix of amino acids to the culture medium and that this effect was mediated by the SPS amino acid sensor system (6). To test if individual amino acids known to be SPS activators have the same negative effect on UGA4 expression, wild-type and ssy1Δ cells were incubated with leucine, phenylalanine, tryptophan, or methionine or not incubated (Table 3). In a wild-type strain, GABA induction of the UGA4-lacZ fusion gene was significantly reduced by the addition of the amino acids tested, whereas in a ssy1Δ mutant, this effect was not observed, indicating that the treatment with each amino acid was sufficient to reduce UGA4 expression and that this decrease was dependent on the activity of the SPS sensor. Similar results were obtained using ptr3Δ and ssy5Δ cells (data not shown).
Table 3.
Effect of amino acids on transcription driven by the UGA4-lacZ construction in wild-type and ssy1Δ cellsa
| Condition | Value for: |
|
|---|---|---|
| Wild type | ssy1Δ | |
| MM | 94 ± 2 | 38 ± 1 |
| MM-GABA | 697 ± 54 | 442 ± 61 |
| MM-Leu-GABA | 232 ± 8 | 407 ± 38 |
| MM-Phe-GABA | 329 ± 25 | 405 ± 47 |
| MM-Met-GABA | 396 ± 46 | 616 ± 68 |
| MM-Trp-GABA | 394 ± 17 | 512 ± 30 |
β-Galactosidase activity was determined for wild type (23344c) and ssy1Δ (30995b) cells carrying the UGA4-lacZ fusion gene. Cells were grown in minimal medium (MM) and preincubated for 30 min, with each amino acid (1.3 mM) added before the addition of 0.1 mM GABA, or not preincubated. After 60 min, samples were taken out and β-galactosidase activity was measured. The results shown, expressed in Miller Units, are the means ± standard deviations for duplicates within a representative assay.
To get further insights into the regulation of UGA4 by amino acids, we tested whether the downstream effectors of the SPS signaling pathway participate in UGA4 regulation by leucine, an amino acid commonly used as an inducer of the SPS sensor. Although in the single mutants stp1Δ and stp2Δ, GABA induction of UGA4 diminished in the presence of extracellular leucine, this effect disappeared in the double mutant stp1Δ stp2Δ (Fig. 1), suggesting that at least one of these two factors is essential for the signaling cascade that is triggered by the extracellular amino acids and that modulates UGA4 expression.
Fig. 1.
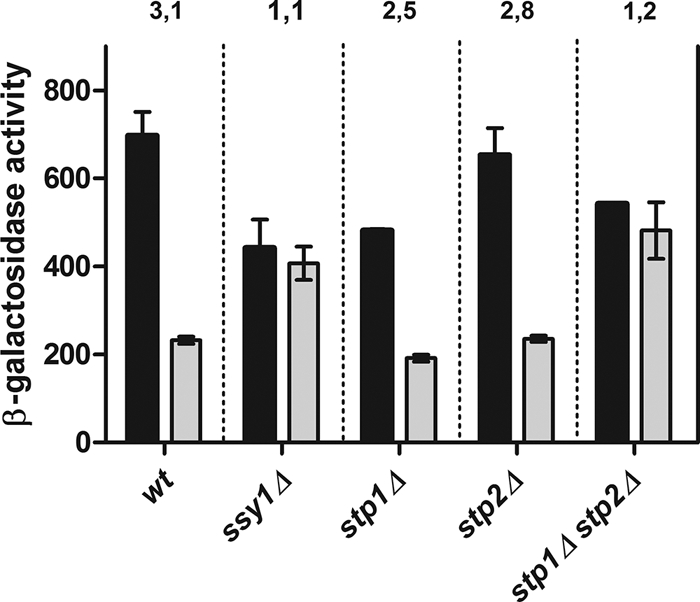
Effect of leucine on the expression of the UGA4-lacZ fusion gene in wild-type, ssy1Δ, stp1Δ, stp2Δ, and stp1Δ stp2Δ cells. β-Galactosidase activity was determined for wild-type (23344c), ssy1Δ (30995b), stp1Δ (KW018), stp2Δ (KW021), and stp1Δ stp2Δ (KW023) cells carrying the UGA4-lacZ fusion gene. Cells were grown in minimal medium and preincubated with 1.3 mM leucine for 30 min (gray bars) or not preincubated (black bars). Then, cells were incubated with 0.1 mM GABA for 60 min, and samples were taken out for β-galactosidase activity measurements. The results shown, expressed in Miller Units, are the means ± standard deviations for duplicates within a representative assay. The numbers above the bars are the ratios between the Miller units calculated for untreated cells and the Miller units calculated for treated cells of each strain.
