

Abstract
MicroRNAs (miRNAs) are a class of noncoding RNAs of lengths ranging from 18 to 23 nucleotides (nt) that play critical roles in a wide variety of biological processes. There is a growing amount of evidence that miRNAs play critical roles in intricate host-pathogen interaction networks, but the involvement of miRNAs during influenza viral infection is unknown. To determine whether the cellular miRNAs play an important role in H1N1 influenza A viral infections, 3′ untranslated region (UTR) reporter analysis was used to identify putative miRNA targets in the influenza virus genome, and virus proliferation analysis was used to detect the effect of the screened miRNAs on the replication of H1N1 influenza A virus (A/WSN/33) in MDCK cells. The results showed that miRNA 323 (miR-323), miR-491, and miR-654 inhibit replication of the H1N1 influenza A virus through binding to the PB1 gene. Moreover mutational analysis of the predicted miRNA binding sites showed that the three miRNAs bind to the same conserved region of the PB1 gene. Intriguingly, despite the fact that the miRNAs and PB1 mRNA binding sequences are not a perfect match, the miRNAs downregulate PB1 expression through mRNA degradation instead of translation repression. This is the first demonstration that cellular miRNAs regulate influenza viral replication by degradation of the viral gene. Our findings support the notion that any miRNA has antiviral potential, independent of its cellular function, and that the cellular miRNAs play an important role in the host, defending against virus infection.
MicroRNAs (miRNAs) are small RNA molecules with lengths of 21 to 23 nucleotides (nt) (21, 41). They have been detected in many plant and animal species and even in some animal viral RNA genomes (3, 27, 39). MiRNAs regulate many cellular processes, including cellular proliferation, apoptosis, homeostasis, and tumor formation by binding to the target mRNAs, causing target cleavage or translational block (6, 36). Currently, it is believed that the choice of posttranscriptional mechanisms is determined by the extent to which the miRNAs and their target transcripts are complementary to one another (13, 20, 47). Perfect or near-perfect matches, as is common in plant microRNAs and in a small class of animal microRNAs, cause target cleavage and degradation analogous to the action of small interfering RNAs (siRNAs) (42). However, in most animal cells, miRNA-mRNA base pairing is imperfect, and the mRNA is not cleaved. Instead, the translational efficiency of the mRNA is reduced (27, 34). In general, the 5′ portion of the miRNA (2 to 8 nt, termed the 5′ seed region) is perfectly complementary to 3′-UTR elements in the mRNA and is thought to be important in mediating posttranscriptional repression (23, 24).
An increasing number of studies suggest that viral miRNAs are key in controlling viral infection in mammalian hosts via several distinct mechanisms (9, 18, 31, 43). Simian virus 40, a member of the polyomavirus family, encodes miRNAs that target the gene encoding a major viral protein, the T antigen. The T antigen is a dominant target of the cytotoxic T lymphocyte (CTL) response, and downregulation of its expression decreases CTL-mediated lysis of infected cells (38). Another DNA virus, herpes simplex virus type 1 (HSV-1), is an example of a viral miRNA that targets a cellular gene. A remarkable feature of HSV-1 is the fact that it can establish latent infections and can remain undetected in cells for years. The viral latency-associated transcript (LAT) plays a critical role in this phenomenon by inhibiting apoptosis of infected cells. A miRNA produced from LAT, miR-LAT, targets the cellular mRNAs encoding two components of the transforming growth factor β (TGF-β) pathway (TGF-β and the transcription factor SMAD3) that regulate cell proliferation and programmed cell death (15). Therefore, it is clear that viral miRNAs can control expression of viral or cellular genes in order to interfere with antiviral host defense.
In addition to the role of viral RNAs in the host-pathogen interaction, some reports suggest that cellular miRNAs can also regulate viral infections. For example, miR-32 has been shown to target a sequence in the genome of the primate foamy virus type 1 (PFV-1) (25). Two other cellular miRNAs, miR-24 and miR-93, target the viral large protein (L protein) and phosphoprotein (P protein) genes, and decreased miR-24 and miR-93 expression has been shown to lead to increased vesicular stomatitis virus (VSV) replication (32). Furthermore, Huang et al. reported that cellular miRNAs potently inhibit HIV-1 production in resting primary CD4+ T cells (19). They found that the 3′ ends of HIV-1 mRNAs are targeted by a cluster of cellular miRNAs that include miR-28, miR-125b, miR-150, miR-223, and miR-382, which are enriched in resting CD4+ T cells compared to activated CD4+ T cells. Their data indicate that cellular miRNAs are pivotal to HIV-1 latency and suggest that manipulation of cellular miRNAs could represent a novel approach for purging the HIV-1 reservoir. Another miRNA, miR-122, is specifically expressed and is highly abundant in the human liver. Sequestration of miR-122 in liver cells results in a marked loss of autonomously replicating hepatitis C viral RNAs. Therefore, miR-122 likely facilitates replication of the viral RNA, suggesting that miR-122 may represent a potential therapeutic target for antiviral intervention (10, 22).
The H1N1 influenza A viruses (IAV) continue to pose serious threats to public health, as exemplified by the ongoing 2009 H1N1 influenza pandemic (14, 30). Li et al. reported that cellular miRNA expression can be altered during influenza virus infection, irrespective of the lethality of the virus (26). However, the involvement of miRNAs during IAV infection or replication is still unclear. Since there is strong evidence that cellular miRNAs can be used by host cells to resist the viral infection, we hypothesized that one or more cellular miRNAs could be involved in the replication of H1N1 IAV. In this report, we screened miR-323, miR-491, and miR-654, which inhibit the replication of the H1N1 IAV in MDCK cells through binding to the conserved region of the PB1 gene. To determine the action model of these miRNAs, we used real-time PCR to detect the PB1 mRNA levels in infected MDCK cells overexpressing miR-323, miR-491, and miR-654. The results showed that despite the fact that the miRNAs and PB1 mRNA binding sequences are not a perfect match, the miRNAs downregulate PB1 expression through mRNA degradation. In addition, real-time PCR analysis showed that endogenous miR-323, miR-491, and miR-654 are expressed in different cell lines and expression of them can be altered during influenza virus infection. Our results suggest that cellular miRNAs can inhibit the replication of the H1N1 IAV through downregulating the viral gene expression in infected MDCK cells. Our results are consistent with the emerging notion that miRNAs might be broadly implicated in viral infection of mammalian cells, with either a positive or negative effect on replication.
MATERIALS AND METHODS
Cell culture.
