

Abstract
The pathway of electrons required for the reduction of sulfate in sulfate-reducing bacteria (SRB) is not yet fully characterized. In order to determine the role of a transmembrane protein complex suggested to be involved in this process, a deletion in Desulfovibrio vulgaris Hildenborough was created by marker exchange mutagenesis that eliminated four genes putatively encoding the QmoABC complex and a hypothetical protein (DVU0851). The Qmo (quinone-interacting membrane-bound oxidoreductase) complex is proposed to be responsible for transporting electrons to the dissimilatory adenosine-5′-phosphosulfate reductase in SRB. In support of the predicted role of this complex, the deletion mutant was unable to grow using sulfate as its sole electron acceptor with a range of electron donors. To explore a possible role for the hypothetical protein in sulfate reduction, a second mutant was constructed that had lost only the gene that codes for the DVU0851 protein. The second constructed mutant grew with sulfate as the sole electron acceptor; however, there was a lag that was not present with the wild-type or complemented strain. Neither deletion strain was significantly impaired for growth with sulfite or thiosulfate as the terminal electron acceptor. Complementation of the Δ(qmoABC-DVU0851) mutant with all four genes or only the qmoABC genes restored its ability to grow by sulfate respiration. These results confirmed the prediction that the Qmo complex is in the electron pathway for sulfate reduction and revealed that no other transmembrane complex could compensate when Qmo was lacking.
Sulfate-reducing bacteria (SRB) are a diverse group of organisms with the common ability to gain energy through the delivery of electrons to sulfate during anaerobic respiration. The in vivo mechanism of sulfate reduction has not been fully elucidated, but the biochemistry of several of the reductases has been studied in detail (23, 31, 32) and a crystal structure has been determined for the adenosine phosphosulfate (APS) reductase (24). The reduction of sulfate has been observed to follow at least a three-step process: activation of intracellular sulfate to APS through consumption of the equivalent of two ATPs, reduction of APS to sulfite, and reduction of sulfite to sulfide (28).
Genes for the proteins involved in each of the three primary steps have been annotated in sequenced genomes of sulfate reducers (1). Also, a number of transmembrane complexes have been predicted to be involved in the sulfate reduction pathway (29, 31). QmoABC seemed the most likely to be linked to sulfate reduction given the proximity of qmoABC to the apsBA genes (Fig. 1). The existence of a conduit for electrons from the periplasm to sulfate was a prediction of the hydrogen cycling model proposed by Odom and Peck (26). In their elegant but controversial model, electrons and protons from substrate oxidation were proposed to be used by cytoplasmic hydrogenases to make hydrogen in the cytoplasm. The hydrogen would diffuse through the cytoplasmic membrane to the periplasm, where it would be oxidized by the periplasmic hydrogenases. The protons generated would contribute to the gradient that drives the ATP synthase, generating ATP. The electrons would then be channeled through the periplasmic c-type cytochrome matrix to transmembrane complexes that deliver the electrons to cytoplasmic enzymes able to reduce APS to sulfite or sulfite to sulfide.
FIG. 1.

Diagram of the genome region of D. vulgaris containing the predicted six-gene operon apsBA-qmoABC-DVU0851. The locations of the probe used for Northern blot analyses (A), amplified regions from D. vulgaris genomic DNA and cDNA (B to D), deleted segments (E and F), and template regions for probes for Southern confirmation of deletions (G and H) are shown. Arrows represent ORFs, and arrowheads indicate the direction of transcription. Genes in gray are predicted to be in neighboring operons. COG, clusters of orthologous genes.
Several transmembrane complexes in Desulfovibrio vulgaris have been proposed to have involvement in the sulfate reduction pathway: the high-molecular-mass cytochrome c complex (Hmc, encoded by DVU0531 to DVU0536) (10, 19), the type II cytochrome c3 complex (Tmc or TpII-c3, encoded by DVU0263 and DVU0264) (38), the heterodisulfide reductase-like menaquinol-oxidizing complex (DsrMKJOP or HmeCDEAB, encoded by DVU1290 to DVU1286) (13, 14, 22, 25, 32), the NADH-quinone oxidoreductase complex (RnfCDGEAB, encoded by DVU2792 to DVU2797) (30), and the quinone-interacting membrane-bound oxidoreductase complex (Qmo, encoded by DVU0848 to DVU0850) (13, 14, 25, 31). The Hmc and Qmo complexes have received the most attention as conduits for electrons used in the reduction of APS to sulfite (31). Because of the proximity of the qmo genes to the aps genes (Fig. 1) in 12 sequenced SRB (Desulfotalea psychrophila, D. vulgaris [strains Hildenborough, DP4, and Miyazaki], Desulfovibrio desulfuricans [27774 and ND132], Desulfovibrio strain G20, Desulforudis audaxviator, Desulfotomaculum reducens, Desulfovibrio africanus, Desulfococcus oleovorans, and Chlorobium tepidum) (1, 25, 30; S. Brown, personal communication), the Qmo proteins have been suggested to provide the primary pathway of electrons to the APS reductase. By comparison, the hmc operon is located more than 300 kb from the aps genes. However, D. vulgaris strains deleted of the hmc genes had a ca. 50% decrease in growth rate and yield with hydrogen as the electron donor and sulfate as the electron acceptor (10). An increase of about 50% in the expression of hmc resulting from a mutation in a regulator (19) increased D. vulgaris growth with hydrogen by a similar amount but caused a slight decrease in growth rate with lactate. Therefore, the Hmc complex might serve as a component of the electron transport system from hydrogen to sulfate.
Three subunits of the Qmo complex from Desulfovibrio desulfuricans 27774 were the first Qmo proteins biochemically studied (31). Homologs of the encoding genes qmoABC were recognized in the D. vulgaris Hildenborough genome (31), DVU0848 to DVU0850, respectively (15). QmoC was identified as the likely transmembrane subunit that interacts with the menaquinone pool and with QmoA and/or QmoB located in the cytoplasm (31). Additionally, genome sequence availability showed that an open reading frame (ORF) encoding a hypothetical protein (DVU0851) was present at the 3′ end of the putative operon containing the aps and qmo genes (15). Homologs of that predicted gene are found in all genomes of the Desulfovibrio strains available (D. vulgaris [Hildenborough, DP4, and Miyazaki], D. desulfuricans [27774 and ND132], Desulfovibrio africanus, Desulfovibrio strain G20, and Desulfovibrio salexigens) (1; S. Brown, personal communication) and in two additional SRB (Desulfohalobium retbaense and Desulfomicrobium baculatum) (Joint Genome Institute). However, a homolog to DVU0851 was not identified in other related SRB: Desulfotalea psychrophila, Desulfobacterium autotrophicum HRM2, Desulforudis audaxviator MP104C, and Desulfococcus oleovorans HxD3. The DVU0851 protein was identified on a two-dimensional protein gel when D. vulgaris was grown on lactate-sulfate medium (12), but putative functions have not been suggested.
In order to establish the roles of the Qmo complex and DVU0851 in the reduction of sulfate, we constructed two deletion mutants: (i) a single deletion of the qmoABC and DVU0851 genes and (ii) a deletion of the gene coding for the DVU0851 protein. The mutant lacking all four genes was unable to grow on defined medium with sulfate as the sole electron acceptor. Additionally, the mutant lacking only DVU0851 was fully capable of growth by sulfate respiration. From these observations, we infer that the transmembrane QmoABC complex is the unique channel for electron delivery to the APS reductase.
MATERIALS AND METHODS
Strains and media.