In order to establish the regions of the UGA4 promoter responsible for the effect of leucine on UGA4 transcription, we analyzed the promoter activity of different DNA fragments covering the region comprising positions −583 to +1 of this gene (Fig. 2 A). The induction driven by both the complete promoter of UGA4 and the promoter lacking the UASGATA element was repressed in the presence of leucine (Fig. 2B and D). In a ssy1Δ strain, the induction profiles of the UGA4-lacZ fusion gene were similar in the presence and absence of leucine (Fig. 2C). These results indicate that the UASGATA element is not a target sequence of the signal triggered by leucine. The construct lacking both the UASGATA and the UASGABA elements was unable to produce any significant expression (Fig. 2E). The transcription levels directed by the constructs without the central core of the UASGABA element were high and independent of GABA, but they were still sensitive to the presence of leucine (Fig. 2F and G). Altogether, these results indicate that neither the UASGATA element nor the central core of the UASGABA element participates in the regulation by leucine of the UGA4 gene. The positive effect of altering or deleting the central core of the UASGABA element on the transcriptional activity of UGA4 (Fig. 2, compare panel B to panels F and G) suggested the existence of a negative factor acting on this element.
Fig. 2.

Effect of leucine on transcription driven by different promoter constructions in wild-type and ssy1Δ cells. (A) Scheme of the fusion genes used. (B to G) β-Galactosidase activity was determined for wild-type (23344c) (B, D, E, F, and G) and ssy1Δ (30995b) (C) cells carrying the UGA4-lacZ (B and C), the UASGATAΔ-lacZ (D), the UASΔ-lacZ (E), the UASGABAmut-lacZ (F), or the UASGABAdel-lacZ (G) fusion gene. Cells were grown in minimal medium and preincubated with 1.3 mM leucine for 30 min before the addition of 0.1 mM GABA (squares) or not preincubated (circles). Then, samples were taken out at the indicated time points, and β-galactosidase activity was measured. The results shown, expressed in Miller Units, are the means for duplicates within a representative assay, with the deviation being less than 15%.
In silico analysis using the databases YEASTRACT (http://www.yeastract.com) (41) and SCPD (http://rulai.cshl.edu/SCPD) revealed that there is a consensus binding site for the transcription factor Leu3 within the UASGABA region, as was already mentioned (38, 39). These findings and the whole-genome transcriptional profiles reported by Boer and collaborators (9) prompted us to postulate that Leu3 was a putative repressor of the UGA4 gene acting on the UASGABA region.
In order to determine whether or not Leu3 modulates UGA4 expression, cells deficient in LEU3 were transformed with the plasmid containing the full-length promoter region of UGA4 fused to lacZ. The results depicted in Fig. 3 A supported our hypothesis proposing Leu3 as a negative regulator of the UGA4 gene since high levels of UGA4 expression in leu3Δ cells were detected. The transcription of UGA4 in the absence of Leu3 did not depend on GABA, suggesting that this factor is involved in the induction process, probably by maintaining low basal levels of UGA4 expression. On the other hand, Leu3 seems to be participating in the regulation of UGA4 by leucine, although it might be remarked that in cells lacking Leu3, some effect of leucine on UGA4 expression was still detectable (Fig. 3A).
Fig. 3.

Effect of Leu3 on UGA4 regulation. (A) β-Galactosidase activity was determined for leu3Δ (SBCY01) cells carrying the UGA4-lacZ fusion gene. Cells were grown in minimal medium and preincubated with 1.3 mM leucine for 30 min before the addition of 0.1 mM GABA (squares) or not preincubated (circles). Then, samples were taken out at the indicated time points. The results shown, expressed in Miller Units, are the means for duplicates within a representative assay, with the deviation being less than 15%. (B) Wild-type cells expressing the Leu3-HA fusion protein (SBCY02) were grown in minimal medium (MM) preincubated for 30 min with 1.3 mM leucine or not preincubated. ChIP assays were carried out using antibodies against the HA epitope. qPCR was performed with specific primers that amplify the UGA4 promoter (F/R UGA4qPCR) (black bars), a region 2.5 kb downstream of the UGA4 promoter (F/R UGA4 UCqPCR) (white bars) (used as a negative control), and the LEU2 promoter (F/R LEU2qPCR) (gray bars) (used as a positive control). ChIP DNA was normalized to input DNA and calculated as a signal-to-noise ratio over an IgG control ChIP. The ΔΔCT method was used to calculate the fold change of binding to the promoter of interest.