293T and MDCK cells (purchased from ATCC) were cultured in Earle's modified Eagle's medium containing 10% fetal bovine serum (HyClone), 2 mM l-glutamine, 100 units/ml penicillin, and 100 g/ml streptomycin at 37°C under 5% CO2 with 95% air atmosphere.
Plasmid construction.
The luciferase expression vector pRL-TK (Promega) was used as the parent vector for 3′-UTR reporter analysis experiments. The eight segments of the H1N1 IAV (the HA, NA, NP, NS, PA, PB1, PB2, and M genes) and subfragments of the PB1 gene (the PB1-1, PB1-2, PB1-3, PB1-4, and PB1-5 genes) were amplified by PCR from the 12-plasmid IAV reverse-genetics system (H1N1 [A/WSN/33]) and were directionally cloned into the 3′ UTR of the luciferase gene in the pRL-TK vector as shown in Fig. 1 a. These constructed vectors were named pRL-TK-HA, pRL-TK-NA, pRL-TK-NP, pRL-TK-NS, pRL-TK-PA, pRL-TK-PB1, pRL-TK-PB2, pRL-TK-M, pRL-TK-PB1-1, pRL-TK-PB1-2, pRL-TK-PB1-3, pRL-TK-PB1-4, and pRL-TK-PB1-5. In order to facilitate cloning, an XbaI restriction site was added to the 5′ primer and the 3′ primer. For Western blot assays, luciferase and the luciferase-PB1-5 fragment were amplified by PCR using the pRL-TK-PB1-5 vector as a template. The amplified fragment was then cloned into the EcoRI and XhoI sites of the pcDNA3.0-FLAG vector (Invitrogen), generating the Flag-thymidine kinase (TK) and Flag-TK-PB1-5 vector. The PB1 gene fragment was amplified by PCR using the PB1 vector in the 12-plasmid IAV reverse-genetics system as a template. The amplified fragment was then cloned into the HindIII and XhoI sites of the pcDNA3.0-FLAG vector (Invitrogen), generating the Flag-PB1 vector. All inserts were sequenced in their entirety in order to verify polymerase fidelity. The primer sequences are available from the corresponding author upon request.
FIG. 1.

The PB1 gene may harbor potential binding sites for miRNAs. (a) Circular map of the pRL-TK vector. The IAV genes were inserted into the XbaI sites. (b) The relative luciferase activity from the pRL-TK reporter constructs carrying one of eight fragments of the H1N1 IAV and pGL3-control vector. (c) Schematic map of the PB1 genome. Numbers denote nucleotide positions spanned by fragments of the PB1 gene that were inserted into the 3′ UTR of the luciferase gene in pRL-TK. (d) The relative luciferase activity from pRL-TK reporter constructs carrying fragments from the dissection of the PB1 gene and pGL3-control vector. The relative luciferase activity from pRL-TK and pGL3-control vector was used as control.
Construction of mutant plasmids.
In order to identify the miRNA binding sites in the PB1 gene, the nucleotide sequences of putative binding sites in the PB1 gene were mutated in pRL-TK-PB1-5, Flag-PB1, and PB1 in the 12-plasmid IAV reverse-genetics system using the Muta-Direct kit (SBS) according to the manufacturer's protocol. The primers were designed according to the instructions provided in the kit (primer sequences are available from the corresponding author upon request). Due to the low efficiency of recovery of mutants when four nucleotides are changed at once, the mutant vectors were obtained through two successive rounds of PCR. Mutant plasmids were generated by PCR using 20 ng of the parent vector as template under the following conditions: 95°C for 5 min, followed by 25 cycles of 95°C for 30 s, 58°C for 1 min, and 72°C for 8 min. The resulting mixture was digested with 1 μl of Dpn-1 for 30 min at 37°C in order to remove the parental DNA. The remaining DNA was used to transform DH5α (TaKaRa), and a reasonable number of colonies were obtained. Mutant plasmids were confirmed by sequencing. The mutated pRL-TK-PB1-5, Flag-PB1, and PB1 vectors were named ΔpRL-TK-PB1-5, ΔFlag-PB1, and ΔPB1, respectively. In order to construct expression vectors for the mutant miRNAs, the mutant sequences were synthesized directly. The mutant miRNA vectors were named ΔmiR-323, ΔmiR-491, and ΔmiR-654.
Prediction of miRNA-binding sites.
MiRNA binding sites were predicted according to the principles of miRNA target recognition (5, 7, 33, 44). In brief, target sites for miRNAs were predicted using the MicroInspector algorithm at http://bioinfo.uni-plovdiv.bg/microinspector/. The cutoff values for hybridization temperature and free energy were set to 37°C and −17 kcal/mol, respectively. Identified miRNA-target gene pairs were confirmed using RNAHybrid at http://bibiserv.techfak.uni-bielefeld.de/ and the rna22 miRNA target predictor at http://cbcsrv.watson.ibm.com/rna22.html. Finally, cross-species sequence comparison was used to detect whether the target sequence had been conserved evolutionarily between related species.
MiRNA inhibitors and expression vectors.
MiRNA inhibitors were purchased from Ambion (AM17000). The psiSTRIKE vectors (Promega) were used to construct miRNA expression vectors according to the manufacturer's protocol. Gene sequences for miRNAs were acquired from the Sanger miRNA Registry at http://microrna.sanger.ac.uk/sequences/. In short, the primers were annealed to form a double-stranded DNA and were then inserted into the psiSTRIKE vectors. Bacteria were transformed with the resulting constructs, and a reasonable number of colonies were obtained. The resulting miRNA expression vectors were confirmed by digestion with the restriction enzyme PstI according to the manufacturer's protocol. In addition, we constructed the expression vector of Caenorhabditis elegans miR-239b and siRNA-PB1 as the negative control and the positive control, respectively. Primer sequences are available from the corresponding author upon request.
Transfection.
The plasmids and miRNA inhibitors were transfected into cells using TransFast transfection reagent (Promega).
To determine whether miRNAs play a direct role in repression of luciferase expression from the pRL-TK vector containing the viral gene, 293T cells were plated in 24-well plates. When the cells reached 60 to 70% confluence, they were cotransfected with the pRL-TK vector containing the appropriate viral gene (0.5 μg) and the pGL3-control vector (0.01 μg). The empty pRL-TK vector and pGL3-control vector were used as a negative control. After 30 h, the cells were harvested for relative luciferase assay analysis.