The strains used in this study are listed in Table 1 (see also Table S1 in the supplemental material). Escherichia coli strains were cultured in SOC medium (components per liter of medium: 5 g yeast extract, 9 g tryptone, 0.5 g sodium chloride, 0.19 g potassium chloride, 3.6 g glucose, 10 ml of 1 M magnesium chloride, and 10 ml of 1 M magnesium sulfate) or LC medium (components per liter of medium: 10 g tryptone, 5 g sodium chloride, and 5 g yeast extract). Where indicated, kanamycin or spectinomycin was added to LC medium to a final concentration of 50 μg/ml or 100 μg/ml, respectively. Chemicals and antibiotics were obtained from Fisher Scientific (Pittsburgh, PA), Sigma-Aldrich (St. Louis, MO), or RPI Corp. (Mt. Prospect, IL).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristicsb | Source and/or reference |
|---|---|---|
| E. coli strains | ||
| TOP10 (both chemically competent and electrocompetent) | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen (catalog no. C4040-10 and C4040-50) |
| α-Select (Bronze efficiency) | F−deoR endA1 recA1 relA1 gyrA96 hsdR17(rk−mk+) phoA supE44 thi-1 Δ(lacZYA-argF)U169 φ80δlacZΔM15 λ− | Bioline |
| GM272 | F−fhuA2 or fhuA31, lacY1 or lacZ4, tsx-1 or tsx-78, glnV44(AS) galK2(Oc) λ− dcm-6 dam-3 mtlA2 metB1 thi-1? hsdS21 | CGSCa (no. 6478); 17 |
| D. vulgaris strains | ||
| ATCC 29579 | WT D. vulgaris Hildenborough | ATCC |
| JW801 | WT ΔpDV1 | 37 |
| JW9021 | WT Δ(qmoABC-DVU0851) Kmr | This study |
| JW9063 | WT ΔDVU0851 Kmr | This study |
| Plasmids | ||
| pCR8/GW/TOPO | TOPO cloning vector; Spr | Invitrogen |
| pCR-XL-TOPO | TOPO cloning vector; Kmr | Invitrogen |
| pSC27 | Desulfovibrio shuttle vector; source of aph(3′)-II; Kmr | 34 |
| pMO719 | pCR8/GW/TOPO containing SRB replicon (pBG1); Spr | 18 |
| pMO9020 | pCR8/GW/TOPO with 977 bp upstream and 951 bp downstream of aph(3′)-II cassette to delete qmoABC-DVU0851; Spr Kmr | This study |
| pMO9021 | pCR-XL-TOPO containing aph(3′)-IIp::qmoABC-DVU0851; Kmr | This study |
| pMO9040 | pMO719 with aph(3′)-IIp::qmoABC; Spr | This study |
| pMO9042 | pMO719 with aph(3′)-IIp::qmoABC-DVU0851; Spr | This study |
| pMO9062 | pCR8/GW/TOPO with 964 bp upstream and 951 bp downstream of aph(3′)-II cassette to delete DVU0851; Kmr Spr | This study |
| pMO9070 | pCR8/GW/TOPO; Kmr Spr | This study |
| pMO9071 | pMO719 with aph(3′)-II; Kmr Spr | This study |
| pMO9072 | pMO719 with aph(3′)-IIp and MCS; Spr; for complementation constructs | This study |
| pMO9074 | pMO9072 with DVU0851 in MCS; Spr | This study |
| pMO9116 | pMO9040 with aph(3′)-IIp::RBS::qmoABC; Spr | This study |
| pMO9117 | pMO9042 with aph(3′)-IIp::RBS::qmoABC-DVU0851; Spr | This study |
| pMO9118 | pMO9074 with aph(3′)-IIp::RBS::DVU0851; Spr | This study |
CGSC, Coli Genetic Stock Center.
WT, wild type; aph(3′)-IIp, promoter from kanamycin resistance gene aph(3′)-II; MCS, multicloning site; RBS, ribosomal binding site (TGCAGTCCCAGGAGGTACCAT).
All D. vulgaris strains were grown at 30°C in an anaerobic growth chamber (Coy Laboratory Products, Inc., Grass Lake, MI) in MO medium. MO basal medium was pH adjusted to 7.2 with 5 M HCl after addition of all medium components: 8 mM magnesium chloride, 20 mM ammonium chloride, 0.6 mM calcium chloride, 2 mM potassium phosphate (dibasic), 60 μM ferrous chloride, 120 μM EDTA, 30 mM Tris (pH 7.4), 1 ml Thauer's vitamin solution (4), and 6 ml trace element solution per liter. Trace element solution contains 2.5 mM manganese chloride, 1.26 mM cobaltous chloride, 1.47 mM zinc chloride, 210 μM sodium molybdate, 320 μM boric acid, 380 μM nickel sulfate, 11.7 μM cupric chloride, 35 μM sodium selenite, and 24 μM sodium tungstate. Where noted for MO medium, sodium lactate (60 mM), sodium pyruvate (60 mM), sodium formate (60 mM), ethanol (60 mM), or hydrogen (23 mM) was added as the electron donor and sodium sulfate (30 mM), sodium thiosulfate (30 mM), or sodium sulfite (40 mM) was added as the terminal electron acceptor. Sodium acetate (10 mM and 20 mM, respectively) was included in formate and hydrogen media, and cysteine hydrochloride (1 mM) was included in pyruvate fermentation medium. Antibiotics were added to the MO medium as follows: G418 (RPI Corp.) at 400 μg/ml or spectinomycin at 100 μg/ml. G418 was routinely used in place of kanamycin because it proved to be more effective for selection of the kanamycin resistance marker [aph(3′)-II] in D. vulgaris. Because G418 and kanamycin are similar to one another, the same antibiotic resistance gene provided resistance to both antibiotics. For solidified MO medium, 15 g agar per liter was added and the sterile molten medium was amended with sodium thioglycolate (1.2 mM final concentration) and titanium citrate (380 μM) as reductants. Yeast extract (1 g/liter) was added where noted, and the medium was designated MOY medium.
Protein determination.
Protein yield was determined with the Bradford assay (3), and bovine serum albumin (Sigma, St. Louis, MO) was used as the standard.
Plasmid construction.
The pMO9020 plasmid (Fig. 2A) for the qmoABC-DVU0851 deletion was constructed by splicing by overlap extension (SOE) PCR (16). Three regions were amplified (see Table S2 in the supplemental material): 977 bp upstream of qmoA, 951 bp downstream of DVU0851, and the Tn5 kanamycin resistance gene with its cognate promoter. The primers for amplifying the kanamycin resistance gene each contained a common sequence (TAGATTCGAGGTACGGCTACAGTCTT [5′ forward] and CGGTCTATGAACTTAGTGAGCGGATT [5′ reverse]) external to unique bar codes (CTCTTTCTAAGTGAGTCGAG and CACCTGAGAAGACGTAGTAC) that placed these sequences on each side of the antibiotic resistance gene. The bar codes were included for future experimentation (for an example, see reference 11). Amplification of the kanamycin resistance gene was from plasmid pSC27 (34).
FIG. 2.

Plasmids used in this study. (A) Diagram of mutagenic plasmid pMO9020. The region from nucleotides 683 to 3715 is the mutagenic cassette for deletion of qmoABC and DVU0851 genes by marker exchange mutagenesis. NcoI sites were used for Southern confirmation of the deletion. (B) pMO9072 vector constructed for complementation studies in SRB and pMO9072. The promoter aph(3′)-IIp is constitutively expressed and drives the expression of a gene placed in the multicloning site. The pBG1 segment is an endogenous cryptic plasmid from Desulfovibrio strain G20 (34) that allows for stable replication of plasmids in SRB. Unique restriction sites that can be used for the introduction of complementing DNA are shown. SpR, spectinomycin resistance.