Gene regulation by Leu3 depends on the levels of α-IPM (29). To determine whether the negative effect of Leu3 on UGA4 expression is also modulated by intracellular levels of α-IPM, we measured the expression of our UGA4-lacZ fusion gene in strains with different capacities for synthesizing α-IPM (Table 4). In both a wild-type strain and a mutant strain producing feedback-resistant α-IPM synthase (LEU4fbr), the levels of GABA-dependent expression of the UGA4-lacZ fusion gene were high. When these two strains were grown in the presence of leucine, a condition expected to lower the α-IPM production in the wild type but not in the LEU4fbr strain (24), the levels of UGA4-lacZ induction were reduced. It is noteworthy that when a similar experiment was performed with a ssy1Δ strain, this reduction was not observed, indicating that such effect was caused by the signal mediated by the SPS sensor in response to leucine rather than by changes in the α-IPM levels. In a strain devoid of α-IPM synthase activity and hence α-IPM, where it is known that Leu3 acts as a strong repressor (29), the expression of UGA4-lacZ was 4-fold lower than in a wild-type strain, showing that the repressing activity of this factor in leu4Δ leu5Δ cells is stronger than in wild-type cells. However, the fact that the expression of the two transcription factors responsible for UGA4 induction, Uga3 and Uga35, was almost undetectable in leu4Δ leu5Δ cells (Table 5) suggested that the regulation of UGA4 by Leu3 might be indirect.
Table 4.
Expression of the UGA4-lacZ fusion gene in cells with different α-IPM-synthesizing capacitiesa
| Condition | Value for: |
|||
|---|---|---|---|---|
| Wild type | ssy1Δ | leu4Δ leu5Δb | LEU4fbr | |
| MM | 90 ± 9 | 51 ± 7 | 39 ± 2 | 60 ± 5 |
| MM-GABA | 544 ± 41 | 454 ± 26 | 131 ± 2 | 475 ± 44 |
| MM-Leu (2 mM)-Ile (1 mM)c | 64 ± 1 | 18 ± 1 | 14 ± 1 | 76 ± 5 |
| MM-Leu (2 mM)-Ile (1 mM)c-GABA | 274 ± 13 | 547 ± 9 | 67 ± 5 | 240 ± 3 |
β-Galactosidase activity was determined for wild-type (23344c), ssy1Δ (30995b), leu4Δ leu5Δ (SBCY05), and LEU4fbr (SBCY08) cells carrying the UGA4-lacZ fusion gene. Cells were grown in minimal medium (MM) containing or not containing leucine and isoleucine. Each culture was divided in two, and only one half was induced with 0.1 mM GABA for 60 min. Samples were taken out, and β-galactosidase activity was measured. The results shown, expressed in Miller Units, are the means ± standard deviations for duplicates within a representative assay.
Strain SBCY05 requires leucine (0.23 mM) for growth.
Since higher concentrations of leucine alone can cause growth retardation, isoleucine was added to alleviate that effect (24).
Table 5.
Expression of the UGA35-lacZ and UGA3-lacZ fusion genes in wild type, leu3Δ, and leu4Δ leu5Δ cellsa
| Gene | Value for: |
||
|---|---|---|---|
| Wild type | leu3Δ | leu4Δ leu5Δ | |
| UGA35-lacZ | 47.2 ± 2.8 | ≤10 | ≤10 |
| UGA3-lacZ | 128.3 ± 8.0 | 33.6 ± 1.6 | ≤10 |
β-Galactosidase activity was determined for wild-type (23344c), leu3Δ (SBCY01), and leu4Δ leu5Δ (SBCY05) cells carrying the UGA35-lacZ and UGA3-lacZ fusion genes. Cells were grown in minimal medium. Samples were taken out, and β-galactosidase activity was measured. The results shown, expressed in Miller Units, are the means ± standard deviations for duplicates within a representative assay.
We confirmed that α-IPM levels were being effectively modulated in leu4Δ leu5Δ and LEU4fbr strains by measuring the expression of BAP2, known to be regulated by α-IPM levels through Leu3 activity (data not shown).
UGA4 expression basal levels (i.e., before the addition of GABA) driven by the promoters lacking the central core of the UASGABA element in wild-type cells (Fig. 2F and G) and by the full-length UGA4 promoter in leu3Δ cells (Fig. 3A) were very high. These data and the fact that the consensus binding site for Leu3 is within the UASGABA element pointed to Leu3 as a transcription factor negatively regulating UGA4 transcription and probably acting through the UASGABA element. Therefore, we decided to investigate the in vivo binding of Leu3 to the regulatory region of UGA4. For this purpose, we used chromatin immunoprecipitation (ChIP) assays with cells expressing Leu3 with its C-terminal end fused to the HA epitope. We were not able to detect any significant in vivo binding of Leu3-HA to the UGA4 promoter either in cells incubated in minimal medium or in cells treated with leucine (Fig. 3B). Similar results were obtained with the use of a strain that expresses an N-terminal tagged version of Leu3 (data not shown). This result supported the idea that Leu3 acts negatively on UGA4 expression in an indirect way as mentioned above. The LEU2 promoter, a well-known Leu3 target (29), was used as a positive control for Leu3-HA binding. The functionality of both C- and N-tagged fusion proteins was checked by measuring UGA4 expression in these strains (data not shown).