To determine the effect of miRNAs on the luciferase expression from pRL-TK-PB1-5, 293T cells were plated in 24-well plates. When the cells reached 50 to 60% confluence, they were transfected with the indicated miRNA expression vectors (0.5 μg). After 24 h, the cells were cotransfected with the pGL3-control vector (0.01 μg) and the pRL-TK-PB1-5 vector (0.5 μg). The cells were harvested at 30 h posttransfection and were analyzed for relative luciferase activity.
To determine whether miR-323, miR-491, and miR-654 could downregulate the luciferase expression in Flag-TK-PB1-5, 293T cells were plated in 6-well plates. When the cells reached a confluence of 50 to 60%, they were either transfected with the appropriate miRNA expression vector alone (2.5 μg) or were cotransfected with the appropriate miRNA expression vector (1.5 μg) and the appropriate miRNA inhibitor (150 pmol). At 24 h posttransfection, cells were transfected with the Flag-TK-PB1-5 vector or Flag-TK (2.5 μg). After 48 h, the cells were collected for Western blot analysis.
To investigate the miRNA binding sites in the PB1 gene, 293T cells were plated in 24-well plates. When the cells reached 50 to 60% confluence, they were transfected with wild-type or mutant miRNA expression vectors (0.5 μg). After 24 h, the cells were cotransfected with the pGL3-control vector (0.01 μg) and the pRL-TK-PB1-5 vector or the ΔpRL-TK-PB1-5 vector (0.5 μg). The cells were harvested at 30 h posttransfection and were analyzed for relative luciferase activity.
To determine the effect of miR-323, miR-491, and miR-654 on the PB1 expression from Flag-PB1, MDCK cells were plated in 6-well plates. When the cells reached a confluence of 50 to 60%, they were transfected with the miRNA expression vector (2.5 μg). At 24 h posttransfection, cells were transfected with the Flag-PB1 or ΔFlag-PB1 vector (2.5 μg). After 48 h, the cells were collected and analyzed by a Western blot assay and real-time PCR.
In order to determine the effects of miRNAs on the replication of H1N1 IAV, MDCK cells were plated in 6-well plates. When the cells reached a confluence of 70 to 80%, the miRNA expression vectors (0.5 μg) or specific miRNA inhibitors were transfected into cells. After 24 h, the cells were infected with H1N1 IAV or with mutant H1N1 IAV at a hemagglutination (HA) value of 4. The virus replication was measured by an HA assay, fluorescence, and real-time PCR.
Luciferase assay.
Luciferase assays were performed using the dual-luciferase reporter assay system kit (Promega) according to the manufacturer's protocol. Collected cells were washed once with cold phosphate-buffered saline (PBS). Passive lysis buffer (100 μl) was then added to the cells. After 10 min, the supernatants were collected by centrifugation at 12,000 × g for 30 s, and the relative luciferase expression values were analyzed using the Modulus single-tube multimode reader (Promega).
The relative luciferase expression equals the expression of Renilla luciferase (pRL-TK) divided by the expression of firefly luciferase (pGL3-control vector).
Western blot assay.
After transfection, the cells were collected and washed with cold PBS three times. The PBS was decanted, and the cell pellet was resuspended in 100 μl of lysis buffer (Promega) for 10 min on ice. The resulting solution was then combined with 25 μl 5× SDS sample buffer and boiled for 10 min. The samples were resolved on a 12% SDS-PAGE gel and transferred to a polyvinylidene difluoride (PVDF) membrane (Amersham Bioscience). The membranes were blocked using 5% nonfat dry milk for 1 h. Membranes were then incubated overnight at 4°C in a purified primary mouse anti-Flag antibody and an anti-β-actin antibody (BD Biosciences), each at a 1:200 dilution in 5% nonfat milk. After three washes with Tris-buffered saline containing 0.05% Triton X-100 (TBST), the membranes were incubated for 1 h at room temperature with the appropriate horseradish peroxidase-conjugated secondary antibody (Santa Cruz) at a 1:5,000 dilution in TBST. Protein bands were visualized using the Super ECL Plus system (Applygen Technologies). β-Actin was used as a loading control.
Real-time reverse transcription-PCR analysis.
In order to determine the level of expression of PB1 mRNA or ΔPB1 mRNA, total RNA was prepared, and 2 μg of total RNA was reverse transcribed into cDNA using the QuantiTect reverse transcription kit (Qiagen) according to the manufacturer's protocol. The level of β-actin mRNA was measured as a control. PCR amplification of β-actin and PB1 or ΔPB1 cDNAs was carried out.
In order to detect the cellular expression level of miRNAs, total RNA was prepared, and 2 μg of the total RNA was reverse transcribed into cDNA using the QuantiMir Complete kit (SBI) according to the manufacturer's protocol. PCR amplification of the miRNA cDNAs was carried out. The expression of U6 was measured as a control using the primers provided in the kit.
Real-time PCR was conducted using 2 μl of cDNA diluted 1:50 and SYBR premix Ex Taq II (TaKaRa). Cycling conditions for real-time PCR were as follows: 95°C for 1 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Real-time PCR was conducted using the ABI Prism 7300 sequence detection system, and the data were analyzed with ABI Prism 7300 SDS software (Applied Biosystems). The primer sequences used in real-time PCR are available from the corresponding author upon request.
Virus infection.
H1N1 IAV (A/WSN/33) or mutant H1N1 IAV was used to infect the MDCK cells. The cells were washed with PBS three times and infected with the virus in infection medium (culture medium lacking fetal calf serum [FCS] and containing 2.5 μg/ml trypsin). The HA value of the viruses was 4. After incubation for 1 h, the cells were washed three times with PBS, and then infection medium was added to the cells. The infected cells were cultured at 35°C in 5% CO2.
HA assay.
The HA assay was carried out in V-bottom 96-well plates. Serial 2-fold dilutions of virus samples were mixed with an equal volume of a 0.5% (vol/vol) suspension of chicken erythrocytes and incubated for 20 min. Wells containing an adherent, homogeneous layer of erythrocytes were scored as positive.
Fluorescence microscopy.
Infected MDCK cells were grown on coverslips. The cells were fixed by incubation in cold acetone for 10 min. The coverslips were washed three times with PBS. The cells were then incubated overnight at 4°C with an anti-HA mouse monoclonal antibody at 1:50 (Santa Cruz). The coverslips were washed three times with PBS. The cells were then incubated in the secondary antibody, goat anti-mouse-fluorescein isothiocyanate (FITC) (Santa Cruz) at 1:200, for 1 h at room temperature. Following this incubation, the cells were washed three times with PBS, mounted onto slides in 90% glycerin, and sealed with nail polish. Stained cells were observed by fluorescence microscopy (Olympus; MT6000 Fluorescent).
