

Abstract
Three genes, nadA, nadB, and nadC, involved in NAD de novo biosynthesis are broadly conserved in the genomes of numerous bacterial species. In the genome of Corynebacterium glutamicum, nadA and nadC but not nadB are annotated. The nadA and nadC genes are located in a gene cluster containing two other genes, designated ndnR and nadS herein. ndnR encodes a member of the Nudix-related transcriptional regulator (NrtR) family. nadS encodes a homologue of cysteine desulfurase involved in Fe-S cluster assembly. The gene cluster ndnR-nadA-nadC-nadS is genetically characterized herein. Mutant strains deficient in nadA, nadC, or nadS required exogenous nicotinate for growth, and the nicotinate auxotrophy was complemented by introduction of the corresponding gene in trans, indicating that each of these genes is essential for growth in the absence of an exogenous source of NAD biosynthesis. The results of reverse transcriptase PCR analyses and ndnR promoter-lacZ expression analyses revealed that the expression of ndnR, nadA, nadC, and nadS genes was markedly and coordinately repressed by nicotinate. The expression of these genes was enhanced by the disruption of ndnR, resulting in the loss of the nicotinate-responsive regulation of gene expression. These results suggest that NdnR acts as a transcriptional repressor of NAD de novo biosynthesis genes and plays an essential role in the regulation of the response to nicotinate.
NAD plays a crucial role in living organisms because it acts as a cofactor for numerous redox reactions in cellular metabolism. The biochemistry and genetics of NAD metabolism have been well studied in the enteric bacteria Salmonella enterica serovar Typhimurium and Escherichia coli (1-3, 7, 16, 22, 27, 30). NAD is synthesized from l-aspartate through the intermediate metabolite nicotinate mononucleotide (Fig. 1). The NAD de novo biosynthesis pathway converting l-aspartate to nicotinate mononucleotide proceeds via three enzymatic reactions catalyzed by l-aspartate oxidase, quinolinate synthetase, and quinolinate phosphoribosyltransferase. l-Aspartate oxidase encoded by nadB catalyzes the conversion of l-aspartate to iminoaspartate. Quinolinate synthetase encoded by nadA catalyzes the condensation of iminoaspartate with dihydroxyacetone phosphate. Quinolinate phosphoribosyltransferase encoded by nadC catalyzes the reaction between quinolinate and 5-phosphoribosyl-1-pyrophosphate to give nicotinate mononucleotide, pyrophosphate, and CO2. Nicotinate mononucleotide is also generated from nicotinate. Extracellular nicotinate and its derivatives, which are derived from pyridine nucleotide degradation, are taken up by bacteria and utilized for NAD biosynthesis. Thus, the salvage pathway complements the de novo biosynthesis of NAD, and the relevant pathways must be coordinately regulated for maintaining homeostasis of the NAD cofactor pool. While the NAD de novo biosynthesis genes are found in the genomes of a number of bacterial species, some microbes lack these genes and depend entirely on salvage of NAD precursors (4).
FIG. 1.
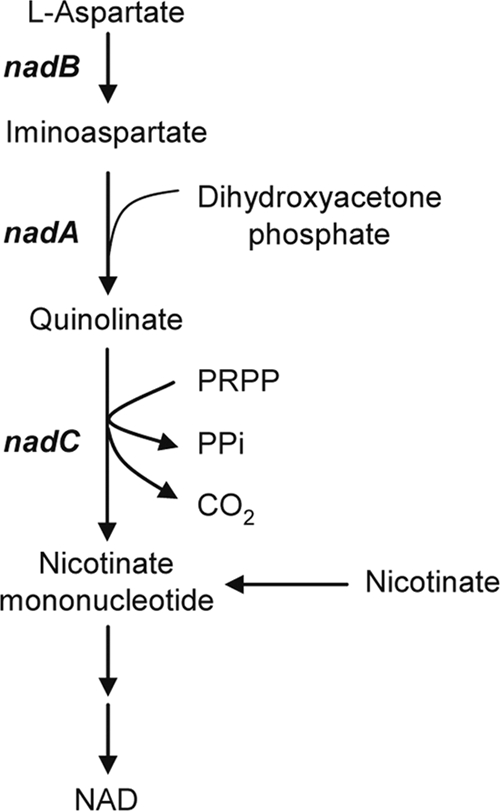
The NAD de novo biosynthesis pathway. l-Aspartate is converted to nicotinate mononucleotide through the three reactions catalyzed by l-aspartate oxidase, encoded by nadB, quinolinate synthetase, encoded by nadA, and quinolinate phosphoribosyltransferase, encoded by nadC. Nicotinate mononucleotide, which is also generated from nicotinate, is subsequently converted to NAD. PRPP, 5-phosphoribosyl-1-pyrophosphate; PPi, inorganic pyrophosphate.
In S. enterica serovar Typhimurium, the expression of nadB and nadA but not nadC is regulated by the transcriptional regulator NadR in response to the intracellular NAD level (6, 23). However, NadR-mediated regulation is limited to a compact phylogenetic group of enterobacteria (5). In Bacillus subtilis, a different transcriptional regulator, NiaR (YrxA), regulates the expression of the nadABC operon in response to the intracellular nicotinate level (26). NiaR homologues are broadly found in the bacterial genomes of the Bacillus/Clostridium group and in the deeply branching Fusobacteria and Thermotogales lineages (25). A recent comparative genomics study based on several hundred bacterial species anticipates that members of a novel family of transcriptional regulators, the Nudix-related transcriptional regulator (NrtR) family, are responsible for the regulation of various aspects of NAD metabolism, including the de novo biosynthesis, and that the regulatory networks are highly diverse among bacteria (24). However, the physiological role of the NrtR family regulators in vivo remains unclear.
Corynebacterium glutamicum, a Gram-positive soil bacterium, is widely used for the industrial production of amino acids, such as glutamate and lysine (13, 15). We have developed a bioprocess for lactate, succinate, and ethanol production using C. glutamicum (9, 10, 19-21). It is important to understand the regulation of NAD metabolism to enable optimal engineering based on the genome sequence of this industrially important microorganism (8, 12, 36). In the genome of C. glutamicum, putative nadA and nadC genes have been annotated, but no nadB orthologue has been identified to date. Thus, the functionality of C. glutamicum nadA and nadC is of particular interest.