Considering that the Uga35/Dal81 transcription factor is required for the full induction of several amino acid permeases in response to signals triggered by the SPS sensor (1, 8) and that this factor is also required for the induction of UGA genes by GABA (42), we decided to study the in vivo binding of Uga35/Dal81 to the UASGABA region of the UGA4 promoter. For this, we performed ChIP assays using a strain expressing the HA-Uga35/Dal81 fusion protein. We found that HA-Uga35/Dal81 bound to the UGA4 promoter in a GABA-dependent manner and that this binding was impaired by preincubation with leucine (Fig. 4 A). These observations correlate with the low levels of UGA4 induction measured in the presence of leucine (Table 3 and Fig. 2B).
Fig. 4.

Binding of HA-tagged Uga35/Dal81 and Uga3 to the UGA4 promoter in wild-type and ssy1Δ cells. Wild-type (A and B) and ssy1Δ (C and D) cells expressing the HA-Uga35 (SBCY10 and SBCY24) (A and C) and HA-Uga3 (SBCY13 and SBCY26) (B and D) fusion proteins were grown in minimal medium (MM), preincubated for 30 min with 1.3 mM leucine or not preincubated, and then incubated with 0.1 mM GABA or not incubated. ChIP assays were carried out using antibodies against the HA epitope. qPCR was performed with specific primers that amplify the UGA4 promoter (F/R UGA4qPCR) (black bars) and a region 2.5 kb downstream of the UGA4 promoter (F/R UGA4 UCqPCR) (white bars), used as a negative control. ChIP DNA was normalized to input DNA and calculated as a signal-to-noise ratio over an IgG control ChIP. The ΔΔCT method was used to calculate the fold change of binding to the promoter of interest.
In both inducible processes (transcription of genes controlled by the SPS pathway and induction by GABA of UGA genes), Uga35/Dal81 acts together with an inducer-specific transcription factor (1). In the case of the response to GABA of UGA4, this factor is Uga3. For this reason, we decided to investigate Uga3 binding to the UGA4 promoter in vivo under the same conditions used to test the binding for Uga35/Dal81. Our results showed that HA-Uga3 interacted with the UGA4 promoter similarly to the way that HA-Uga35/Dal81 did (Fig. 4B), suggesting that Uga3 bound to the UGA4 promoter in a GABA-dependent manner and that leucine weakened this interaction.
To determine whether the effect of leucine on HA-Uga3 and HA-Uga35/Dal81 binding to the UGA4 promoter is dependent on the SPS sensor, we performed ChIP assays using strains deficient in SSY1 and expressing tagged versions of Uga3 or Uga35/Dal81. We detected both transcription factors bound to the UGA4 promoter even in the presence of leucine (Fig. 4C and D), confirming that the lower binding capacity of HA-Uga3 and HA-Uga35/Dal81 in the presence of leucine observed in wild-type cells (Fig. 4A and B) was caused by the signal triggered by this amino acid through the SPS sensor system.
In our attempt to understand the events that caused the high levels of UGA4 expression in a LEU3-deficient strain, we decided to study the recruitment of HA-Uga3 and HA-Uga35/Dal81 to the promoter of UGA4 in the absence of Leu3. Although the expression level of the UGA3-lacZ fusion gene in a leu3Δ strain was significantly lower than that in a wild-type strain (Table 5), this transcription factor appeared bound to the UGA4 promoter after the addition of GABA (Fig. 5, left panel), as was already observed in the wild-type strain (Fig. 4B). On the other hand, HA-Uga35/Dal81 binding seemed to be impaired by the leu3 deficiency (Fig. 5, right panel). In this strain, UGA35-lacZ fusion gene expression was almost undetectable, explaining the low level of recruitment observed (Table 5). These results did not explain the high basal levels of UGA4 expression observed in the absence of Leu3 (Fig. 3A); however, the lack of UGA4 induction in this strain would be explained by the low level of availability of Uga35/Dal81. The expression levels of UGA4-lacZ, UASGABAmut-lacZ (Fig. 6), and UASGABAdel-lacZ (data not shown) in a leu3Δ uga35Δ strain were significantly lower than those in a leu3Δ strain, indicating that the Uga35/Dal81 factor is in some way involved in the high levels of expression observed under both conditions (i.e., UGA4-lacZ expression in a leu3Δ strain and expression driven by the UASGABAmut-lacZ construction in a wild-type strain) in the absence of an inducer.
Fig. 5.

Binding of HA-tagged Uga35/Dal81 and Uga3 to the UGA4 promoter in leu3Δ cells. leu3Δ cells expressing the HA-Uga3 (SBCY22) (left panel) and HA-Uga35 (SBCY23) (right panel) fusion proteins were grown in minimal medium (MM) and then incubated with 0.1 mM GABA or not incubated. ChIP assays were carried out using antibodies against the HA epitope. qPCR was performed with specific primers that amplify the UGA4 promoter (F/R UGA4qPCR) (black bars) and a region 2.5 kb downstream of the UGA4 promoter (F/R UGA4 UCqPCR) (white bars), used as a negative control. ChIP DNA was normalized to input DNA and calculated as a signal-to-noise ratio over an IgG control ChIP. The ΔΔCT method was used to calculate the fold change of binding to the promoter of interest.