The three regions were PCR amplified with Herculase polymerase (Stratagene, La Jolla, CA) and the primers found in Table S2 (obtained from IDT, Coralville, IA). Herculase polymerase was used for all PCRs unless otherwise stated.
The resulting mutagenic PCR product (consisting of the upstream region, the kanamycin resistance gene, and the downstream region) was captured in the cloning vector pCR8/GW/TOPO (Invitrogen, Carlsbad, CA) by following the manufacturer's suggested protocol, generating plasmid pMO9020 (Fig. 2A). The recombinant plasmid was transformed via heat shock into accompanying chemically competent TOP10 cells (Invitrogen). Plasmid was harvested from 1.5 ml of a transformant culture by using a QIAprep spin miniprep kit (Qiagen, Valencia, CA). Sequencing of both strands of the mutagenic cassette was performed at the University of Missouri DNA Core Facility (http://www.biotech.missouri.edu/dnacore/). The sequences obtained were aligned with the published D. vulgaris sequence (GenBank accession no. AE017285.1; http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nucleotide&val=AE017285.1) to verify that no mutations were introduced during PCR amplifications (data not shown). Following verification, a single construct was designated pMO9020 (Fig. 2A) and used for subsequent experiments.
Construction of the deletion cassette for the DVU0851 gene encoding a hypothetical protein was similar to that for the deletion of qmoABC-DVU0851. The three PCR products were combined in a single SOE PCR, captured in the pCR8/GW/TOPO vector, and transformed into chemically competent TOP10 cells. The captured product was sequenced, and its sequence was compared with the published sequence (data not shown). One base change was observed resulting in a silent mutation in the gene coding for the DVU0852 protein at the 684th base of that gene, a G to an A, converting CGG to CGA, both coding for arginine. Sequence data from the deletion cassette for DVU0851 could not be obtained on both strands for the last 53 bases of qmoC to the first base of the common sequence bar code apparently because of a predicted stable hairpin structure located between qmoC and DVU0851 (see Fig. S1C in the supplemental material). A single mutagenic plasmid construct was designated pMO9062 and used for subsequent experiments.
To complement the deletion strains, three plasmids able to stably replicate in SRB were constructed that used the promoter from the kanamycin resistance gene aph(3′)-II [aph(3′)-IIp, as identified in the pCR4 Zero Blunt TOPO manual (Invitrogen)] to drive the expression of complementing genes: qmoABC-DVU0851 (pMO9042), qmoABC alone (pMO9040), or DVU0851 alone (pMO9074). For the construction of pMO9040 and pMO9042, an amplicon containing all four genes was obtained by PCR amplification of qmoABC-DVU0851. An A overhang was added to the 5.5-kb product, which was then captured in pCR-XL-TOPO (Invitrogen) (according to the manufacturer's recommendations, with the exception that the gel-extracted fragment was not exposed to UV irradiation). The plasmid was transformed into TOP10 cells via electroporation. One of the transformants was grown, and the plasmid designated pMO9021 was isolated and digested with either EcoRV [to yield a blunt fragment containing aph(3′)-IIp::qmoABC-DVU0851] or EcoRV and PshAI [to yield a blunt fragment containing only aph(3′)-IIp::qmoABC]. Each fragment was then ligated into EcoRV-digested and dephosphorylated pMO719, generating pMO9042 and pMO9040, respectively. A ribosomal binding site, TGCAGTCCCAGGAGGTACCAT (9), was introduced into each plasmid via the sequence and ligation-independent cloning (SLIC) (21) method with pMO9040 and pMO9042 as template DNAs for PCR amplification (primers qmoA-SLIC-RBS-F and pMO9075-R2). The products from these amplifications were transformed into E. coli α-select (Bioline), and successful transformants were isolated on spectinomycin-containing agar plates. Plasmids pMO9116 and pMO9117 were isolated from the two transformations. The promoter and complementing genes from each plasmid were sequenced.
For complementation of ΔDVU0851, the DVU0851 gene was amplified and ligated into SnaBI-digested and dephosphorylated pMO9072 (see construction below) for constitutive expression from aph(3′)-IIp, yielding pMO9074. A ribosomal binding site was added via the SLIC method with pMO9074 as the template DNA in PCR amplification (primers DVU0851-SLIC-RBS-F and pMO9075-SLIC-R2). The product from the PCR was transformed into E. coli α-select, and transformants were isolated on spectinomycin-containing agar plates. The plasmid isolated from one of these colonies was designated pMO9118, and the promoter and DVU0851 were sequenced.
Construction of pMO9072 for complementation studies.
To create a vector containing aph(3′)-IIp followed by cloning sites for insertion of a complementing gene, aph(3′)-IIp and the aminoglycoside phosphotransferase gene [aph(3′)-II; Kmr] were amplified from pCR-XL-TOPO, an A overhang was added to the fragment with Taq DNA polymerase, and the modified fragment was captured in pCR8/GW/TOPO to produce pMO9070. aph(3′)-IIp and aph(3′)-II were released from pMO9070 by digestion with HpaI and EcoRV and ligated into EcoRV-digested and dephosphorylated pMO719, generating plasmid pMO9071. Plasmid pMO9071 was then transformed into a chemically competent E. coli GM272 strain carrying the dam mutation (35) to allow the use of a restriction enzyme that is sensitive to dam methylation. The plasmid was purified from GM272, and the aph(3′)-II gene [not including aph(3′)-IIp] was removed with the restriction enzyme BsaBI (New England Biolabs). The remaining plasmid was religated, thereby leaving aph(3′)-IIp and eight unique restriction enzyme recognition sites (XcmI, BsaBI, FspI, ScaI, SnaBI, PmeI, SphI, and NspI) to create pMO9072 (Fig. 2B). However, it should be noted that this plasmid does not contain a ribosomal binding site that must be included with the gene to be expressed.
Transformation of D. vulgaris strains.
To prepare D. vulgaris for electroporation, a freezer stock was used to inoculate 5 ml MO medium containing lactate-sulfite and 0.1% (wt/vol) yeast extract (MOYLS3) and grown overnight at 30°C. The 5-ml overnight culture was diluted to 50 ml of the same medium and grown to an optical density at 600 nm (OD600) of ca. 0.35 at 30°C. The culture was harvested by centrifugation at 4°C for 12 min at 3,000 × g and washed with 50 ml of chilled, sterile wash buffer (30 mM Tris-HCl buffer, pH 7.2 [not anaerobic]). The cells were spun at 4°C for another 12 min at 3,000 × g, the pellet was resuspended in 0.5 ml wash buffer, and 50-μl aliquots were used for each electroporation. Approximately 700 ng of plasmid DNA was added to the cells and mixed, and the mixture was transferred to a 1-mm gapped electroporation cuvette (Molecular BioProducts, San Diego, CA). The cuvette and the safety stand were transferred into an anaerobic chamber, and electroporation was carried out at 1,750 V, 250 Ω, and 25 μF with an ECM 630 electroporator (BTX, Holliston, MA). The electroporated cells were diluted into 1 ml MOYLS3 medium and allowed to recover overnight at 30°C. Transformants were selected as G418- or spectinomycin-resistant colonies from aliquots of electroporated cells mixed into molten MOYLS3 medium and poured into empty petri dishes for solidification. Colonies were seen after ca. 4 days of incubation at 30°C in the anaerobic chamber.