In this study, we genetically characterized the nad gene cluster cgR_1153 (ndnR)-cgR_1152 (nadA)-cgR_1151 (nadC)-cgR_1150 (nadS) in the genome of C. glutamicum (Fig. 2). ndnR encodes an NrtR family protein. nadS encodes a homologue of cysteine desulfurase, which is believed to be required in E. coli and B. subtilis for the maturation of NadA, an Fe-S protein (14, 29, 31). Each of the nadA, nadC, and nadS genes was shown to be essential for growth when C. glutamicum cells are incubated in the absence of an exogenous source of NAD. Disruption of ndnR enhanced the levels of nadA, nadC, and nadS mRNAs, as well as the expression of the ndnR promoter-lacZ reporter gene fusion. As a result, the marked repression of these genes by nicotinate which was exhibited in the wild-type strain was completely eliminated, indicating that the regulation of the NAD de novo biosynthesis genes is primarily mediated by NdnR.
FIG. 2.
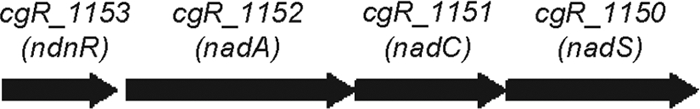
The nad gene cluster of C. glutamicum strain R. cgR_1153 (ndnR) encodes an NrtR family transcriptional regulator. cgR_1152 (nadA) encodes quinolinate synthetase. cgR_1151 (nadC) encodes quinolinate phosphoribosyltransferase. cgR_1150 (nadS) encodes a cysteine desulfurase-like protein.
MATERIALS AND METHODS
Bacterial strains.
C. glutamicum R (36) was used as a wild-type strain for our experiments. The cgR_1150 (nadS)-deficient strain was obtained from a single-gene-disruptant mutant library constructed by transposon-mediated mutagenesis (32). In this strain, the transposon is inserted 449 bases downstream of the 5′ end of the initiation codon of the nadS gene.
Culture conditions.
For genetic manipulations, E. coli and C. glutamicum strains were grown as described previously (33).
For analytical purposes, the C. glutamicum cell starter culture was grown aerobically in 10 ml nutrient-rich A medium (10) containing 4% glucose at 33°C in a 100-ml test tube overnight. The cells were harvested by centrifugation at 4,000 g for 10 min at 4°C. The cell pellet was subsequently washed twice with minimal BT medium (10). The washed cells were suspended with 100 ml BT medium containing glucose at 40 mM and supplemented with nicotinate at the concentrations indicated below and then aerobically cultured at 33°C in a 500-ml flask. To assess nicotinate auxotrophy, minimal BTM medium (BT medium supplemented with 1.0 mg liter−1 ZnSO4·7H2O, 0.2 mg liter−1 CuSO4·5H2O, 10 mg liter−1 CaCl2·H2O, 0.02 mg liter−1 NiCl2·6H2O, and 20 μM protocatechuate) was used.
Cell growth was monitored by measuring the optical density at 610 nm using a spectrophotometer (DU640; Beckman Coulter, CA).
DNA techniques.
Chromosomal DNA and plasmid DNA were prepared from C. glutamicum and the target DNA regions were amplified by PCR as described previously (33).
C. glutamicum cells were transformed by electroporation as described previously (34). E. coli cells were transformed by the CaCl2 procedure (28).
DNA sequencing was performed with an ABI Prism 3100xl genetic analyzer (Applied Biosystems, Foster City, CA). DNA sequence data were analyzed with the Genetyx program (Software Development, Tokyo, Japan).
Construction of mutants.
For gene deletion, the upstream and downstream regions of the gene to be deleted were amplified using the sets of primers summarized in Table 1. The resultant amplicons were fused and cloned into pCRA725 (9), a suicide vector for markerless gene disruption. The resultant plasmids, pCRC303, pCRC304, and pCRC305, were used for the deletion of nadA, nadC, and ndnR, respectively. C. glutamicum was subsequently transformed with the respective plasmid DNA, and screening for deletion mutants was performed as previously described (9). Deletion of the target genes was checked by PCR. In all cases, it was observed that each open reading frame (ORF) was deleted except for the 30-bp 5′- and 3′-terminal regions.
TABLE 1.
Primers used in this study
| Primer and purpose | Target gene | Sequence (5′-3′)a | Overhanging restriction site |
|---|---|---|---|
| Plasmid construction for gene deletion | |||
| nadA-U-Fw | nadA | AAACTGCAGCCAAAAAGGAAATAAAAGAC | PstI |
| nadA-U-Rv | GGGGTACCGTTGACAGATGGGGTGATTG | KpnI | |
| nadA-D-Fw | GGGGTACCGTTACTCCTAGCTCCTCGAA | KpnI | |
| nadA-D-Rv | CTCTGAGCTCGCAGAGGCGAGTCTTCCCAC | SacI | |
| nadC-U-Fw | nadC | CTCTGTCGACCTCTTTCCAACTTGCCCGCG | SalI |
| nadC-U-Rv | GGGGTACCGCCAACGATACGGTCAGTAT | KpnI | |
| nadC-D-Fw | GGGGTACCGCACTTGACCTAGGACTCGA | KpnI | |
| nadC-D-Rv | CTCTGAGCTCCTGATCCTCCTCAAACCCCAA | SacI | |
| ndnR-U-Fw | ndnR | CTCTGTCGACGGCTGGCCAACATCATGCCC | SalI |
| ndnR-U-Rv | TCCCCCGGGGGCCATCTGGATTTCAGGTG | SmaI | |
| ndnR-D-Fw | TCCCCCGGGCGCCCACCCAAACTGTTCAG | SmaI | |
| ndnR-D-Rv | CTCTGAGCTCGGGATTCAGGGTGCACGATG | SacI | |
| Plasmid construction for gene expression | |||
| nadA-Fw | nadA | CTCTGAGCTCTAGAAGAAAAGACCCCAATC | SacI |
| nadA-Rv | GGGGTACCTTACGCATCCTTCGAGGAGC | KpnI | |
| nadC-Fw | nadC | GGAATTCCCTCGAAGGATGCGTAATTT | EcoRI |
| nadC-Rv | CTCTGAGCTCTGCATTATCAAGGTAGAGCA | SacI | |
| nadS-Fw | nadS | CTCTGAGCTCCTAGGACTCGATATTTTCTA | SacI |
| nadS-Rv | GGGGTACCGCGTAAACCTCTGACTAGCG | KpnI | |
| ndnR-Fw | ndnR | GGAATTCCTTGCCCGCTTCACCTGAAAT | EcoRI |
| ndnR-Rv | GGGGTACCTTATCTTTGGAATCTGAACA | KpnI | |
| Plasmid construction for chromosomal integration of lacZ fusion | |||
| PndnR-Fw | ndnR promoter | CTCTTTTAAATCCACGTTGCAACCAGGAGT | DraI |
| PndnR-Rv | CTCTTTTAAATGAAGCGGGCAAGAAACCAC | DraI | |
| For RT-PCR analyses | |||
| 16S-F | 16S rRNA | TCGATGCAACGCGAAGAAC | |
| 16S-R | GAACCGACCACAAGGGAAAAC | ||
| nadA-F | nadA | TCACCTCAATTTATGGCGATGACAC | |
| nadA-R | GCGTTCAAACGCCCACTCA | ||
| nadC-F | nadC | TGGTGACAGCTTTGAGAC | |
| nadC-R | CTCTGAATGAAGTTGAGAG | ||
| nadS-F | nadS | GTGCGCCTAAAGGGATTGGAGT | |
| nadS-R | AGGCAGTGGCAAAGGCGATAG | ||
| ndnR-F | ndnR | AGCTATCTAGAACAGCTCTACACT | |
| ndnR-R | CGGACAAGTGCCCAATACAC | ||
| 1149-F | cgR_1149 | CTCATTCCAGCGTCACGA | |
| 1149-R | GCATGTCGTAATCAAATGTTCCTAA |
The restriction site overhangs used in the cloning procedure are underlined.