Fig. 6.

Effect of leucine on the expression of the UGA4-lacZ and the UASGABAmut-lacZ fusion genes in wild-type, leu3Δ, uga35Δ and leu3Δ uga35Δ cells. β-Galactosidase activity was determined for wild-type (23344c), leu3Δ (SBCY01), uga35Δ (SBCY17), and leu3Δ uga35Δ (SBCY20) cells carrying the UGA4-lacZ (left panel) or the UASGABAmut-lacZ (right panel) fusion gene. Cells were grown in minimal medium. White bars correspond to untreated cells, black bars correspond to cells treated with 0.1 mM GABA for 60 min, and gray bars correspond to cells treated with 1.3 mM leucine for 30 min and 0.1 mM GABA for 60 min. The results shown, expressed in Miller Units, are the means ± standard deviations for duplicates within a representative assay.
DISCUSSION
The aim of this work was to elucidate the mechanisms by which leucine regulates UGA4 induction. Here, we demonstrate for the first time that leucine affects the GABA-mediated binding of the Uga3 and Uga35/Dal81 transcription factors to the UGA4 promoter and that this effect depends on the SPS sensor pathway.
We previously reported that UGA4 expression is regulated by amino acids through the plasma membrane sensor SPS (6). In this paper, we studied the effect of leucine, one of the most potent known elicitors of signaling through SPS (15, 19), on UGA4. The effect of leucine on UGA4 expression was detected shortly after the addition of this amino acid, suggesting a sensor mediated response that was strictly dependent on the components of the SPS sensor.
In the stp1Δ and stp2Δ single mutants, the effect of leucine was almost indistinguishable from that observed in the wild type, while no effect of leucine was detected in the stp1Δ stp2Δ double mutant, indicating that these two factors are involved in UGA4 regulation by leucine and confirming that they are functionally redundant (31).
On the basis of the proposal of Abdel-Sater and collaborators (1) and the findings of Boban and Ljungdahl (8), demonstrating that the binding of Stp1 to the AGP1 promoter was dependent on the presence of Uga35/Dal81, we studied the participation of Uga35/Dal81 in UGA4 regulation by leucine. In this work, we demonstrated that GABA induced the interaction between HA-Uga35/Dal81 and the UGA4 promoter and that leucine impaired this interaction.
The behavior observed in the binding of HA-Uga3 to the UGA4 promoter in response to leucine was similar to that observed for HA-Uga35/Dal81. This was an unexpected result, since Uga3 is an inducer-specific transcription factor of UGA genes and there were no previous reports relating Uga3 with the amino acid-responsive pathway. One possible explanation is that Uga3 would need Uga35/Dal81 to some extent to properly bind to the UGA4 promoter. In consequence, the binding of Uga3 depending on Uga35/Dal81 would respond to leucine. In vitro electromobility shift assays using an Escherichia coli-produced version of Uga3 protein showed a Uga35/Dal81- and GABA-independent binding of Uga3 to the UASGABA element (25). These results, apparently in contrast with ours, came from an in vitro assay, and the aim of our work was to elucidate the in vivo mechanism, which is probably more complex and highly regulated.
A hierarchy has been proposed for different induction processes mediated by the Uga35/Dal81 factor (1), i.e., SPS amino acid-regulated genes and GABA-induced genes. Our results showing that the decrease caused by leucine in the recruitment of HA-Uga35/Dal81 to the UGA4 promoter depended on Ssy1 support this hypothesis. The signal triggered by the SPS sensor in response to extracellular leucine activates Stp1/Stp2, which would be recruiting Uga35/Dal81 to promote transcriptional leucine induction of other permeases and decreasing the availability of Uga35/Dal81, and consequently of Uga3, for GABA induction of the UGA4 gene (Fig. 7).
Fig. 7.

Schematic representation of the molecular events triggered by leucine and GABA affecting UGA4 gene expression. (Adapted from reference 31 with permission of the publisher.)
Therefore, the element in the UGA4 promoter involved in the response to leucine seems to be the UASGABA element, since this element is in this regulatory region where both the Uga3 and the Uga35/Dal81 factors act.
Bricmont and collaborators (10) have demonstrated that the Zn(II)2-Cys6 cluster-type DNA domain of Uga35/Dal81 is not required for its role in allophanate-induced transcription. Our results showed that HA-Uga35/Dal81 bound to the UGA4 promoter. Two possible explanations may account for this. The first one is that Uga35/Dal81 directly interacts with the UGA4 promoter. The second one is that Uga35/Dal81 might be associated with the UGA4 promoter via an interaction with another protein, such as Uga3.