One of the plasmids for complementation (pMO9117) did not yield spectinomycin-resistant colonies when transformed into the deletion strain JW9021. Therefore, it was first transformed into wild-type D. vulgaris, isolated, and then used to transform JW9021. The transformation efficiency of pMO9117 into wild-type D. vulgaris was 155 transformant CFU/μg. The plasmid was isolated and then used to transform JW9021, with an increased efficiency of 9.9 × 103 CFU/μg.
Storage of D. vulgaris mutants.
For freezer stocks, sterile glycerol was added to fully grown cultures to a final concentration of 10% (vol/vol), samples were aliquoted into cryogen vials, and filled vials were stored at −80°C.
Southern blots.
In order to verify that qmoABC-DVU0851 was deleted from putative JW9021 isolates, a Southern blot analysis was performed by standard procedures (5). Gels of NcoI-digested genomic DNA were probed with a PCR fragment of apsA, which was located on the D. vulgaris genome immediately upstream of the qmoA gene. A DNA band of 8,450 bp showed hybridization in the wild-type sample, in contrast to a fragment of 2,760 bp from a correctly constructed marker exchange deletion in JW9021 (data not shown).
For confirmation of JW9063 deleted of DVU0851, genomic DNA was digested with BsrBI and probed with a PCR product from qmoC. Fragment sizes of 2,613 bp for the wild-type strain and 1,666 bp for JW9063 were confirmed by Southern analysis (data not shown).
Northern blots.
RNA was isolated from D. vulgaris strains (wild type and JW9021) by following a protocol supplied with RNAwiz reagent (Ambion, Austin, TX). Cells were grown in 50 ml of MO medium containing either lactate-sulfate (wild type) or lactate-sulfite (wild type and JW9021) and harvested at mid-exponential growth (OD600, ∼0.3). An RNA ladder (Promega) and equal masses of RNA samples (∼3.5 μg each) were prepared, subjected to electrophoresis, transferred, and probed using a protocol similar to that described in reference 6.
Making cDNA.
A 10-μg sample of RNA was DNase treated with a Turbo DNA-free kit (Ambion). To determine whether genomic DNA was eliminated, PCR amplification in the absence of reverse transcriptase was performed. The resulting sample was freeze-dried overnight and resuspended in RNase-free deionized water to a final concentration of 0.5 μg/μl. A 2-μg aliquot of DNase-treated RNA was used to make cDNA with an ImProm-II kit (Promega) according to the manufacturer's suggestions.
Growth curves.
In an anaerobic growth chamber, 5-ml aliquots of medium were added to 15-ml culture tubes (path length, 15 mm), inoculated with a 2% volume of stationary-phase D. vulgaris strains (OD600, ∼0.8), and sealed with a rubber stopper, leaving 10 ml of headspace. The cultures were grown at 37°C, and the optical density (at 600 nm) was read at various time points with a Genesys 20 spectrophotometer (Thermo Spectronic, Waltham, MA).
Sulfide determination.
Culture samples were taken approximately every 50 h (at time of inoculation, 50 h, 100 h, and 150 h), and a colorimetric assay was performed to determine the amount of dissolved sulfide generated (8). We used a slightly modified version of the protocol by Cord-Ruwisch (8). Samples of 100 μl were taken instead of 50 μl, and 4 ml of copper reagent was used instead of 1.95 ml.
RESULTS
Construction of deletion strains in sulfate-reducing bacteria.
QmoABC proteins are hypothesized to function as a transmembrane electron conduit providing a reductant for APS reductase, encoded by apsBA (32). The qmoABC genes are downstream of apsBA (Fig. 1) and have been predicted to be a part of the same operon (http://www.microbesonline.org/cgi-bin/fetchLocus.cgi?locus=206275&disp=1). By deleting qmoABC, we sought to test the hypothesis that these genes encode a complex essential for sulfate reduction. These three genes and DVU0851, predicted to be the promoter-distal gene of the putative operon (33), were selected for deletion (Fig. 1). As a standard procedure when deleting genes encoding transmembrane complexes, we have initially chosen not to leave orphan genes that might produce proteins that could interact/interfere with other transmembrane complexes.
The deletion strategy used in this study was adapted from a marker exchange strategy developed for Saccharomyces cerevisiae (11) and has previously been used for making other deletions in D. vulgaris (2). In short, regions upstream and downstream of the genes to be deleted were cloned on either side of a kanamycin resistance marker, aph(3′)-II. The antibiotic resistance cassette contained two sets of flanking oligonucleotides referred to as bar codes, first those unique to this deletion (20 bp each) and the outermost bar codes (26 bp each) common to most mutants constructed in this lab (2, 11). For deleting qmoABC-DVU0851, the constructed segment of DNA—upstream, downstream, and kanamycin cassette regions—was cloned into a Spr plasmid, creating pMO9020 (Fig. 2A), and confirmed by sequencing. This mutagenic plasmid cannot be stably maintained in D. vulgaris as a separate replicon. Two homologous recombination events were necessary for marker replacement with elimination of the vector sequences in the transformants. In D. vulgaris, double recombination events following the introduction of mutagenic plasmids by the electroporation procedure described above were at least 10-fold more frequent than simple plasmid integration into the chromosome. pMO9020 was transformed into wild-type D. vulgaris by electroporation (transformation efficiency with pMO9020 was 7.8 × 103 CFU/μg plasmid). Successful marker replacement was first tested by screening of JW9021 G418-resistant isolates for sensitivity to spectinomycin, the antibiotic resistance encoded on the vector. Putative deletion isolates were confirmed by Southern blot analysis (data not shown). In order to determine whether DVU0851 contributed to sulfate respiration in D. vulgaris, a strain deleted of this gene alone was also constructed and designated JW9063. The construction, transformation, colony screening, and Southern verification of this strain were similar to those of JW9021 (transformation efficiency with pMO9063 was 5.3 × 103 CFU/μg plasmid).
Growth characteristics of deletion and complementation strains.
Growth rates of JW9021, JW9063, and wild-type D. vulgaris were compared in defined MO medium with various electron donor and acceptor combinations. Figure 3 shows the results of these tests with lactate (Fig. 3A and B) or pyruvate (Fig. 3C and D) as the electron donor and sulfate (Fig. 3A and C) or sulfite (Fig. 3B and D) as the electron acceptor. Growth of JW9021, lacking qmoABC-DVU0851, was undetectable with sulfate as the terminal electron acceptor regardless of the electron donor when growth was measured as a change in optical density (Fig. 3A and C), whole-cell protein concentration increases (data not shown), or dissolved sulfide production (Table 2). In contrast, when the electron acceptor was sulfite with either lactate or pyruvate as the electron donor, growth of JW9021 was comparable to that of the wild type (Fig. 3B and D). The wild type and JW9021 were also tested for growth in defined lactate-thiosulfate medium, and no significant differences were observed between the two strains (data not shown). Additionally, growth tests on formate-sulfate, formate-sulfite, ethanol-sulfate, ethanol-sulfite, hydrogen-sulfate, and hydrogen-sulfite media confirmed that sulfate could not be respired by JW9021 regardless of the electron donor but that all donors supported growth with sulfite as the electron acceptor (data not shown).
FIG. 3.

Growth of D. vulgaris and two deletion mutants on sulfate- and sulfite-containing media. Growth comparisons of D. vulgaris Hildenborough (wild type) and deletion constructs JW9021 [Δ(qmoABC-DVU0851)] and JW9063 (ΔDVU0851) on lactate-sulfate (60 mM-30 mM) (A), lactate-sulfite (60 mM-40 mM) (B), pyruvate-sulfate (60 mM-30 mM) (C), and pyruvate-sulfite (60 mM-40 mM) (D) media are shown. Wild type, white circles; JW9021, black circles; JW9063, gray circles. Readings reflect averages of three samples, and error bars show standard deviations.