Construction of ndnR promoter-lacZ fusion.
A DNA fragment containing the ndnR promoter region was amplified by PCR using the C. glutamicum chromosomal DNA as a template and a set of primers with appropriate restriction sites (Table 1). The amplified DNA was digested with the appropriate restriction enzyme and inserted into the corresponding site of pCRA741 (11) to construct the ndnR promoter-lacZ fusion. The lacZ fusion was integrated into strain-specific island 7 (SSI7) on the chromosome of C. glutamicum R by markerless gene insertion methods as described previously (9).
Plasmids for gene expression.
Plasmids for the expression of the nadA, nadC, nadS, and ndnR genes were obtained as follows. The region for each of the ORFs was amplified by PCR using the C. glutamicum chromosomal DNA as a template and a set of primers with appropriate restriction sites (Table 1). The amplified ORF region was digested with the restriction enzymes and was inserted into the corresponding sites of the Escherichia coli-Corynebacterium shuttle vector pCRB1 (18), yielding pCRC306, pCRC307, pCRC308, and pCRC309 for the expression of nadA, nadC, nadS, and ndnR, respectively, under the control of the lac promoter.
Quantitative RT-PCR.
Total RNA was prepared from C. glutamicum cells using an RNeasy minikit and RNAprotect bacteria reagent (Qiagen, Hilden, Germany) and quantitative reverse transcriptase PCR (RT-PCR) was performed using an Applied Biosystems 7500 fast real-time PCR system as described previously (33). The primers used are listed in Table 1. Specific amplification of the targeted DNA was confirmed by electrophoresis and sequencing of the PCR product. The relative abundance of the targeted mRNAs was quantified based on the cycle threshold value, which is defined as the cycle number required to obtain a fluorescence signal above the background. To standardize the results, the relative abundance of 16S rRNA was used as the internal standard.
β-Galactosidase assay.
C. glutamicum cells were harvested, washed once with Z buffer (17), resuspended with the same buffer, and treated with toluene. The permeabilized cells were then incubated with o-nitrophenyl-β-d-galactopyranoside, and activity was measured in Miller units as previously described (17).
RESULTS
A cluster of NAD de novo biosynthesis genes in the genome of C. glutamicum.
In order to examine the functionality of the nadA and nadC genes, in-frame deletion mutants (ΔnadA and ΔnadC) were constructed. A mutant strain deficient in nadS (nadS::Tn) was obtained from a mutant library previously constructed by transposon-based insertion of a selection marker (32). When ΔnadA, ΔnadC, and nadS::Tn were cultured on plates containing minimal medium, a severe growth defect was observed compared to the growth of the wild-type strain. The growth defect was suppressed by supplementation with nicotinate. A plasmid carrying the nadA, nadC, or nadS gene under the control of a constitutive promoter was introduced into the respective mutant strain, ΔnadA, ΔnadC, or nadS::Tn. Each of the complemented strains was cultured in liquid minimal medium, and their growth was compared to that of the mutant strain and the wild-type strain, both of which were transformed with a vector plasmid without the target gene. When the ΔnadA cells cultured in nutrient-rich medium were transferred to minimal medium, growth tended to slow down. When an aliquot of the cell culture was subsequently transferred to new minimal medium, the growth was severely defective (Fig. 3A). In contrast, when an aliquot of the cell culture was transferred to medium supplemented with nicotinate, the ΔnadA strain grew in a manner comparable to the wild type. The nadA-complemented strain in either the presence or absence of nicotinate (Fig. 3B) grew as well as the wild-type strain (data not shown). Similar effects of disruption and complementation on nicotinate auxotrophy were observed in the case of nadC (Fig. 3C and D) and nadS (Fig. 3E and F). These results indicate that nadA, nadC, and nadS are each essential for growth in the absence of an exogenous source of NAD biosynthesis.
FIG. 3.

Effect of disruption of nadA, nadC, or nadS on growth of C. glutamicum cells. Strains deficient in nadA, nadC, or nadS containing either a control vector plasmid pCRB1 (A, C, and E, respectively) or a plasmid carrying nadA, nadC, and nadS under the control of a constitutive promoter (B, D, and F, respectively) were grown in minimal BTM medium with (circles) or without (squares) nicotinate supplementation. The optical density at 610 nm (OD610) was monitored. Similar results were obtained from two independent cultivations, and representative results are shown.
Repression of the expression of NAD de novo biosynthesis genes in response to nicotinate.