The Uga35/Dal81 factor seems to be participating in two opposite processes. On the one hand, it mediates the negative effect of leucine on UGA4 induction. On the other hand, the high basal levels of UGA4 expression observed in leu3Δ cells and the high levels of expression of the UASGABAmut-lacZ fusion gene in wild-type cells seem to depend on Uga35/Dal81. This dual function of the Uga35/Dal81 transcription factor needs to be further investigated.
Furthermore, in this work, we also demonstrated that the transcription factor Leu3 negatively regulated the UGA4 gene, irrespective of the presence or absence of α-IPM. In addition, this negative effect was stronger in cells devoid of α-IPM synthase activity, probably due to the low expression levels of Uga3 and Uga35/Dal81 factors under this condition.
The repression of UGA4 by leucine was barely detected in cells deficient in the LEU3 gene, suggesting that UGA4 regulation by leucine was mediated at least in part by Leu3. However, we were not able to detect significant binding of Leu3-HA to the UGA4 promoter. Tang and collaborators, comparing expression and binding under both low and high levels of Leu3 activity, showed no detectable binding of Leu3 to several genes whose expression was affected by LEU3 deletion. They proposed that this could be due to the low sensitivity of the ChIP technique or due to an indirect regulation of these genes by Leu3. Moreover, these authors were not able to detect Leu3 binding to the UGA4 promoter even when experiments were performed under conditions of high levels of activity of Leu3 (40). It is worth remarking that the growth conditions they used are different from those used in the present report. The facts that the effect of Leu3 on UGA4 was negative under all conditions, in contrast to what was described for the BAP2 and LEU2 genes (29, 37), and that we were not able to detect Leu3-HA bound to the UGA4 promoter suggested an indirect regulation of UGA4 by Leu3. Transcription driven by the UASGABAmut and the UASGABAdel constructs in a uga35Δ/dal81Δ strain was still quite sensitive to leucine, and this effect was not observed when LEU3 was also deleted. This suggests that Leu3 is further regulating UGA4 in response to leucine besides its effect on UGA35/DAL81.
In summary, we demonstrated that the UGA4 gene is under the regulation of the SPS signaling pathway and that its induction was inhibited by amino acids, as was the expression of the other genes under both NCR and SPS regulation (16). Leu3, Uga3, and Uga35/Dal81 play an important role in the regulation of UGA4 transcription, and these three factors are responsible for the negative effect of leucine on GABA induction. The mechanism by which Leu3 negatively regulates UGA4 transcription still remains unclear, although evidence presented here suggests that this effect is indirect rather than caused by the direct interaction of this factor with the UGA4 promoter. Uga3 and Uga35/Dal81 bind to the UGA4 promoter in a GABA-dependent manner, and this binding is impaired by extracellular leucine through the SPS sensor (Fig. 7). Some interplay between SPS signaling molecules and Leu3 has been proposed (37) since Stp1 and Leu3 are both dependent on Ssy1. However, this connection is not completely elucidated yet.
ACKNOWLEDGMENTS
This research was supported by the University of Buenos Aires (UBA) and Consejo Nacional de Investigaciones Científicas y Tecnológicas (CONICET), Argentina.
Footnotes
Published ahead of print on 25 June 2010.
REFERENCES
- 1.Abdel-Sater F., Iraqui I., Urrestarazu A., Andre B. 2004. The external amino acid signaling pathway promotes activation of Stp1 and Uga35/Dal81 transcription factors for induction of the AGP1 gene in Saccharomyces cerevisiae. Genetics 166:1727–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andre B., Hein C., Grenson M., Jauniaux J. C. 1993. Cloning and expression of the UGA4 gene coding for the inducible GABA-specific transport protein of Saccharomyces cerevisiae. Mol. Gen. Genet. 237:17–25 [DOI] [PubMed] [Google Scholar]
- 3.Andre B., Talibi D., Soussi Boudekou S., Hein C., Vissers S., Coornaert D. 1995. Two mutually exclusive regulatory systems inhibit UASGATA, a cluster of 5′-GAT(A/T)A-3′ upstream from the UGA4 gene of Saccharomyces cerevisiae. Nucleic Acids Res. 23:558–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andreasson C., Heessen S., Ljungdahl P. O. 2006. Regulation of transcription factor latency by receptor-activated proteolysis. Genes Dev. 20:1563–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bermudez Moretti M., Correa Garcia S., Ramos E., Batlle A. 1996. delta-Aminolevulinic acid uptake is mediated by the gamma-aminobutyric acid-specific permease UGA4. Cell. Mol. Biol. (Noisy-le-grand) 42:519–523 [PubMed] [Google Scholar]