Generation of mutant infectious influenza virus particles.
The 12-plasmid IAV reverse-genetics system was kindly provided by George F. Gao (Institute of Microbiology, Chinese Academy of Sciences, Beijing, China). Using this system, the PB1 gene nucleotide sequence was mutated without changing the amino acid sequence. To generate the mutant IAV, 293T cells were plated in a 100-mm dish. When the cells reached 80 to 90% confluence, the 12-plasmid IAV system containing ΔPB1 (2 μg/vector, totaling 24 μg of vectors in 6 ml of culture medium) was transfected into 293T cells using TransFast transfection reagent (Promega). After 72 h, the influenza viruses were harvested and propagated in MDCK cells. The mutations in ΔPB1 were confirmed by sequencing.
RESULTS
The PB1 gene of the H1N1 IAV may harbor potential miRNA binding sites.
Recently, an increasing number of reports suggest that cellular miRNAs play an important role in viral replication. Therefore, we utilized a 3′-UTR reporter assay to determine whether cellular miRNAs have a direct role in repression of H1N1 IAV gene expression in cells. Eight fragments of the H1N1 IAV were subcloned into the luciferase reporter plasmid (pRL-TK) between the luciferase open reading frame (ORF) and the polyadenylation signal (Fig. 1a). As shown in Fig. 1b, the relative luciferase activity in 293T cells cotransfected with pRL-TK-PB1 and pGL3-control vector (bar 1) was approximately 30% less than that of cells cotransfected with pRL-TK and pGL3-control vector (bar 9). We speculated that the decreased expression of the reporter gene may be attributed to miRNA-mediated repression. In order to better characterize the location of this element, five smaller fragments of the PB1 gene (Fig. 1c) were individually inserted into the 3′ UTR of the luciferase gene in the pRL-TK vector (Fig. 1a). As shown in Fig. 1d, the relative luciferase activity in cells cotransfected with pRL-TK-PB1-5 and pGL3-control vector (bar 5) was substantially decreased in comparison to the control (bar 6). These results suggest that the PB1-5 gene might harbor potential binding sites for miRNAs that inhibit expression of the luciferase gene.
miR-323, miR-491, and miR-654 effectively inhibit luciferase expression in Flag-TK-PB1-5.
To determine whether miRNAs are functional inhibitors of luciferase expression in pRL-TK-PB1-5, we searched for putative miRNA-binding sites in the PB1-5 gene fragment using the MicroInspector program. The putative binding sites were further verified using the RNA22 and RNAHybrid programs. It was determined through these analyses that the PB1-5 gene harbors putative binding sites for miR-323, miR-491, miR-654, miR-639, miR-591, miR-608, miR-601, miR-26a, miR-509, miR-378, miR-541, and miR-939. In order to determine whether these miRNAs inhibit expression of luciferase in pRL-TK-PB1-5, 293T cells were transfected with the miRNA expression vectors which could express the miRNAs effectively (data not shown) and cotransfected with pRL-TK-PB1-5 and pGL3-control vector. The relative luciferase activity was determined using the dual-luciferase reporter assay system kit. As shown in Fig. 2 a, the relative luciferase activity was downregulated in the cells with overexpressing miR-323, miR-654, miR-639, miR-591, miR-608, miR-601, miR-26a, miR-509, miR-939, or miR-491 in comparison to the control (P < 0.01). The miRNAs which downregulated luciferase expressions in pRL-TK-PB1-5 by approximately 50% or less, compared to the control sample, were selected for further research. According to this criterion, miR-323, miR-654, miR-608, miR-601, and miR-491 were selected. The complementary sequences of the 5′ seed region of miRNAs in the target gene were thought to be the putative binding sites of miRNAs, so we blasted the putative binding sites in the NCBI database. The results showed that the putative binding sites of miR-323, miR-491, and miR-654 are highly conserved across a variety of influenza viral strains (Table 1). So, miR-323, miR-491, and miR-654 were thought more likely to target and regulate the expression of the PB1 gene.
FIG. 2.

miR-323, miR-491, and miR-654 inhibit expression of luciferase through binding to the PB1-5 gene fragment. (a) The relative luciferase activity from pRL-TK-PB1-5 and pGL3-control vector in 293T cells with overexpressing miRNAs. The control is the expression vector of C. elegans miR-239b. (b) Western blotting was used to detect the effects of miR-323, miR-491, and miR-654 on luciferase expression in 293T cells transfected with Flag-TK-PB1-5 or Flag-TK. (c) Western blotting was used to further detect the effects of miRNA inhibitors on luciferase expression in Flag-TK-PB1-5. The expressions of β-actin were analyzed as a control in Western blotting.
TABLE 1.
The conserved binding sites of miR-323, miR-491, and miR-654 in the PB1 gene compared across a selection of influenza viral strainsa
| Virus type and NCBI sequence no. | Sequenceb |
|---|---|
| H1N1 FLAP1MB | GAGGAGTTCACTGAGATCATGAAGATCTGTTCCACCATTG |
| H2N1 FJ686720 | GAGGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTG |
| H3N1 CY039618 | GGAGGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTG |
| H4N1 CY034651 | GGAGGAATTTGCTGAGATCATGAAGATCTGTTCCACCATTG |
| H4N4 CY004720 | AGAGGAGTTCGCTGAGATCATGAAGATCTGTTCCACCATTG |
| H5N1 FJ573466 | AGGGTTTGCTGAGATCATGAAGATCTGTTCCACCATTGAAG |
| H6N1 CY042630 | GGAAGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTG |
| H7N1 AB268552 | AGAAGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTG |
| H9N1 CY005104 | AGAGGAGTTTGCTGAGATCATAAAGATCTGTTCCACCATT |
| H10N1 CY032866 | AGGGGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTG |
| H11N1 FJ517280 | AAGGAGGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTGA |
| H12N1 CY005349 | AGAGGAGTTTGCTGAGATCATAAAGATCTGTTCCACCATTGA |
| H1N2 GQ405858 | GAAGACTTCGCTGAGATCATGAAGATCTGTTCCACCATTG |
| H1N3 CY042123 | GAGGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTGA |
| H1N4 CY042541 | GAGGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTGA |
| H1N5 CY020867 | GAGGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTGA |
| H1N7 CY042483 | GAAGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTG |
| H2N8 CY004560 | GAGGAGTTCGCTGAGATCATGAAGATCTGTTCCACCATTG |
| H1N9 CY042159 | GAGGAGTTTGCTGAGATCATGAAGATCTGTTCCACCATTGA |
Collected from NCBI 22 January 2010.