Previously, we performed DNA microarray analyses to examine the transcriptional profiles of densely packed C. glutamicum cells under oxygen-deprived conditions in minimal medium compared to those under normal aeration conditions in nutrient-rich medium at physiological cell densities (11). Under the former conditions, applicable for a high-productivity bioprocess, 161 genes were upregulated. Among them, the induction levels of all four genes in the NAD de novo biosynthesis gene cluster, ndnR, nadA, nadC, and nadS, are particularly prominent. This observation suggests that their induction is due to the absence of an exogenous source of NAD biosynthesis. Here, we examined the expression of these genes during aerobic growth of the C. glutamicum wild-type strain cultured in minimal medium and in nutrient-rich medium (Fig. 4A). Total RNA was prepared from cells cultured for 2 h, 4 h, and 6 h and was subjected to quantitative RT-PCR analyses. The level of ndnR mRNA was markedly upregulated during growth in minimal medium relative to the level in nutrient-rich medium (Fig. 4B). The expression pattern of nadS under these conditions was the same as that of ndnR (Fig. 4C).
FIG. 4.
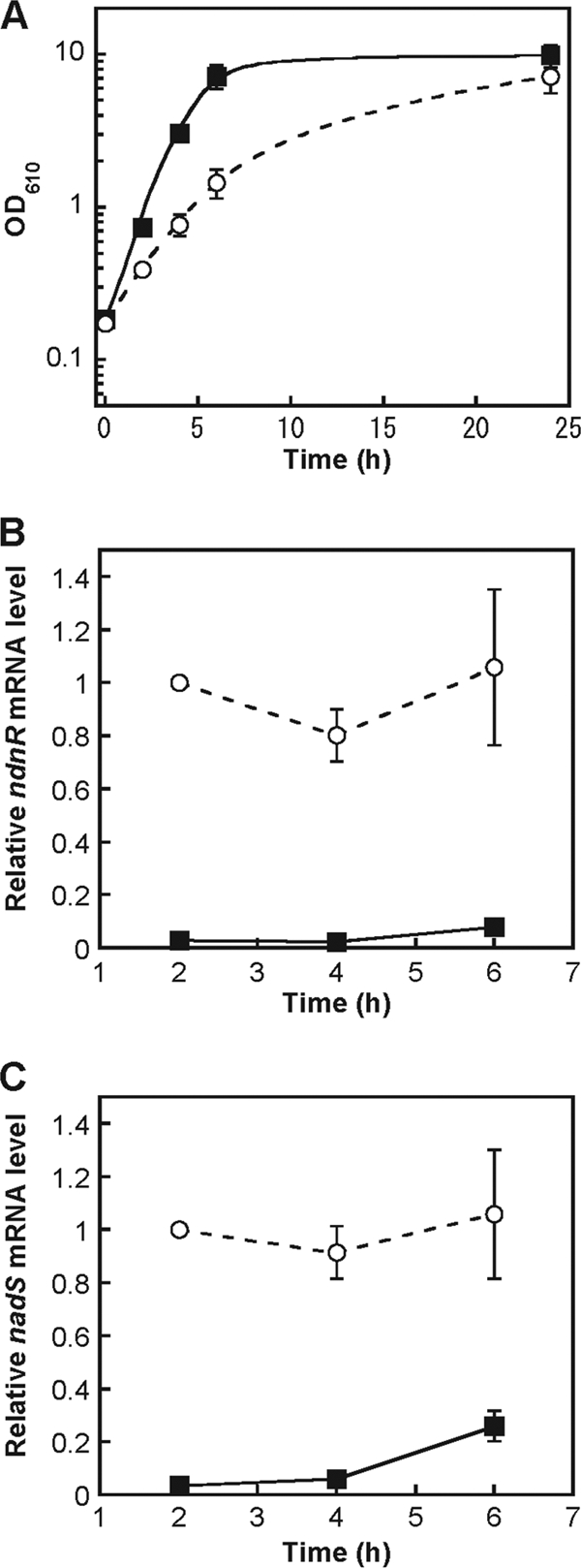
Expression of the ndnR and nadS genes during growth in nutrient-rich medium or in minimal medium. The C. glutamicum wild-type strain was cultured in nutrient-rich A medium (black rectangles) or in minimal BT medium (white circles). The optical density at 610 nm (OD610) was monitored (A). The levels of ndnR (B) and nadS (C) mRNAs in the cells were determined by quantitative RT-PCR. The mRNA levels are presented relative to the value from the 2-h culture in minimal medium. The values represent the mean results from three independent cultivations, with standard errors.
In order to examine the expression of the NAD de novo biosynthesis genes in response to nicotinate, C. glutamicum wild-type cells were cultured in minimal medium without nicotinate for 4 h and then were supplemented with nicotinate to a final concentration of 41 μM. Quantitative RT-PCR analyses revealed that ndnR, nadA, nadC, and nadS mRNAs rapidly decreased to extremely low levels within 12 min of nicotinate supplementation (Fig. 5 A). The ndnR-nadA and nadA-nadC intergenic regions are 48 bp and 3 bp long, respectively. nadC and nadS overlap by 1 bp. Therefore, it is likely that the ndnR-nadA-nadC-nadS genes are transcribed as an operon. The downregulation of the downstream genes in the cluster in response to nicotinate was relatively slow (Fig. 5A). This may be a reflection of a 5′ to 3′ direction of the mRNA degradation process. As a control, the expression of cgR_1149, encoding a major facilitator superfamily protein of unknown function, which is located immediately downstream of nadS in the opposite direction, was also analyzed in these experiments. In contrast to the ndnR-nadA-nadC-nadS genes, the level of cgR_1149 mRNA was little affected by nicotinate supplementation (Fig. 5A).
FIG. 5.