- 6.Bermudez Moretti M., Perullini A. M., Batlle A., Correa Garcia S. 2005. Expression of the UGA4 gene encoding the delta-aminolevulinic and gamma-aminobutyric acids permease in Saccharomyces cerevisiae is controlled by amino acid-sensing systems. Arch. Microbiol. 184:137–140 [DOI] [PubMed] [Google Scholar]
- 7.Bernard F., Andre B. 2001. Ubiquitin and the SCF(Grr1) ubiquitin ligase complex are involved in the signalling pathway activated by external amino acids in Saccharomyces cerevisiae. FEBS Lett. 496:81–85 [DOI] [PubMed] [Google Scholar]
- 8.Boban M., Ljungdahl P. O. 2007. Dal81 enhances Stp1- and Stp2-dependent transcription necessitating negative modulation by inner nuclear membrane protein Asi1 in Saccharomyces cerevisiae. Genetics 176:2087–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boer V. M., Daran J. M., Almering M. J., de Winde J. H., Pronk J. T. 2005. Contribution of the Saccharomyces cerevisiae transcriptional regulator Leu3p to physiology and gene expression in nitrogen- and carbon-limited chemostat cultures. FEMS Yeast Res. 5:885–897 [DOI] [PubMed] [Google Scholar]
- 10.Bricmont P. A., Daugherty J. R., Cooper T. G. 1991. The DAL81 gene product is required for induced expression of two differently regulated nitrogen catabolic genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 11:1161–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christianson T. W., Sikorski R. S., Dante M., Shero J. H., Hieter P. 1992. Multifunctional yeast high-copy-number shuttle vectors. Gene 110:119–122 [DOI] [PubMed] [Google Scholar]
- 12.Cunningham T. S., Dorrington R. A., Cooper T. G. 1994. The UGA4 UASNTR site required for GLN3-dependent transcriptional activation also mediates DAL80-responsive regulation and DAL80 protein binding in Saccharomyces cerevisiae. J. Bacteriol. 176:4718–4725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis M. A., Small A. J., Kourambas S., Hynes M. J. 1996. The tamA gene of Aspergillus nidulans contains a putative zinc cluster motif which is not required for gene function. J. Bacteriol. 178:3406–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Boer M., Nielsen P. S., Bebelman J. P., Heerikhuizen H., Andersen H. A., Planta R. J. 2000. Stp1p, Stp2p and Abf1p are involved in regulation of expression of the amino acid transporter gene BAP3 of Saccharomyces cerevisiae. Nucleic Acids Res. 28:974–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Didion T., Grausland M., Kielland-Brandt C., Andersen H. A. 1996. Amino acids induce expression of BAP2, a branched-chain amino acid permease gene in Saccharomyces cerevisiae. J. Bacteriol. 178:2025–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckert-Boulet N., Nielsen P. S., Friis C., dos Santos M. M., Nielsen J., Kielland-Brandt M. C., Regenberg B. 2004. Transcriptional profiling of extracellular amino acid sensing in Saccharomyces cerevisiae and the role of Stp1p and Stp2p. Yeast 21:635–648 [DOI] [PubMed] [Google Scholar]
- 17.Forsberg H., Gilstring C. F., Zargari A., Martinez P., Ljungdahl P. O. 2001. The role of the yeast plasma membrane SPS nutrient sensor in the metabolic response to extracellular amino acids. Mol. Microbiol. 42:215–228 [DOI] [PubMed] [Google Scholar]
- 18.Forsberg H., Ljungdahl P. O. 2001. Genetic and biochemical analysis of the yeast plasma membrane Ssy1p-Ptr3p-Ssy5p sensor of extracellular amino acids. Mol. Cell. Biol. 21:814–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaber R. F., Ottow K., Andersen H. A., Kielland-Brandt M. C. 2003. Constitutive and hyperresponsive signaling by mutant forms of Saccharomyces cerevisiae amino acid sensor Ssy1. Eukaryot. Cell 2:922–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gauss R., Trautwein M., Sommer T., Spang A. 2005. New modules for the repeated internal and N-terminal epitope tagging of genes in Saccharomyces cerevisiae. Yeast 22:1–12 [DOI] [PubMed] [Google Scholar]
- 21.Gietz R. D., Woods R. A. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350:87–96 [DOI] [PubMed] [Google Scholar]
- 22.Goldstein A. L., McCusker J. H. 1999. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15:1541–1553 [DOI] [PubMed] [Google Scholar]
- 23.Guldener U., Heck S., Fielder T., Beinhauer J., Hegemann J. H. 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 24:2519–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Y., Cooper T. G., Kohlhaw G. B. 1995. The Saccharomyces cerevisiae Leu3 protein activates expression of GDH1, a key gene in nitrogen assimilation. Mol. Cell. Biol. 15:52–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Idicula A. M., Blatch G. L., Cooper T. G., Dorrington R. A. 2002. Binding and activation by the zinc cluster transcription factors of Saccharomyces cerevisiae. Redefining the UASGABA and its interaction with Uga3p. J. Biol. Chem. 277:45977–45983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iraqui I., Vissers S., Bernard F., de Craene J. O., Boles E., Urrestarazu A., Andre B. 1999. Amino acid signaling in Saccharomyces cerevisiae: a permease-like sensor of external amino acids and F-Box protein Grr1p are required for transcriptional induction of the AGP1 gene, which encodes a broad-specificity amino acid permease. Mol. Cell. Biol. 19:989–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobs P., Jauniaux J. C., Grenson M. 1980. A cis-dominant regulatory mutation linked to the argB-argC gene cluster in Saccharomyces cerevisiae. J. Mol. Biol. 139:691–704 [DOI] [PubMed] [Google Scholar]