Underlined sequences indicate conserved binding sites. Some influenza viruses are not shown in the table.
To further investigate whether miR-323, miR-491, and miR-654 could downregulate luciferase expression in pRL-TK-PB1-5, two additional experiments were carried out. As shown in Fig. 2b, luciferase expression from Flag-TK in 293T cells with overexpressing miR-323, miR-491, or miR-654 was similar to that of the control, while the luciferase expression from Flag-TK-PB1-5 was less than that of the control. The results indicated that miRNAs downregulate the luciferase expression due to the PB1-5 gene fragment. The second experiment utilized miRNA inhibitors purchased from Ambion to further verify the function of the miRNAs in the inhibition of luciferase expression in Flag-TK-PB1-5. Consistent with the role of miRNA inhibitors in blocking miRNA function, expression of luciferase in 293T cells cotransfected with both the miRNA expression vector and its specific inhibitor was higher than that of cells cotransfected with miRNA expression vector and the negative-control miRNA inhibitor (Fig. 2c). These results demonstrate that miR-323, miR-491, and miR-654 downregulate the luciferase expression when the PB1-5 gene fragment was inserted into the 3′ UTR of luciferase. Therefore, the PB1-5 gene fragment likely harbors binding sites of miR-323, miR-491, and miR-654.
miR-323, miR-491, and miR-654 bind to the same conserved region in the PB1 gene.
The results imply that miR-323, miR-491, and miR-654 downregulate the expression of luciferase-PB1-5 through binding to a sequence in the PB1-5 gene fragment; however, the precise binding sites of the miRNAs are unknown. Sequence analysis indicates that the 5′ seed sequence of these miRNAs all contain the same nucleotide sequence, 5′-GUGG-3′, which perfectly matches a 5′-CCAC-3′ sequence found in the PB1 gene (Fig. 3 a). The modes of hybridization between the miRNAs and the PB1 mRNA were predicted using RNAHybrid software as shown in Fig. 3b. Based on the similarity between the three 5′ seed sequences, the three miRNAs were thought to bind to the same region of the PB1 gene. To investigate the miRNA binding sites in the PB1 gene, the predicted 5′-CACC-3′ miRNA binding site in the PB1 gene was mutated to 5′-GGAA-3′ in the pRL-TK-PB1-5 vector as shown in Fig. 3c, generating the ΔpRL-TK-PB1-5 vector. In order to analyze the effects of this mutation, 293T cells were transfected with miRNA expression vectors and cotransfected with pGL3-control vector and either ΔpRL-TK-PB1-5 or pRL-TK-PB1-5. The results from this experiment showed that miRNA expression does not result in repression of the relative luciferase activity in cells transfected with ΔpRL-TK-PB1-5 (Fig. 3e). To further confirm these results, the 5′ seed sequences of the miRNAs were mutated in their respective expression vectors in a manner that complements the mutations in ΔpRL-TK-PB1-5 as shown in Fig. 3d. The mutant miRNAs were named ΔmiR-323, ΔmiR-491, and ΔmiR-654. In order to test whether the mutant miRNAs could inhibit luciferase expression from ΔpRL-TK-PB1-5, 293T cells were transfected with vectors expressing the ΔmiRNAs and cotransfected with ΔpRL-TK-PB1-5 and pGL3-control vector. The results showed that the ΔmiRNAs inhibit the relative luciferase activity from ΔpRL-TK-PB1-5 (Fig. 3e). These results demonstrate that all three miRNAs bind to the same sequence, 5′-CCACC-3′ in the PB1 gene, and the binding sites are critical for miRNA-mediated repression of the relative luciferase activity from pRL-TK-PB1-5 and pGL3-control vector. In addition, the binding sites of the miRNAs are located in the 3′ coding region of the PB1 gene and are highly conserved across a variety of influenza viral strains through alignment of strain genomes from the NCBI database (Table 1).
FIG. 3.

miR-323, miR-491, and miR-654 bind to the same region in the PB1 gene. (a) The sequences of miR-323, miR-491, miR-654, and PB1-5 mRNA. The same nucleotide sequence in miRNAs 5′-GUGG-3′ perfectly matches the 5′-CCAC-3′ sequence in the PB1 mRNA. (b) The model of hybridization between the miRNAs and PB1-5 mRNA were predicted using RNAHybrid software. (c) The mutant nucleotide sequence of pRL-TK-PB1-5 and sequencing analysis. (d) The mutant nucleotide sequences of the miRNAs. The mutant 5′-UUCC-3′ sequences in ΔmiRNAs perfectly match the 5′-GGAA-3′ sequence in ΔpRL-TK-PB1-5 mRNA. (e) Luciferase assay to analyze the importance of binding sites of miRNAs in the PB1 gene for miRNAs to inhibit the relative luciferase activity from pRL-TK-PB1-5 and pGL3-control vector.
miR-323, miR-491, and miR-654 downregulate PB1 expression by degrading mRNA in MDCK cells.
Since miR-323, miR-491, and miR-654 can downregulate the luciferase expression by binding to the PB1-5 gene fragment, we investigated whether these miRNAs directly regulate PB1 expression through the binding sites. As shown in Fig. 4 a, the binding sites of miRNAs in the PB1 gene were mutated, and the mutant vector was termed ΔFlag-PB1. The results showed that the PB1 expression in MDCK cells overexpressing miR-323, miR-491, or miR-654 was less than that of the control. When the binding sites of miRNAs in the PB1 gene were mutated, three miRNAs could not inhibit PB1 expression (Fig. 4b). The results indicate that miR-323, miR-491, and miR-654 can inhibit PB1 expression and that the specific binding sites on the PB1 gene are very important for inhibition.
FIG. 4.

miR-323, miR-491, and miR-654 downregulate PB1 expression through mRNA degradation in MDCK cells. (a) The mutant PB1 gene. The nucleotide sequence, but not the amino acid sequence, of the PB1 gene was altered. (b) Western blotting was used to detect the effects of miR-323, miR-491, and miR-654 on PB1 expression or mutant PB1 expression in MDCK cells. Lanes 1 to 5 are the samples in MDCK cells and MDCK cells overexpressing C. elegans miR-239b, miR-323, miR-491, and miR-654, respectively. (c) Real-time PCR was used to detect the expression level of the PB1 mRNA and mutant PB1 mRNA in MDCK cells and MDCK cells overexpressing C. elegans miR-239b, siRNA-PB1, miR-323, miR-491, and miR-654. The expression vectors of C. elegans miR-239b and siRNA-PB1 were used as a miRNA negative control and positive control, respectively. *, the results were significantly different (P < 0.01).