TABLE 2.
Dissolved sulfide generated by wild-type D. vulgaris, mutants, and complemented mutants when grown on lactate-sulfate or lactate-sulfite mediuma
| Strain (genotype) | Plasmid (complementing gene[s]) | Medium | Dissolved sulfide (mM) generated atb: |
|
|---|---|---|---|---|
| 0 h | 100 h | |||
| Wild type | No plasmid | Lac-SO4 | −0.4 ± 0.2 | 14.0 ± 2.5 |
| Lac-SO3 | −1.1 ± 1.5 | 24.7 ± 0.5 | ||
| JW9021 [Δ(qmoABC-DVU0851)] | No plasmid | Lac-SO4 | −0.3 ± 0.2 | −0.1 ± 0.2 |
| Lac-SO3 | −0.2 ± 0.2 | 20.9 ± 1.1 | ||
| pMO9116 (qmoABC) | Lac-SO4 | −0.4 ± 0.1 | 13.8 ± 1.2 | |
| Lac-SO3 | −0.3 ± 0.1 | 22.0 ± 0.2 | ||
| pMO9117 (qmoABC-DVU0851) | Lac-SO4 | −0.3 ± 0.1 | 10.4 ± 7.7 | |
| Lac-SO3 | −0.3 ± 0.1 | 23.6 ± 1.0 | ||
| pMO9118 (DVU0851) | Lac-SO4 | 0.0 ± 0.0 | 0.0 ± 0.1 | |
| Lac-SO3 | 0.0 ± 0.1 | 21.0 ± 3.7 | ||
| JW9063 (ΔDVU0851) | No plasmid | Lac-SO4 | 0.2 ± 0.1 | 14.7 ± 0.4 |
| Lac-SO3 | 0.5 ± 0.4 | 22.6 ± 0.8 | ||
| pMO9118 (DVU0851) | Lac-SO4 | 0.2 ± 0.1 | 13.6 ± 0.2 | |
| Lac-SO3 | 0.3 ± 0.1 | 23.1 ± 1.8 | ||
Lactate-sulfate, Lac-SO4 (60 mM-30 mM); lactate-sulfite, Lac-SO3 (60 mM-40 mM).
Values are averages of three biological replicates ± standard deviations.
To establish that the growth phenotype of the deletion strain JW9021 [Δ(qmoABC-DVU0851)] resulted from the absence of the Qmo complex, complementation analyses were performed. JW9021 containing a stable plasmid carrying the four deleted genes or the qmoABC genes alone was found to grow at about the same rate as the wild type and had a protein yield similar to that of the wild type (Fig. 4A). Controls showed that complementing plasmids did not alter yields of protein or rates of growth during sulfite respiration (data not shown).
FIG. 4.

Growth of D. vulgaris deletion and complementation strains on sulfate-containing media. (A) Growth of wild-type D. vulgaris (white circles), JW9021 [Δ(qmoABC-DVU0851)] (black circles), and three complemented strains, JW9021(pMO9116) [Δ(qmoABC-DVU0851)] complemented with qmoABC (white squares), JW9021(pMO9117) [Δ(qmoABC-DVU0851)] complemented with qmoABC-DVU0851 (black squares), and JW9021(pMO9118) [Δ(qmoABC-DVU0851)] complemented with DVU0851 (gray squares), on defined lactate-sulfate (60 mM-30 mM) medium. (B) Growth of these strains on defined pyruvate-sulfate (60 mM-30 mM) medium. (C) Growth of wild-type D. vulgaris (white circles), JW9063 (ΔDVU0851) (gray circles), and JW9063(pMO9118) (ΔDVU0851) complemented with DVU0851 (▵) on defined lactate-sulfate medium. Optical density readings are averages of three samples, and error bars show standard deviations.
A possible role for the hypothetical protein DVU0851 in sulfate reduction was then further explored through examination of JW9063, lacking only DVU0851. Growth of JW9063 with sulfite as the terminal electron acceptor was comparable to that of the wild type (179 ± 24.7 μg protein/ml for the mutant versus 227.2 ± 23.2 μg protein/ml for the wild type). However, when sulfate was the electron acceptor, JW9063 grew well and to cell densities equivalent to those of the wild type following a slight lag when lactate was the electron donor (Fig. 3A). The lag was observed when growth was monitored by optical density (Fig. 3A) or by dissolved sulfide production (data not shown); nevertheless, the final protein (110 μg/ml) and soluble sulfide (12.3 mM) produced were comparable to those obtained with the wild-type strain (132 μg/ml and 11.5 mM at 150 h). Thus, deletion of DVU0851 alone did not inhibit growth with sulfate.
Addition of plasmid pMO9118, supplying only DVU0851 to JW9021, did not correct the growth defect with sulfate (Fig. 4), demonstrating that this gene alone was not sufficient to restore the missing function of the QmoABC complex. Complementation of JW9021 with qmoABC (pMO9117) did restore respiration of sulfate. The DVU0851 protein is apparently not essential but may play a secondary role in the formation or stabilization of the Qmo complex. Additional support for a minor role for DVU0851 was the observation that the complemented deletion of the DVU0851 gene no longer exhibited a growth lag on lactate-sulfate medium (Fig. 4C).
Measuring hydrogen sulfide.
To confirm that JW9021 was unable to reduce sulfate, the mutant was grown in sulfate- or sulfite-containing medium and dissolved sulfide accumulated at the stationary phase was measured (Table 2). After 100 h of incubation, dissolved sulfide concentrations above those carried over from the inoculum were not detected in JW9021 or JW9021 containing the DVU0851 complementing plasmid with sulfate as the terminal electron acceptor. By comparison, all other constructs in lactate-sulfate medium and all strains in lactate-sulfite medium contained detectable levels of sulfide.
Northern blot analysis for apsA expression.
The proximity of apsBA, encoding the APS reductase, to qmoABC-DVU0851 and the prediction that these genes could form a single operon raised a question regarding the stability of mRNA for apsBA in the deletion strain. To ensure that transcripts from apsBA could be observed in the Δ(qmoABC-DVU0851) strain, JW9021, a Northern blot analysis was performed. Wild-type cells cultured in MO medium containing lactate-sulfate or lactate-sulfite as well as JW9021 cells cultured with lactate-sulfite were grown to mid-exponential phase, and the RNA from each was isolated and probed for apsA (Fig. 5). The apsA-containing transcripts in both the wild type and JW9021 in lactate-sulfite medium were present at low levels but were quite similar in concentration. It was noted that apsA expression in the wild type in the presence of sulfate was much higher than in the presence of sulfite. Additionally, the transcript size obtained with the apsA probe, from all three samples, was about 2.5 kb (Fig. 5). This is shorter than 8.2 kb, the size expected if apsBA and qmoABC-DVU0851 were transcribed as a single mRNA (http://www.microbesonline.org/cgi-bin/fetchLocus.cgi?locus=206275&disp=1) (1). Even in the mRNA from wild-type cells that had abundant apsA transcripts, no messages longer than ca. 2.5 kb were detected when probed with apsA (Fig. 5), suggesting that apsBA may be transcribed independently of qmoABC-DVU0851 or that any longer transcripts are readily processed.
FIG. 5.