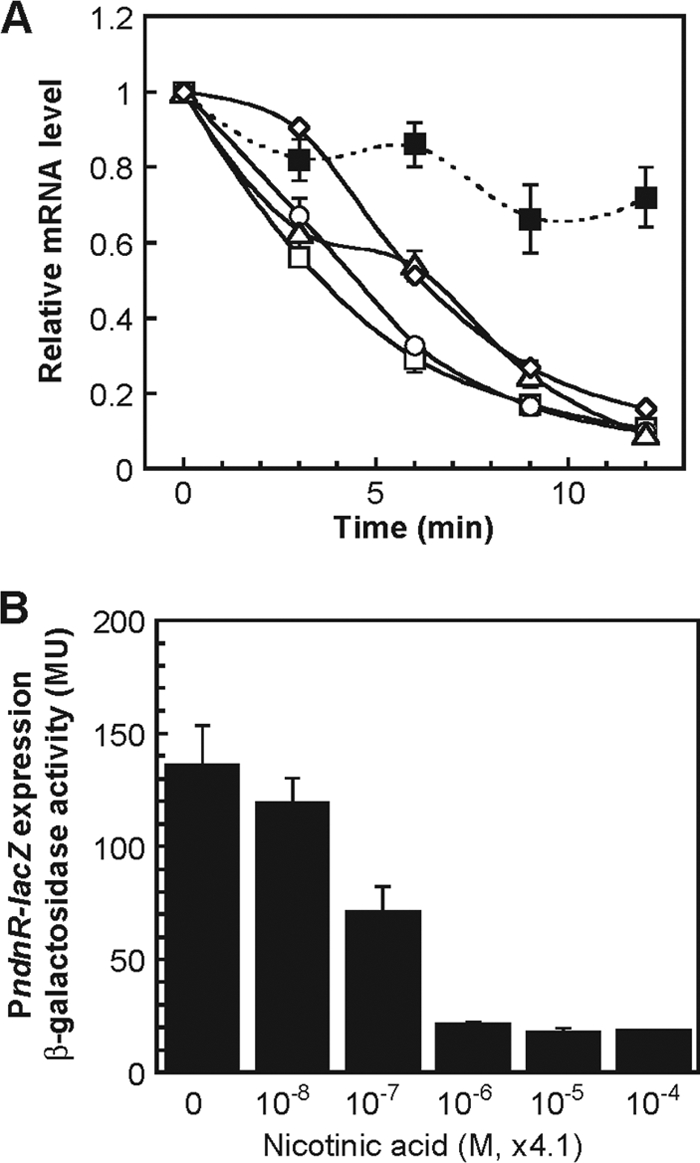
Expression of the ndnR, nadA, nadC, nadS, and cgR_1149 genes in response to nicotinate. (A) The C. glutamicum wild-type strain was cultured in minimal BT medium for 4 h, and then the culture was supplemented with nicotinate to the final concentration of 41 μM. The levels of ndnR (white rectangles), nadA (white circles), nadC (white triangles), nadS (white diamonds), and cgR_1149 (black rectangles) mRNAs in the cells 0, 3, 6, 9, and 12 min after the nicotinate supplementation were determined by quantitative RT-PCR. The mRNA levels are presented relative to the value attained using cells immediately before nicotinate supplementation (0 min). The values represent the means from three independent cultivations with standard errors. (B) C. glutamicum wild-type strain carrying the ndnR promoter-lacZ (PndnR-lacZ) fusion in the chromosome was cultured in minimal BT medium supplemented with nicotinate at 0, 0.041, 0.41, 4.1, 41, and 410 μM. The β-galactosidase activity in the exponentially growing cells was measured in Miller units (MU). The values represent the mean results from three independent cultivations, with standard errors.
The ndnR promoter-lacZ reporter gene (PndnR-lacZ) fusion was integrated into the genome of the C. glutamicum wild-type strain. The resultant strain was cultured in minimal medium supplemented with nicotinate at 0, 0.041, 0.41, 4.1, 41, and 410 μM. Cell growth in the presence of these various concentrations of nicotinate was mostly similar (data not shown). On the other hand, the β-galactosidase activity of exponentially growing cells was markedly enhanced as the concentration of nicotinate in the medium decreased, with nicotinate levels greater than 4.1 μM resulting in marked repression of the expression of PndnR-lacZ (Fig. 5B).
Involvement of NdnR in control of NAD de novo biosynthesis genes.
In order to examine the involvement of NdnR, an NrtR family transcriptional regulator, in the expression of ndnR, nadA, nadC, and nadS, its in-frame deletion mutant was constructed. The growth of the resultant ΔndnR strain was comparable to that of the wild-type strain in minimal medium in either the presence or absence of nicotinate (data not shown). Analyses of the levels of nadA mRNA in exponentially growing cells by quantitative RT-PCR revealed that, in the wild-type strain, nadA expression was severely repressed in the presence of nicotinate (Fig. 6). In contrast, in the ΔndnR strain, the level of nadA mRNA was markedly high in either the presence or absence of nicotinate. The nadA expression in the ΔndnR strain even in the absence of nicotinate was 3 times higher than the induced level in the wild-type strain, and the response to nicotinate was minimal in the ΔndnR strain. The effects of the disruption of ndnR on nadA, nadC, and nadS mRNAs were almost the same (Fig. 6).
FIG. 6.

Effects of disruption of ndnR on the expression of the nadA, nadC, and nadS genes. The C. glutamicum wild-type strain (WT) and the ndnR-deficient mutant strain (ΔndnR) were grown in minimal BT medium with (NA) or without nicotinate supplementation at 41 μM. The levels of ndnR, nadA, nadC, and nadS mRNAs in the exponentially growing cells were determined by quantitative RT-PCR. The mRNA levels are presented relative to the value for the wild-type strain grown in the absence of nicotinate. The values represent the mean results from three independent experiments, with standard errors.
Moreover, an in-frame deletion mutant of ndnR was constructed from a strain chromosomally integrated with PndnR-lacZ, and its β-galactosidase activity was compared to that of the parental strain. These strains grew to similar extents in minimal medium in either the presence or absence of nicotinate (data not shown). In the wild-type background, PndnR-lacZ expression in exponentially growing cells was markedly repressed in the presence of nicotinate relative to that in its absence, whereas in the ndnR-deficient mutant background, PndnR-lacZ expression was relatively high in either the presence or absence of nicotinate (Fig. 7A). Disruption of ndnR eliminated the response of PndnR-lacZ expression to nicotinate. The effects of disruption of ndnR on PndnR-lacZ expression were consistent with those on the levels of nadA, nadC, and nadS mRNAs, shown in Fig. 6, suggesting that these genes are probably cotranscribed as an operon under the control of the same promoter. A plasmid carrying the ndnR gene under the control of a constitutive promoter was introduced into the ndnR-deficient mutant strain containing PndnR-lacZ in the chromosome. The growth of the ndnR-complemented strain in the medium tested was comparable to that of the parental strain carrying a vector plasmid without ndnR (data not shown). In the ndnR-complemented strain, the nicotinate-repressible expression of PndnR-lacZ was restored (Fig. 7B).
FIG. 7.