In general, miRNAs downregulate the target genes through mRNA degradation or translation inhibition. In order to characterize the mechanism of inhibition in the present study, we utilized real-time PCR to detect the level of PB1 mRNA in MDCK cells overexpressing miRNAs. The results showed that the expression of PB1 in MDCK cells with overexpressing siRNA-PB1, miR-323, miR-491, or miR-654 was reduced (P < 0.01) compared to the negative control (Fig. 4c). These observations indicate that the miRNAs downregulate the expression of PB1 through mRNA degradation. Additional, real-time PCR analysis was utilized to further assess the importance of the miRNA binding sites in the PB1 gene in suppression of gene expression. The results showed that none of the three miRNAs inhibited expression of the mutated PB1 mRNA, while siRNA-PB1 still downregulated the expression level of the mutated PB1 mRNA (Fig. 4c). The results further confirmed that the miRNA binding sites 5′CCACC3′ in the PB1 gene are critical for miRNA function.
miR-323, miR-491, and miR-654 inhibit replication of the H1N1 IAV through binding to the PB1 gene.
To determine whether miR-323, miR-491, and miR-654 regulate replication of the H1N1 IAV in MDCK cells, the three miRNA expression vectors were transfected individually and collectively as a mixture into MDCK cells. Twenty-four hours after transfection, the cells were infected with the H1N1 IAV, and the HA value was measured at different time points. The HA measurements increased dramatically between 0 and 24 h and continued to increase until 48 h, but the value began to fall after 60 h of infection. All of the miRNAs inhibited viral replication individually, and the miRNA mixture had no greater antiviral capability than the individual miRNAs (Fig. 5 a). These results were verified by fluorescence microscopy (Fig. 5b).
FIG. 5.

The miRNAs inhibit the replication of H1N1 IAV through binding to the PB1 gene in MDCK cells. (a) The HA values were used to assess the effects of miRNAs on replication of H1N1 IAV in MDCK cells; the HA values were determined in triplicate at different time points. (b) Fluorescence microscopy was used to verify the effect of miRNAs on replication of H1N1 IAV in MDCK cells. (c) Real-time PCR was used to detect the effects of miRNA expression vectors and miRNA inhibitors on the relative expressions of the PB1 gene in virus-infected MDCK cells. (d) The HA values were used to determine the effect miRNAs on replication of mutant H1N1 IAV in MDCK cells; the HA values were determined in triplicate at different time points. The expression vector of C. elegans miR-239b was used as negative control.
To further confirm that these miRNAs inhibit replication of the H1N1 IAV, specific miRNA inhibitors were cotransfected into MDCK cells with the miRNA expression vectors, and the transfected cells were infected by H1N1 IAV. After 24 h, the infected cells were collected to analyze the expression of PB1 by real-time PCR. The results of real-time PCR showed that the relative expressions of PB1 in infected MDCK cells with overexpressing miRNAs and negative miRNA inhibitors (Fig. 5c, bars 2, 4, and 6) were lower than those for the infected cells with overexpressing miRNAs and specific miRNA inhibitors (Fig. 5c, bars 3, 5 and 7).
In order to investigate whether the miRNA binding sites in the PB1 gene are important for inhibition of replication of the H1N1 IAV, we mutated the PB1 gene in the 12-plasmid influenza virus system as shown in Fig. 4a. The mutant viruses were generated by transfecting the 12-plasmid system containing the mutant PB1 gene into 293T cells. To analyze the effects of miRNAs on replication of the mutant virus, MDCK cells were transfected with vectors expressing C. elegans miR-239b, miR-323, miR-491, or miR-654. At 24 h posttransfection, the cells were infected with the mutant IAV. The HA values were measured at 24 h, 36 h, 48 h, and 60 h after infection. The results showed that the miRNAs did not inhibit replication of the mutant influenza virus as effectively as the wild-type virus (Fig. 5d). These observations indicate that the specific miRNA binding sites in the PB1 gene are critical for inhibition of replication of the influenza virus.
Endogenous miR-323, miR-491, and miR-654 inhibit the replication of H1N1 IAV in MDCK cells.
Since exogenous miR-323, miR-491, and miR-654 can inhibit the replication of H1N1 IAV in MDCK cells, we investigated whether endogenous miR-323, miR-491, and miR-654 are involved in the virus replication. To evaluate the possibility of endogenous miRNA involvement in the replication of IAV, the endogenous expression levels of miR-323, miR-491, and miR-654 were detected using real-time PCR in different cell lines. As shown in Fig. 6, endogenous miR-323, miR-491, and miR-654 were expressed in A549, HLF, QSG, MCF-7, 293T, MDCK, HeLa, 16HBE, and WISN cells. Expression of miR-491 was not detected in HLF cells, and miR-654 was particularly highly expressed in MDCK cells. The results suggest that endogenous miR-323, miR-491, and miR-654 could be utilized to regulate the replication of H1N1 IAV in MDCK cells.
FIG. 6.

The relative expression levels of miR-323, miR-491, and miR-654 in different cell lines. The relative expression of miR-323 was 10 × 2(CT1 −CT2). The relative expression of miR-491 was 100 × 2(CT1 − CT2). The relative expression of miR-654 was 1,000 × 2(CT1 −CT2). CT1, the cycle threshold value of U6; CT2: the CT value of miRNAs.
We then investigated whether endogenous miR-323, miR-491, and miR654 were involved in virus replication. The MDCK cells transfected with the specific miRNA inhibitors were infected by H1N1 IAV, and the HA values were determined 24 h after infection. The results showed that blocking the function of these three endogenous miRNAs, by their specific inhibitors, resulted in increased IAV production from MDCK cells (Fig. 7). The results suggest that the three endogenous miRNAs have antiviral potential independent of their cellular function.
FIG. 7.