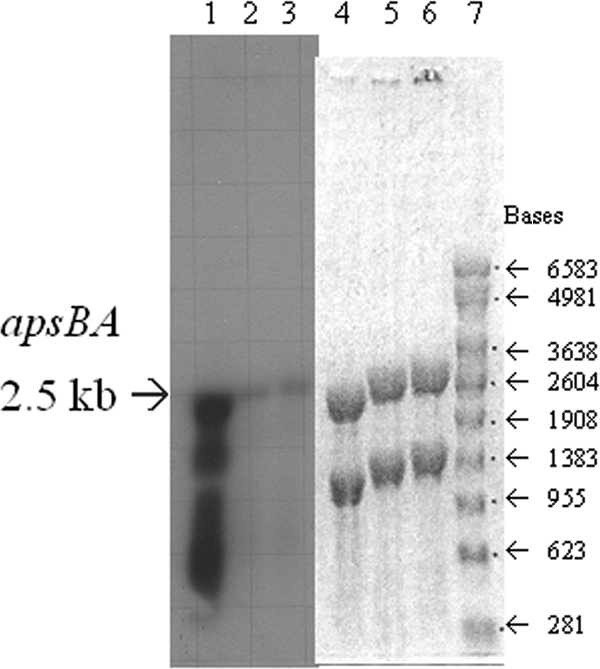
Northern blot of D. vulgaris and the Δ(qmoABC-DVU0851) mutant probed for apsA-containing transcript. Determination of apsA-containing transcript from RNA samples of D. vulgaris (lanes 1, 2, 4, and 5) and deletion mutant JW9021 [Δ(qmoABC-DVU0851)] (lanes 3 and 6) grown on lactate-sulfate (60 mM-30 mM) (lanes 1 and 4) or lactate-sulfite (60 mM-40 mM) (lanes 2, 3, 5, and 6) medium. The RNA was probed with an internal fragment of apsA. The agarose gel (lanes 4 to 7) and developed film (lanes 1 to 3) are shown. An RNA ladder (Promega) was also included (lane 7).
To further investigate the transcript size found for apsA in the Northern blot analysis, we attempted to find evidence of longer transcripts derived from the predicted six-gene operon (Fig. 1). Genomic DNA and cDNA prepared from RNA of wild-type cells grown with lactate-sulfate were used as templates for PCR with primers specific for six regions: an internal region of the apsA gene, an internal portion of the qmoA gene, a region spanning the 3′ end of apsA through the 5′ end of qmoA, an internal portion of qmoC, an internal portion of DVU0851, and a region spanning the 3′ end of qmoC through the 5′ end of DVU0851 (Fig. 1). The genomic DNA was expected to show all six products. The cDNA was also expected to show the six PCR products if the predicted single-operon structure had been correct and unprocessed messages had been accumulated. If apsBA, qmoABC, and DVU0851 are each transcribed separately, the cDNA would yield PCR products for the internal products of apsA, qmoA, qmoC, and DVU0851 but they would not contain the intergenic regions. Bands were observed for all primer sets with the exception of PCR products spanning the intergenic region between apsA and qmoA and that between qmoC and DVU0851, with cDNA as the template (Fig. 1 and 6). These results suggest that apsBA, qmoABC, and DUV0851 are transcribed as three operons or that any inclusive transcripts are readily processed.
FIG. 6.

PCR amplification of apsA, qmoA, qmoC, and DVU0851 and the region spanning the junctions between apsA and qmoA and between qmoC and DVU0851 from D. vulgaris genomic DNA and cDNA. PCR amplification was performed on D. vulgaris genomic DNA (lanes 1 to 6) and cDNA from D. vulgaris grown in lactate-sulfate medium (lanes 8 to 13) for an internal fragment of apsA (expected size, 440 bp) (lanes 1 and 8), an internal fragment of qmoA (expected size, 610 bp) (lanes 2 and 9), a region spanning the intergenic region that includes coding sequences for the C terminus of ApsA and the N terminus of QmoA (expected size, 1,510 bp) (lanes 3 and 10), qmoC (expected size, 875 bp) (lanes 4 and 11), DVU0851 (expected size, 747 bp) (lanes 5 and 12), and a region spanning the intergenic region that includes coding sequences for the C terminus of QmoC and the N terminus of DVU0851 (expected size, 1,711 bp) (lanes 6 and 13). A 1-kb Plus DNA ladder (Fermentas) is in lanes 7 and 14. Sizes of observed bands are labeled on the left-hand side of the gel.
Two possible strong hairpin structures were observed between the apsA and qmoA genes, a predicted intergenic region of 145 bp. One hairpin is a 30-base sequence with a perfect 13-bp intrastrand stem (bold) and 4-base loop (AGGGCGGTTGCGGGGTACCGCAACCGCCCT) (see Fig. S1A in the supplemental material) with a melting temperature of 84.2°C, and the second is a 51-base sequence with a perfect 12-bp stem (bold) and an imperfect 10-base stem (underlined) (ACGGCCTAAGCCGGGGCAGTAAGCACGCCTTATTGTCTGGGCTTAGGCCGT) (Fig. S1B) with a melting temperature of 64.6°C (predictions from mFold; http://www.idtdna.com/analyzer/Applications/OligoAnalyzer/). In addition, in the 89-bp intergenic region between qmoC and DVU0851, there is potentially a 10-bp stem (bold) with a 4-base loop (GTCGGCGCCGCTGCGGCGCGCCGAC) (Fig. S1C) that could form a transcription terminator with a melting temperature of 84.2°C as well. These highly stable hairpin structures support the possibility that multiple transcripts are generated from this region.
DISCUSSION
Role of qmoABC in sulfate reduction.
The exact pathway of electron flow for reduction of sulfate to sulfide in sulfate-reducing bacteria has not yet been completely elucidated. The evidence identifying specific proteins involved in electron transfer to APS reductase or sulfite reductase has been considerable, but it is often circumstantial. This evidence includes the conservation of genes in the genomes of sequenced sulfate-reducing bacteria and their absence in organisms not able to respire sulfate (25), frequent colocalization of genes implicated or known to be involved in sulfate reduction (13, 25), homology to existing transmembrane electron-transporting complexes involved in energy generation (22, 23, 31, 32, 38), observations of correlated changes in the expression levels of such genes in different media (13, 14, 30, 36), and reduced growth of mutants deleted of particular genes (10, 19, 27). In those studies where growth was altered on sulfate, the results were dependent upon the electron donor (10, 19, 27), which might indicate that the step affected was the one producing the electrons rather than the step of delivery of electrons to sulfate or sulfite. However, in this study, we observed the complete cessation of growth on medium containing sulfate as the sole electron acceptor, with lactate, pyruvate, formate, ethanol, or hydrogen as the reductant source, in a mutant of D. vulgaris, JW9021, lacking qmoABC-DVU0851. Second, we showed restoration of growth on lactate-sulfate and pyruvate-sulfate media when the deletion strain was complemented with these genes. The role of the qmoABC-DVU0851 genes is undoubtedly involvement in the reduction of sulfate to sulfite since growth with sulfite was not substantially altered in JW9021 (Fig. 3B and D; Table 2). It should also be pointed out that during extended incubation of JW9021 with the sulfate available (>150 h), growth of suppressors was not observed. We interpret this to mean that other membrane-bound, electron-accepting/donating protein complexes (Hmc, DsrMKJOP, Rnf, Tmc, and Hdr), despite their structural similarities (31), could not readily compensate for the loss of QmoABC. In addition, although NADH oxidase was reported to deliver electrons to APS reductase from NADH in vitro (7), this enzyme was apparently not able to compensate for the lack of a Qmo complex in vivo. Experiments to evaluate possible compensation for the loss of individual components of the Qmo complex, i.e., only the soluble components (qmoAB) or only the membrane-spanning component (qmoC), have not been carried out.