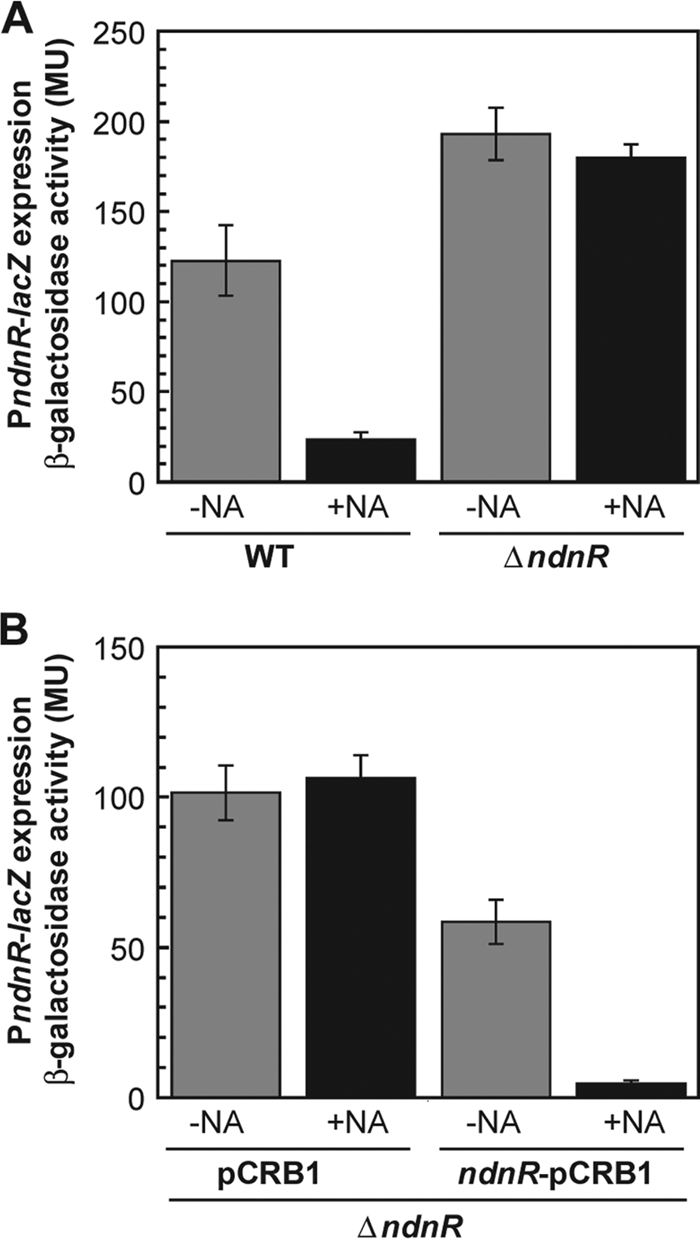
Effect of disruption of ndnR on the expression of the ndnR promoter-lacZ (PndnR-lacZ) fusion. (A) The C. glutamicum wild-type strain (WT) and the ndnR-deficient mutant strain (ΔndnR), both of which carry the PndnR-lacZ gene fusion in the chromosome, were cultured in minimal BT medium with (+NA) or without (-NA) nicotinate supplementation at 41 μM. The β-galactosidase activity in the exponentially growing cells was measured in Miller units (MU). The values represent the mean results from three independent cultivations, with standard errors. (B) The ndnR-deficient mutant strain (ΔndnR) with the PndnR-lacZ gene fusion containing a control vector plasmid (pCRB1) or a plasmid carrying the constitutive promoter-ndnR (ndnR-pCRB1) was grown in minimal BT medium with (+NA) or without (-NA) nicotinate supplementation at 41 μM. The β-galactosidase activity in the exponentially growing cells was measured. The values represent the mean results from three independent cultivations, with standard errors.
DISCUSSION
In this study, we showed that, in C. glutamicum, either nadA or nadC is essential for growth in the absence of an exogenous source of NAD, indicating that these genes are functional for NAD de novo biosynthesis in vivo. Although nadA and nadC, along with nadB, which are involved in the NAD biosynthesis pathway from l-aspartate, are broadly conserved in numerous bacterial species, nadB is not found in C. glutamicum (25). In the genome of C. glutamicum, nadC and nadA are located adjacent to each other in the same cluster that also includes two other genes, ndnR and nadS (Fig. 2). It is noted that, in the genomes of the related species belonging to Actinobacteria, such as Kineococcus radiotolerans and Leifsonia xyli, nadB occurs between nadA and nadC in the same cluster as in C. glutamicum. Among Corynebacterium species, the gene cluster in C. efficiens is the same as that in C. glutamicum, while in C. diphtheriae, nadB occurs between nadA and nadC but a cysteine desulfurase gene (an nadS homologue) does not exist in the cluster. Additionally, in the genome of C. diphtheriae, a gene encoding an NrtR family protein (an ndnR homologue) is located immediately upstream of nadA but in the opposite direction. Interestingly, the same nad gene cluster as in C. diphtheriae is observed in other species of Actinobacteria, such as Mycobacterium species and Nocardia farcinica. In many bacteria, it is likely that l-aspartate oxidase encoded by nadB catalyzes the conversion from l-aspartate to iminoaspartate (Fig. 1). However, the conversion seems to be replaced by a reaction catalyzed by a different type of enzyme, l-aspartate dehydrogenase, the gene of which is located in a gene cluster along with nadA and nadC in the thermophilic bacterium Thermotoga maritima (35). It is possible that the first reaction of NAD de novo biosynthesis in C. glutamicum is catalyzed by an unidentified enzyme. These findings imply that NAD de novo biosynthesis in bacteria is more diverse than previously thought with respect to not only regulation, as described below, but also the structural gene(s).
Our results in this study suggest that nadS is also involved in NAD de novo biosynthesis in C. glutamicum. NadS exhibits 35% amino acid sequence identity to E. coli IscS, which exhibits cysteine desulfurase activity and facilitates Fe-S cluster assembly (14, 29). Disruption of iscS leads to a decrease in the activity of a number of Fe-S proteins and results in generally poor growth. One of the growth traits of the iscS mutant is auxotrophy for nicotinate, suggesting that IscS is required for the maturation of NadA, an Fe-S protein (14, 22, 29). It is suggested that IscS functions primarily in de novo synthesis of Fe-S clusters rather than in the repair of oxidatively damaged clusters (29). In contrast to the E. coli iscS-deficient mutant strain, the C. glutamicum nadS-deficient mutant strain grew as well as the wild-type strain in nicotinate-supplemented minimal medium, suggesting that NadS is specifically involved in the de novo Fe-S cluster synthesis on the nascent NadA apoenzyme. Similar results are observed in the case of a B. subtilis mutant strain deficient in a cysteine desulfurase gene homologue, although the gene product has not yet been functionally characterized in detail (31). At present, whether or not the cysteine desulfurases specific for NAD de novo biosynthesis are additionally involved in Fe-S cluster repair under oxidative stress conditions remains to be determined.