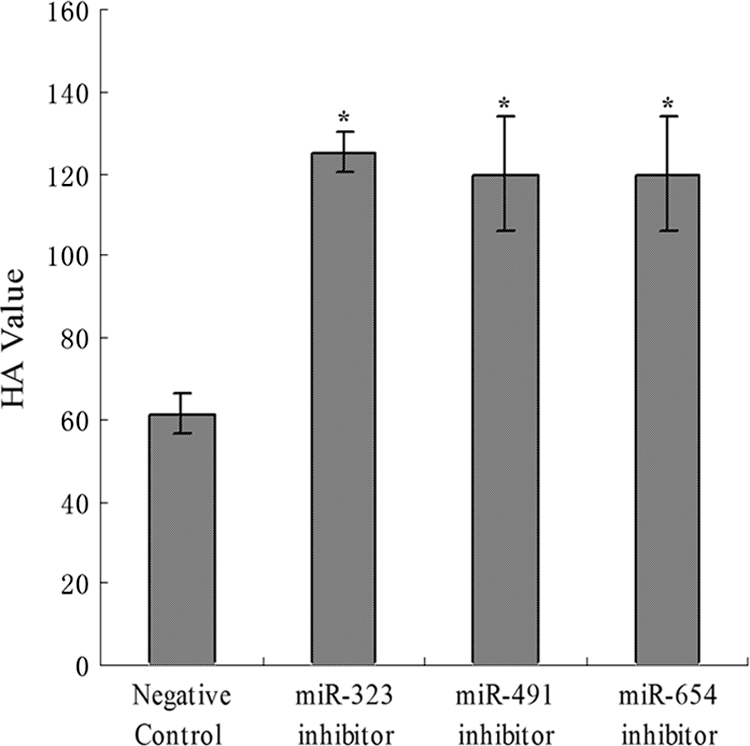
Endogenous miR-323, miR-491, and miR-654 inhibit the replication of HIN1 IAV in MDCK cells. *, results were significantly different (P < 0.01).
DISCUSSION
Studies exploring the interaction between cellular miRNAs and the IAV are critical for providing insight into IAV resistance. In this report, we screened three cellular miRNAs which can inhibit the replication of the H1N1 IAV through the degradation of PB1 mRNA in MDCK cells.
It is well known that IAV has a major impact on both human health and on the global economy. The worst known case was the Spanish flu of 1918 which caused the deaths of 30 to 50 million people (35). Few influenza viruses are sufficiently virulent to directly cause death in humans; instead, most deaths are due to an increased physiologic load in an already compromised host or due to the combined effects of the viral disease and a secondary bacterial infection (29). Segment 2 of the influenza virus gene has two open reading frames (ORFs). One of them encodes PB1, which is involved in both transcription and replication of the RNA genome (40). The second ORF encodes the PB1-F2 protein which is proinflammatory, can contribute to virulence, and facilitates secondary bacterial infections (12, 28). In this study, we have identified three cellular miRNAs which bind to and degrade PB1 mRNA. The low expression of the PB1 protein has been shown to inhibit replication of the influenza virus and downregulate PB1-F2, reducing viral virulence and decreasing the host's susceptibility to secondary bacterial infections (45, 46). Therefore, the three miRNAs identified not only inhibit the replication of H1N1 IAV but also have the potential to reduce the pathogenicity of IAV in humans.
The prevailing wisdom is that miRNAs regulate posttranscriptional processes by binding to the 3′ UTR of target genes. However, Jopling et al. reported that miR-122 regulates viral replication by binding to the 5′ noncoding region (NCR) of the viral genome (22). In the present study, mutational analysis showed that miR-323, miR-491, and miR-654 bind to the same 3′ coding region rather than the 3′ UTR in the PB1 gene. The length of the influenza viral genome is limited, and the virus has to use every nucleotide efficiently. Therefore, it is unlikely that the PB1 gene could contain a 3′-UTR sequence long enough to contain miRNA binding sites. The miRNAs have to bind to the coding region of the viral genes in order to perform their functions. Our finding raises the question of whether the coding regions in other viral or host cell mRNAs can also be targeted by miRNAs.
The extent to which sequences of the miRNA and the target mRNA complement each other is thought to play an important role in the effects caused by miRNAs. In general, if the sequences of the miRNA and the mRNA are perfectly matched, the target mRNA is degraded. Repression of translation is the most common effect if the sequences of the miRNA and the mRNA do not match one another perfectly. However, perplexing observations contrary to this model have come from studies of plant miRNAs. For example, miR-172 appears to regulate APETALA2 via translational repression despite a near-perfect match between the miRNA and its single complementary site in the APETALA2 ORF (2, 11). In this study, we used real-time PCR to detect PB1 mRNA expression levels in the presence of miR-323, miR-491, and miR-654 overexpression. The results showed that despite the fact that the miRNA sequences and the PB1 mRNA do not complement one another very well (Fig. 3b), the miRNAs downregulate expression of the PB1 protein through mRNA degradation. These results indicate that other mechanisms may play a role in directing the action of miRNAs.
An intrinsic property of all viruses is their need to either subvert and/or subdue the host immune response in order to establish a productive replication cycle. The ability to subvert and disable host immunity is directly correlated with increased viral pathogenicity. IAV has many mechanisms to avoid both innate and adaptive human immune responses (12). NS1, an unstructured IAV protein, is responsible for inactivation of the host innate immune response by preventing activation of PKR from alpha/beta interferon (IFN-α/β) signaling, thus allowing replication and viral protein synthesis to proceed unabated in the host cell (17). Additionally, IAV has the ability to escape the host's humoral immunity through a phenomenon known as antigenic drift. Because of this, new flu virus vaccines must be designed every year (37). Since IAV is an eight-segmented minus-sense RNA virus, its segmented nature allows for the swapping and exchange of gene segments between different strains. Specifically, this process occurs through human influenza viruses swapping the HA glycoprotein, neuraminidase (NA) glycoprotein, or polymerase (PB1, PB2, and PA) segments with those of avian and pig IAV. As a result, a rearrangement of gene segments has occurred, creating an entirely novel IAV strain capable of infecting humans (35). While growing evidence has shown that cellular miRNAs could be used by host cells to resist the viral infection, viruses have been found to possess mechanisms to subvert the antiviral function of miRNAs. Several mammalian viruses have been shown to encode viral factors that exhibit RNA silencing suppressor (RSS) activity in animal cells. These factors include the IAV NS1, vaccinia virus E3L, hepatitis C virus core, primate foamy virus type 1 (PFV-1) Tas, and the HIV-1 Tat proteins, as well as the adenovirus virus-associated RNAs I and II (VAI and VAII) (1, 4, 8, 16, 25).
In our study, we found that miR-323, miR-491, and miR-654 can inhibit the replication of the virus in MDCK cells and the three miRNAs are expressed in different cell lines. In addition, the expression levels of the three miRNAs can be altered during H1N1 IAV infection in MDCK cells (Fig. 8). The upregulation of miRNA expression in MDCK cells reflects the mechanism of the host cell's defense against viral infection. The virus has to downregulate the expression of antiviral factors in order to replicate in host cells. The changing model of the three endogenous miRNAs after infection may reflect the resistant procession between the host cells and influenza virus.