Both the reduction of thiosulfate to sulfite and sulfide and its disproportionation to sulfate and sulfide have been proposed as mechanisms for thiosulfate metabolism in sulfate-reducing organisms (20). However, the disproportionation of thiosulfate would then make its respiration dependent on the same enzymes necessary for sulfate reduction, ATP sulfurylase and APS reductase (20). The growth of JW9021 on thiosulfate-containing medium (data not shown) was not significantly different from that of the wild type. Therefore, we infer that D. vulgaris has the capacity to grow on thiosulfate by reduction to sulfite plus sulfide, bypassing the need for sulfate reduction.
Role of the DVU0851 protein in sulfate reduction.
The presence of the DVU0851 protein was not required for sulfate reduction, as observed by the growth of the JW9063 mutant on lactate-sulfate and pyruvate-sulfate media (Fig. 3A and C; Table 2). However, when JW9063 was transferred to sulfate-containing medium, there was a detectable lag when the medium contained lactate as the electron donor compared with that of the wild-type and JW9063(pMO9118) strains (Fig. 4C). Even though the gene coding for the DVU0851 protein is predicted to be in the same operon as the aps and qmo genes in Desulfovibrio strains, it was not a universally conserved gene among the predicted sulfate reduction genes (25), nor was it found in several other sequenced sulfate-reducing organisms.
Expression of apsBA and qmoABC.
The data presented here support the possibility of 3 separate operons, apsBA, qmoABC, and DVU0851, unlike a prediction suggested elsewhere (1). However, we cannot yet eliminate the possibility that a single transcript may be synthesized and rapidly processed. We determined that deletion of the qmoABC-DVU0851 genes did not apparently alter the level of transcription of the apsBA genes. As previously reported (12, 13, 14, 30), our data confirmed that the genes encoding the enzymatic machinery for sulfate reduction are not constitutively expressed in D. vulgaris. Pereira et al. (30) reported that apsBA and qmoABC transcripts were decreased in cells with thiosulfate as the electron acceptor but increased during pyruvate fermentation. A possible regulatory motif and a transcriptional regulatory protein have been proposed (33). Regulation of these genes appears to be complex and is under investigation.
Conclusion.
This study shows that the Qmo complex is essential for sulfate respiration in D. vulgaris. Further experiments are now possible to determine whether all subunits of the Qmo complex are necessary for sulfate reduction, to establish the function of the hypothetical protein DVU0851, and to elucidate the regulation of this process.
Supplementary Material
Acknowledgments
This work was supported through a subcontract between the Lawrence Berkeley National Laboratory and the University of Missouri and is part of the Environmental Stress Pathway Project (ESPP) in the Virtual Institute for Microbial Stress and Survival (VIMSS). The VIMSS ESPP is supported by the Genomics:GTL Program in the Office of Biological and Environmental Research of the U.S. Department of Energy under contract no. DE-AC02-05CH11231 to the Lawrence Berkeley National Laboratory.
Footnotes
Published ahead of print on 25 June 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Alm, E. J., K. H. Huang, M. N. Price, R. P. Koche, K. Keller, I. L. Dubchak, and A. P. Arkin. 2005. The MicrobesOnline web site for comparative genomics. Genome Res. 15:1015-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bender, K. S., H.-C. B. Yen, C. L. Hemme, Z. Yang, Z. He, Q. He, J. Zhou, K. H. Huang, E. J. Alm, T. C. Hazen, A. P. Arkin, and J. D. Wall. 2007. Analysis of a ferric uptake regulator (Fur) mutant of Desulfovibrio vulgaris Hildenborough. Appl. Environ. Microbiol. 73:5389-5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradford, M. M. 1976. A rapid and sensitive for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 4.Brandis, A., and R. K. Thauer. 1981. Growth of Desulfovibrio species on hydrogen and sulphate as sole energy source. J. Gen. Microbiol. 126:249-252. [Google Scholar]
- 5.Brown, T. 1993. Analysis of DNA sequences by blotting and hybridization, p. 2.9.1-2.9.20. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology. John Wiley and Sons, New York, NY.
- 6.Brown, T. 1993. Analysis of RNA by Northern and slot blot hybridization, p. 4.9.1-4.9.14. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology. John Wiley and Sons, New York, NY. [DOI] [PubMed]
- 7.Chen, L., J. LeGall, and A. V. Xavier. 1994. Purification, characterization and properties of an NADH oxidase from Desulfovibrio vulgaris (Hildenborough) and its coupling to adenylyl phosphosulfate reductase. Biochem. Biophys. Res. Commun. 203:839-844. [DOI] [PubMed] [Google Scholar]
- 8.Cord-Ruwisch, R. 1985. A quick method for the determination of dissolved and precipitated sulfides in cultures of sulfate reducing bacteria. J. Microbiol. Methods 4:33-36. [Google Scholar]
- 9.Dolla, A., R. Fu, M. J. Brumlik, and G. Voordouw. 1992. Nucleotide sequence of dcrA, a Desulfovibrio vulgaris Hildenborough chemoreceptor gene, and its expression in Escherichia coli. J. Bacteriol. 174:1726-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dolla, A., B. K. J. Pohorelic, J. K. Voordouw, and G. Voordouw. 2000. Deletion of the hmc operon of Desulfovibrio vulgaris subsp. vulgaris Hildenborough hampers hydrogen metabolism and low-redox-potential niche establishment. Arch. Microbiol. 174:143-151. [DOI] [PubMed] [Google Scholar]
- 11.Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles, S. Véronneau, S. Dow, A. Lucau-Danila, K. Anderson, B. André, A. P. Arkin, A. Astromoff, M. E. Bakkoury, R. Bangham, R. Benito, S. Brachat, S. Campanaro, M. Curtiss, K. Davis, A. Deutschbauer, K.-D. Entian, P. Flaherty, F. Foury, D. J. Garfinkel, M. Gerstein, D. Gotte, U. Güldener, J. H. Hegemann, S. Hempel, Z. Herman, D. F. Jaramillo, D. E. Kelly, S. L. Kelly, P. Kötter, D. LaBonte, D. C. Lamb, N. Lan, H. Liang, H. Liao, L. Liu, C. Luo, M. Lussier, R. Mao, P. Menard, S. L. Ooi, J. L. Revuelta, C. J. Roberts, M. Rose, P. Ross-Macdonald, B. Scherens, G. Schimmack, B. Shafer, D. D. Shoemaker, S. Sookhai-Mahadeo, R. K. Storms, J. N. Strathern, G. Valle, M. Voet, G. Volckaert, C.-Y. Wang, T. R. Ward, J. Wilhelmy, E. A. Winzeler, Y. Yang, G. Yen, E. Youngman, K. Yu, H. Bussey, J. D. Boeke, M. Snyder, P. Philippsen, R. W. Davis, and M. Johnston. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387-391. [DOI] [PubMed] [Google Scholar]