We also showed that the expression of nadA and nadC, along with that of ndnR and nadS, is markedly repressed in cells cultured in the presence of nicotinate. The expression levels decrease rapidly after nicotinate supplementation and remain extremely low during growth in the presence of micromolar levels of nicotinate. These results suggest that C. glutamicum efficiently utilizes the exogenous precursor of NAD and strictly regulates NAD de novo biosynthesis in response to environmental conditions, resulting in the maintenance of NAD homeostasis. In this context, it is noted that mutant strains deficient in NAD biosynthesis genes grow to some extent in nicotinate-depleted medium, but this is perhaps due to carry over of minimal residual NAD precursors after the washing of cells cultured in nutrient-rich medium (Fig. 3). Disruption of ndnR, a gene that encodes a member of the NrtR family of transcriptional regulators, enhances the expression of the ndnR-nadA-nadC-nadS genes, suggesting that its product, NdnR, acts as a transcriptional repressor of the NAD de novo biosynthesis genes. The autoregulation of ndnR may contribute to the fine control of its downstream genes. It should be noted that disruption of ndnR completely eliminates the nicotinate response of the expression of the NAD de novo biosynthesis genes, indicating that the nicotinate-responsive regulation is primarily mediated by NdnR. NdnR is not homologous to either S. enterica serovar Typhimurium NadR or B. subtilis NiaR, which are transcriptional regulators of NAD de novo biosynthesis genes. Comparative genomics analyses suggest that target genes of the NrtR family members are diverse among bacterial species, since the regulators are predicted to be involved in not only various aspects of NAD metabolism but also sugar pentose utilization and phosphoribosyl pyrophosphate biogenesis (24). The NrtR family regulators are composed of an N-terminal domain homologous to the Nudix (nucleoside diphosphate with large variation of residues) hydrolase family and a C-terminal helix-turn-helix (HTH)-like domain. It is predicted that the Nudix and HTH domains bind to an NAD metabolite as an effector molecule and the promoter regions of the target genes, respectively. Further studies of the NdnR-mediated regulation mechanism in C. glutamicum will provide new aspects of the diverse regulation of NAD metabolism in bacteria.
Acknowledgments
We thank Crispinus A. Omumasaba (RITE) for critical reading of the manuscript.
This work was financially supported in part by the New Energy and Industrial Technology Development Organization (NEDO), Japan.
Footnotes
Published ahead of print on 2 July 2010.
REFERENCES
- 1.Bhatia, R., and K. C. Calvo. 1996. The sequencing, expression, purification, and steady-state kinetic analysis of quinolinate phosphoribosyl transferase from Escherichia coli. Arch. Biochem. Biophys. 325:270-278. [DOI] [PubMed] [Google Scholar]
- 2.Flachmann, R., N. Kunz, J. Seifert, M. Gütlich, F. J. Wientjes, A. Läufer, and H. G. Gassen. 1988. Molecular biology of pyridine nucleotide biosynthesis in Escherichia coli. Cloning and characterization of quinolinate synthesis genes nadA and nadB. Eur. J. Biochem. 175:221-228. [DOI] [PubMed] [Google Scholar]
- 3.Foster, J. W., Y. K. Park, T. Penfound, T. Fenger, and M. P. Spector. 1990. Regulation of NAD metabolism in Salmonella typhimurium: molecular sequence analysis of the bifunctional nadR regulator and the nadA-pnuC operon. J. Bacteriol. 172:4187-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gazzaniga, F., R. Stebbins, S. Z. Chang, M. A. McPeek, and C. Brenner. 2009. Microbial NAD metabolism: lessons from comparative genomics. Microbiol. Mol. Biol. Rev. 73:529-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerasimova, A. V., and M. S. Gelfand. 2005. Evolution of the NadR regulon in Enterobacteriaceae. J. Bioinform. Comput. Biol. 3:1007-1019. [DOI] [PubMed] [Google Scholar]
- 6.Grose, J. H., U. Bergthorsson, and J. R. Roth. 2005. Regulation of NAD synthesis by the trifunctional NadR protein of Salmonella enterica. J. Bacteriol. 187:2774-2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes, K. T., A. Dessen, J. P. Gray, and C. Grubmeyer. 1993. The Salmonella typhimurium nadC gene: sequence determination by use of Mud-P22 and purification of quinolinate phosphoribosyltransferase. J. Bacteriol. 175:479-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikeda, M., and S. Nakagawa. 2003. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl. Microbiol. Biotechnol. 62:99-109. [DOI] [PubMed] [Google Scholar]
- 9.Inui, M., H. Kawaguchi, S. Murakami, A. A. Vertès, and H. Yukawa. 2004. Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J. Mol. Microbiol. Biotechnol. 8:243-254. [DOI] [PubMed] [Google Scholar]
- 10.Inui, M., S. Murakami, S. Okino, H. Kawaguchi, A. A. Vertès, and H. Yukawa. 2004. Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J. Mol. Microbiol. Biotechnol. 7:182-196. [DOI] [PubMed] [Google Scholar]
- 11.Inui, M., M. Suda, S. Okino, H. Nonaka, L. G. Puskás, A. A. Vertès, and H. Yukawa. 2007. Transcriptional profiling of Corynebacterium glutamicum metabolism during organic acid production under oxygen deprivation conditions. Microbiology 153:2491-2504. [DOI] [PubMed] [Google Scholar]
- 12.Kalinowski, J., B. Bathe, D. Bartels, N. Bischoff, M. Bott, A. Burkovski, N. Dusch, L. Eggeling, B. J. Eikmanns, L. Gaigalat, A. Goesmann, M. Hartmann, K. Huthmacher, R. Krämer, B. Linke, A. C. McHardy, F. Meyer, B. Möckel, W. Pfefferle, A. Pühler, D. A. Rey, C. Rückert, O. Rupp, H. Sahm, V. F. Wendisch, I. Wiegräbe, and A. Tauch. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J. Biotechnol. 104:5-25. [DOI] [PubMed] [Google Scholar]
- 13.Kinoshita, S., S. Udaka, and M. Shimonō. 1957. Studies on the amino acid fermentation. Part 1. Production of L-glutamic acid by various microorganisms. J. Gen. Appl. Microbiol. 3:193-205. [PubMed] [Google Scholar]
- 14.Lauhon, C. T., and R. Kambampati. 2000. The iscS gene in Escherichia coli is required for the biosynthesis of 4-thiouridine, thiamin, and NAD. J. Biol. Chem. 275:20096-20103. [DOI] [PubMed] [Google Scholar]
- 15.Liebl, W. 2005. Corynebacterium taxonomy, p. 9-34. In L. Eggeling and M. Bott (ed.), Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton, FL.