- 28.Kodama Y., Omura F., Takahashi K., Shirahige K., Ashikari T. 2002. Genome-wide expression analysis of genes affected by amino acid sensor Ssy1p in Saccharomyces cerevisiae. Curr. Genet. 41:63–72 [DOI] [PubMed] [Google Scholar]
- 29.Kohlhaw G. B. 2003. Leucine biosynthesis in fungi: entering metabolism through the back door. Microbiol. Mol. Biol. Rev. 67:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livak K. J., Schmittgen T. D. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 31.Ljungdahl P. O. 2009. Amino-acid-induced signalling via the SPS-sensing pathway in yeast. Biochem. Soc. Trans. 37:242–247 [DOI] [PubMed] [Google Scholar]
- 32.Longtine M. S., McKenzie A., III, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961 [DOI] [PubMed] [Google Scholar]
- 33.Luzzani C., Cardillo S. B., Bermudez Moretti M., Correa Garcia S. 2007. New insights into the regulation of the Saccharomyces cerevisiae UGA4 gene: two parallel pathways participate in carbon-regulated transcription. Microbiology 153:3677–3684 [DOI] [PubMed] [Google Scholar]
- 34.MacPherson S., Larochelle M., Turcotte B. 2006. A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol. Mol. Biol. Rev. 70:583–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marzluf G. A. 1997. Genetic regulation of nitrogen metabolism in the fungi. Microbiol. Mol. Biol. Rev. 61:17–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35a.Miller J. H. 1972. Experiments in molecular genetics, p. 23–56 Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 36.Myers A. M., Tzagoloff A., Kinney D. M., Lusty C. J. 1986. Yeast shuttle and integrative vectors with multiple cloning sites suitable for construction of lacZ fusions. Gene 45:299–310 [DOI] [PubMed] [Google Scholar]
- 37.Nielsen P. S., van den Hazel B., Didion T., de Boer M., Jorgensen M., Planta R. J., Kielland-Brandt M. C., Andersen H. A. 2001. Transcriptional regulation of the Saccharomyces cerevisiae amino acid permease gene BAP2. Mol. Gen. Genet. 264:613–622 [DOI] [PubMed] [Google Scholar]
- 38.Noel J., Turcotte B. 1998. Zinc cluster proteins Leu3p and Uga3p recognize highly related but distinct DNA targets. J. Biol. Chem. 273:17463–17468 [DOI] [PubMed] [Google Scholar]
- 38a.Strachan T., Read A. P. 1999. PCR, DNA sequencing and in vitro mutagenesis. InHuman molecular genetics, 2nd ed.Garland Science, London, United Kingdom [Google Scholar]
- 39.Talibi D., Grenson M., Andre B. 1995. Cis- and trans-acting elements determining induction of the genes of the gamma-aminobutyrate (GABA) utilization pathway in Saccharomyces cerevisiae. Nucleic Acids Res. 23:550–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang L., Liu X., Clarke N. D. 2006. Inferring direct regulatory targets from expression and genome location analyses: a comparison of transcription factor deletion and overexpression. BMC Genomics 7:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teixeira M. C., Monteiro P., Jain P., Tenreiro S., Fernandes A. R., Mira N. P., Alenquer M., Freitas A. T., Oliveira A. L., Sa-Correia I. 2006. The YEASTRACT database: a tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucleic Acids Res. 34:D446–D451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vissers S., Andre B., Muyldermans F., Grenson M. 1990. Induction of the 4-aminobutyrate and urea-catabolic pathways in Saccharomyces cerevisiae. Specific and common transcriptional regulators. Eur. J. Biochem. 187:611–616 [DOI] [PubMed] [Google Scholar]
- 43.Voth W. P., Jiang Y. W., Stillman D. J. 2003. New ‘marker swap’ plasmids for converting selectable markers on budding yeast gene disruptions and plasmids. Yeast 20:985–993 [DOI] [PubMed] [Google Scholar]
- 44.Wach A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12:259–265 [DOI] [PubMed] [Google Scholar]
- 45.Wach A., Brachat A., Pohlmann R., Philippsen P. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793–1808 [DOI] [PubMed] [Google Scholar]
- 46.Wielemans K., Jean C., Vissers S., Andre B. 2010. Amino acid signaling in yeast: post-genome duplication divergence of the Stp1 and Stp2 transcription factors. J. Biol. Chem. 285:855–865 [DOI] [PMC free article] [PubMed] [Google Scholar]