- 12.Haveman, S. A., V. Brunelle, J. K. Voordouw, G. Voordouw, J. F. Heidelberg, and R. Rabus. 2003. Gene expression analysis of energy metabolism mutants of Desulfovibrio vulgaris Hildenborough indicates an important role for alcohol dehydrogenase. J. Bacteriol. 185:4345-4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haveman, S. A., E. A. Greene, C. P. Stilwell, J. K. Voordouw, and G. Voordouw. 2004. Physiological and gene expression analysis of inhibition of Desulfovibrio vulgaris Hildenborough by nitrite. J. Bacteriol. 186:7944-7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haveman, S. A., E. A. Greene, and G. Voordouw. 2005. Gene expression analysis of the mechanism of inhibition of Desulfovibrio vulgaris Hildenborough by nitrate-reducing, sulfide-oxidizing bacteria. Environ. Microbiol. 7:1461-1465. [DOI] [PubMed] [Google Scholar]
- 15.Heidelberg, J. F., R. Seshadri, S. A. Haveman, C. L. Hemme, I. T. Paulsen, J. F. Kolonay, J. A. Eisen, N. Ward, B. Methe, L. M. Brinkac, S. C. Daugherty, R. T. Deboy, R. J. Dodson, A. S. Durkin, R. Madupu, W. C. Nelson, S. A. Sullivan, D. Fouts, D. H. Haft, J. Delengut, J. D. Peterson, T. M. Davidsen, N. Zafar, L. Zhou, D. Radune, G. Dimitrov, M. Hance, K. Tran, H. Khouri, J. Gill, T. R. Utterback, T. V. Feldblyum, J. D. Wall, G. Voordouw, and C. M. Fraser. 2004. The genome sequence of the anaerobic sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat. Biotechnol. 22:554-559. [DOI] [PubMed] [Google Scholar]
- 16.Horton, R. M., Z. Cai, S. N. Ho, and L. R. Pease. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528-535. [PubMed] [Google Scholar]
- 17.Kasher, M. S., and A. Roman. 1988. Alterations in the regulatory region of the human papillomavirus type 6 genome are generated during propagation in Escherichia coli. J. Virol. 62:3295-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keller, K. L., K. S. Bender, and J. D. Wall. 2009. Development of a markerless genetic exchange system in Desulfovibrio vulgaris Hildenborough and its use in generating a strain with increased transformation efficiency. Appl. Environ. Microbiol. 75:7682-7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keon, R. G., R. Fu, and G. Voordouw. 1997. Deletion of two downstream genes alters expression of the hmc operon of Desulfovibrio vulgaris subsp. vulgaris Hildenborough. Arch. Microbiol. 167:376-383. [DOI] [PubMed] [Google Scholar]
- 20.Krämer, M., and H. Cypionka. 1998. Sulfate formation via ATP sulfurylase in thiosulfate- and sulfite-disproportionating bacteria. Arch. Microbiol. 151:232-237. [Google Scholar]
- 21.Li, M., and S. J. Elledge. 2007. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4:251-256. [DOI] [PubMed] [Google Scholar]
- 22.Mander, G. J., E. C. Duin, D. Linder, K. O. Stetter, and R. Hedderich. 2002. Purification and characterization of a membrane-bound enzyme complex from the sulfate-reducing archaeon Archaeoglobus fulgidus related to heterodisulfide reductase from methanogenic archaea. Eur. J. Biochem. 269:1895-1904. [DOI] [PubMed] [Google Scholar]
- 23.Mander, G. J., A. J. Pierik, H. Huber, and R. Hedderich. 2004. Two distinct heterodisulfide reductase-like enzymes in the sulfate-reducing archaeon Archaeoglobus profundus. Eur. J. Biochem. 271:1106-1116. [DOI] [PubMed] [Google Scholar]
- 24.Meyer, B., and J. Kuever. 2008. Homology modeling of dissimilatory APS reductases (AprBA) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS One 3:e1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mussmann, M., M. Richter, T. Lombardot, A. Meyerdierks, J. Kuever, M. Kube, F. Oliver Glöckner, and R. Amann. 2005. Clustered genes related to sulfate respiration in uncultured prokaryotes support the theory of their concomitant horizontal transfer. J. Bacteriol. 187:7126-7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Odom, J. M., and H. D. Peck, Jr. 1981. Hydrogen cycling as a general mechanism for energy coupling in the sulfate-reducing bacteria, Desulfovibrio sp. FEMS Microbiol. Lett. 12:47-50. [Google Scholar]
- 27.Odom, J. M., and J. D. Wall. 1987. Properties of a hydrogen-inhibited mutant of Desulfovibrio desulfuricans ATCC 27774. J. Bacteriol. 169:1335-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peck, H. D., Jr., and J. LeGall. 1982. Biochemistry of dissimilatory sulphate reduction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 298:443-466. [DOI] [PubMed] [Google Scholar]
- 29.Pereira, I. A. C., S. A. Haveman, and G. Voordouw. 2007. Biochemical, genetic and genomic characterization of anaerobic electron transport pathways in sulphate-reducing Deltaproteobacteria, p. 215-240. In L. L. Barton and W. A. Hamilton (ed.), Sulphate-reducing bacteria. Cambridge University Press, Cambridge, United Kingdom.
- 30.Pereira, P., Q. He, F. M. A. Valente, A. V. Xavier, J. Zhou, I. A. C. Pereira, and R. O. Louro. 2008. Energy metabolism in Desulfovibrio vulgaris Hildenborough: insights from transcriptome analysis. Antonie Van Leeuwenhoek 93:347-362. [DOI] [PubMed] [Google Scholar]
- 31.Pires, R. H., A. I. Lourenço, F. Morais, M. Teixeira, A. V. Xavier, L. M. Saraiva, and I. A. C. Pereira. 2003. A novel membrane-bound respiratory complex from Desulfovibrio desulfuricans ATCC 27774. Biochim. Biophys. Acta 1605:67-82. [DOI] [PubMed] [Google Scholar]
- 32.Pires, R. H., S. S. Venceslau, F. Morais, M. Teixeira, A. V. Xavier, and I. A. C. Pereira. 2006. Characterization of the Desulfovibrio desulfuricans ATCC 27774 DsrMKJOP complex—a membrane-bound redox complex involved in the sulfate respiratory pathway. Biochemistry 45:249-262. [DOI] [PubMed] [Google Scholar]
- 33.Rodionov, D. A., I. Dubchak, A. Arkin, E. Alm, and M. S. Gelfand. 2004. Reconstruction of regulatory and metabolic pathways in metal-reducing δ-proteobacteria. Genome Biol. 5:R90.1-R90.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rousset, M., L. Casalot, B. J. Rapp-Giles, Z. Dermoun, P. de Philip, J.-P. Bélaich, and J. D. Wall. 1998. New shuttle vectors for the introduction of cloned DNA in Desulfovibrio. Plasmid 39:114-122. [DOI] [PubMed] [Google Scholar]
- 35.Seidman, C. 1993. Introduction of plasmid DNA into cells, p. 1.8.1-1.8.3. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology. John Wiley and Sons, New York, NY.
- 36.Steger, J. L., C. Vincent, J. D. Ballard, and L. R. Krumholz. 2002. Desulfovibrio sp. genes involved in the respiration of sulfate during metabolism of hydrogen and lactate. Appl. Environ. Microbiol. 68:1932-1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stolyar, S., Q. He, M. P. Joachimiak, Z. He, Z. K. Yang, S. E. Borglin, D. C. Joyner, K. Huang, E. Alm, T. C. Hazen, J. Zhou, J. D. Wall, A. P. Arkin, and D. A. Stahl. 2007. Response of Desulfovibrio vulgaris to alkaline stress. J. Bacteriol. 189:8944-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valente, F. M. A., L. M. Saraiva, J. LeGall, A. V. Xavier, M. Teixeira, and I. A. C. Pereira. 2001. A membrane-bound cytochrome c3: a type II cytochrome c3 from Desulfovibrio vulgaris Hildenborough. Chembiochem 2:895-899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.