- 16.Magni, G., A. Amici, M. Emanuelli, N. Raffaelli, and S. Ruggieri. 1999. Enzymology of NAD+ synthesis. Adv. Enzymol. Relat. Areas Mol. Biol. 73:135-182. [DOI] [PubMed] [Google Scholar]
- 17.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 18.Nakata, K., M. Inui, P. B. Kos, A. A. Vertès, and H. Yukawa. 2003. Vectors for the genetics engineering of corynebacteria., p. 175-191. In B. C. Saha (ed.), Fermentation biotechnology. American Chemical Society, Washington, DC.
- 19.Okino, S., M. Inui, and H. Yukawa. 2005. Production of organic acids by Corynebacterium glutamicum under oxygen deprivation. Appl. Microbiol. Biotechnol. 68:475-480. [DOI] [PubMed] [Google Scholar]
- 20.Okino, S., R. Noburyu, M. Suda, T. Jojima, M. Inui, and H. Yukawa. 2008. An efficient succinic acid production process in a metabolically engineered Corynebacterium glutamicum strain. Appl. Microbiol. Biotechnol. 81:459-464. [DOI] [PubMed] [Google Scholar]
- 21.Okino, S., M. Suda, K. Fujikura, M. Inui, and H. Yukawa. 2008. Production of D-lactic acid by Corynebacterium glutamicum under oxygen deprivation. Appl. Microbiol. Biotechnol. 78:449-454. [DOI] [PubMed] [Google Scholar]
- 22.Ollagnier-de Choudens, S., L. Loiseau, Y. Sanakis, F. Barras, and M. Fontecave. 2005. Quinolinate synthetase, an iron-sulfur enzyme in NAD biosynthesis. FEBS Lett. 579:3737-3743. [DOI] [PubMed] [Google Scholar]
- 23.Penfound, T., and J. W. Foster. 1999. NAD-dependent DNA-binding activity of the bifunctional NadR regulator of Salmonella typhimurium. J. Bacteriol. 181:648-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodionov, D. A., J. De Ingeniis, C. Mancini, F. Cimadamore, H. Zhang, A. L. Osterman, and N. Raffaelli. 2008. Transcriptional regulation of NAD metabolism in bacteria: NrtR family of Nudix-related regulators. Nucleic Acids Res. 36:2047-2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodionov, D. A., X. Li, I. A. Rodionova, C. Yang, L. Sorci, E. Dervyn, D. Martynowski, H. Zhang, M. S. Gelfand, and A. L. Osterman. 2008. Transcriptional regulation of NAD metabolism in bacteria: genomic reconstruction of NiaR (YrxA) regulon. Nucleic Acids Res. 36:2032-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossolillo, P., I. Marinoni, E. Galli, A. Colosimo, and A. M. Albertini. 2005. YrxA is the transcriptional regulator that represses de novo NAD biosynthesis in Bacillus subtilis. J. Bacteriol. 187:7155-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rousset, C., M. Fontecave, and S. Ollagnier de Choudens. 2008. The [4Fe-4S] cluster of quinolinate synthase from Escherichia coli: investigation of cluster ligands. FEBS Lett. 582:2937-2944. [DOI] [PubMed] [Google Scholar]
- 28.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 29.Schwartz, C. J., O. Djaman, J. A. Imlay, and P. J. Kiley. 2000. The cysteine desulfurase, IscS, has a major role in in vivo Fe-S cluster formation in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 97:9009-9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seifert, J., N. Kunz, R. Flachmann, A. Läufer, K. D. Jany, and H. G. Gassen. 1990. Expression of the E. coli nadB gene and characterization of the gene product L-aspartate oxidase. Biol. Chem. Hoppe Seyler 371:239-248. [PubMed] [Google Scholar]
- 31.Sun, D., and P. Setlow. 1993. Cloning, nucleotide sequence, and regulation of the Bacillus subtilis nadB gene and a nifS-like gene, both of which are essential for NAD biosynthesis. J. Bacteriol. 175:1423-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki, N., N. Okai, H. Nonaka, Y. Tsuge, M. Inui, and H. Yukawa. 2006. High-throughput transposon mutagenesis of Corynebacterium glutamicum and construction of a single-gene disruptant mutant library. Appl. Environ. Microbiol. 72:3750-3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teramoto, H., T. Shirai, M. Inui, and H. Yukawa. 2008. Identification of a gene encoding a transporter essential for utilization of C4 dicarboxylates in Corynebacterium glutamicum. Appl. Environ. Microbiol. 74:5290-5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vertès, A. A., M. Inui, M. Kobayashi, Y. Kurusu, and H. Yukawa. 1993. Presence of mrr- and mcr-like restriction systems in coryneform bacteria. Res. Microbiol. 144:181-185. [DOI] [PubMed] [Google Scholar]
- 35.Yang, Z., A. Savchenko, A. Yakunin, R. Zhang, A. Edwards, C. Arrowsmith, and L. Tong. 2003. Aspartate dehydrogenase, a novel enzyme identified from structural and functional studies of TM1643. J. Biol. Chem. 278:8804-8808. [DOI] [PubMed] [Google Scholar]
- 36.Yukawa, H., C. A. Omumasaba, H. Nonaka, P. Kós, N. Okai, N. Suzuki, M. Suda, Y. Tsuge, J. Watanabe, Y. Ikeda, A. A. Vertès, and M. Inui. 2007. Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology 153:1042-1058. [DOI] [PubMed] [Google Scholar]