FIG. 8.

The relative expression of miR-323, miR-491, and miR-654 in MDCK cells with or without H1N1 IAV infection. x axis represents the different infection time points.
Our results suggest that cellular miRNAs are important factors in the host's resistance to viral infection and provide a deeper understanding of the mechanisms underlying the defense system of host cells. The existence of such different defensive tools against new acquisitions in the viral genome underscore the importance of this phenomenon for viral and host interactions.
Acknowledgments
This work was supported by grants from National Basic Research Program 973 (no. 2005CB523010). Support was also provided by the National Natural Science Foundation of China (no. 30900759/C0709) and the Postdoctoral Foundation of China.
We thank George F. Gao for the H1N1 IAV (A/WSN/33) and 12-plasmid influenza virus system and Xin Ye for helpful discussions.
Footnotes
Published ahead of print on 16 June 2010.
REFERENCES
- 1.Andersson, M. G., P. C. Haasnoot, N. Xu, S. Berenjian, B. Berkhout, and G. Akusjarvi. 2005. Suppression of RNA interference by adenovirus virus-associated RNA. J. Virol. 79:9556-9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aukerman, M. J., and H. Sakai. 2003. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 15:2730-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartel, D. P. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281-297. [DOI] [PubMed] [Google Scholar]
- 4.Bennasser, Y., S. Y. Le, M. Benkirane, and K. T. Jeang. 2005. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 22:607-619. [DOI] [PubMed] [Google Scholar]
- 5.Bentwich, I. 2005. Prediction and validation of microRNAs and their targets. FEBS Lett. 579:5904-5910. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharyya, S. N., R. Habermacher, U. Martine, E. I. Closs, and W. Filipowicz. 2006. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125:1111-1124. [DOI] [PubMed] [Google Scholar]
- 7.Brown, J. R., and P. Sanseau. 2005. A computational view of microRNAs and their targets. Drug Discov. Today 10:595-601. [DOI] [PubMed] [Google Scholar]
- 8.Bucher, E., H. Hemmes, P. de Haan, R. Goldbach, and M. Prins. 2004. The influenza A virus NS1 protein binds small interfering RNAs and suppresses RNA silencing in plants. J. Gen. Virol. 85:983-991. [DOI] [PubMed] [Google Scholar]
- 9.Calin, G. A., C. G. Liu, C. Sevignani, M. Ferracin, N. Felli, C. D. Dumitru, M. Shimizu, A. Cimmino, S. Zupo, M. Dono, M. L. Dell'Aquila, H. Alder, L. Rassenti, T. J. Kipps, F. Bullrich, M. Negrini, and C. M. Croce. 2004. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl. Acad. Sci. U. S. A. 101:11755-11760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang, J., J. T. Guo, D. Jiang, H. Guo, J. M. Taylor, and T. M. Block. 2008. Liver-specific microRNA miR-122 enhances the replication of hepatitis C virus in nonhepatic cells. J. Virol. 82:8215-8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen, X. F., M. Yan, D. R. Yang, and Y. Hirose. 2003. A method for predicting critical load evaluating adhesion of coatings in scratch testing. J. Zhejiang Univ. Sci. 4:709-713. [DOI] [PubMed] [Google Scholar]
- 12.Coleman, J. R. 2007. The PB1-F2 protein of influenza A virus: increasing pathogenicity by disrupting alveolar macrophages. Virol. J. 4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doench, J. G., C. P. Petersen, and P. A. Sharp. 2003. siRNAs can function as miRNAs. Genes Dev. 17:438-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge, Q., M. T. McManus, T. Nguyen, C. H. Shen, P. A. Sharp, H. N. Eisen, and J. Chen. 2003. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc. Natl. Acad. Sci. U. S. A. 100:2718-2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta, A., J. J. Gartner, P. Sethupathy, A. G. Hatzigeorgiou, and N. W. Fraser. 2006. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature 442:82-85. [DOI] [PubMed] [Google Scholar]
- 16.Haasnoot, J., W. de Vries, E. J. Geutjes, M. Prins, P. de Haan, and B. Berkhout. 2007. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 3:e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hale, B. G., R. E. Randall, J. Ortin, and D. Jackson. 2008. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89:2359-2376. [DOI] [PubMed] [Google Scholar]
- 18.Hariharan, M., V. Scaria, B. Pillai, and S. K. Brahmachari. 2005. Targets for human encoded microRNAs in HIV genes. Biochem. Biophys. Res. Commun. 337:1214-1218. [DOI] [PubMed] [Google Scholar]
- 19.Huang, J., F. Wang, E. Argyris, K. Chen, Z. Liang, H. Tian, W. Huang, K. Squires, G. Verlinghieri, and H. Zhang. 2007. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 13:1241-1247. [DOI] [PubMed] [Google Scholar]
- 20.Hutvágner, G., and P. D. Zamore. 2002. A microRNA in a multiple-turnover RNAi enzyme complex. Science 297:2056-2060. [DOI] [PubMed] [Google Scholar]
- 21.Hwang, H. W., and J. T. Mendell. 2006. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br. J. Cancer 94:776-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jopling, C. L., M. Yi, A. M. Lancaster, S. M. Lemon, and P. Sarnow. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309:1577-1581. [DOI] [PubMed] [Google Scholar]
- 23.Kuersten, S., and E. B. Goodwin. 2003. The power of the 3′ UTR: translational control and development. Nat. Rev. Genet. 4:626-637. [DOI] [PubMed] [Google Scholar]
- 24.Lai, E. C. 2002. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 30:363-364. [DOI] [PubMed] [Google Scholar]
- 25.Lecellier, C. H., P. Dunoyer, K. Arar, J. Lehmann-Che, S. Eyquem, C. Himber, A. Saib, and O. Voinnet. 2005. A cellular microRNA mediates antiviral defense in human cells. Science 308:557-560. [DOI] [PubMed] [Google Scholar]
- 26.Li, Y., E. Y. Chan, J. Li, C. Ni, X. Peng, E. Rosenzweig, T. M. Tumpey, and M. G. Katze. 2010. MicroRNA expression and virulence in pandemic influenza virus-infected mice. J. Virol. 84:3023-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu, J., F. V. Rivas, J. Wohlschlegel, J. R. Yates III, R. Parker, and G. J. Hannon. 2005. A role for the P-body component GW182 in microRNA function. Nat. Cell Biol. 7:1261-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McAuley, J. L., F. Hornung, K. L. Boyd, A. M. Smith, R. McKeon, J. Bennink, J. W. Yewdell, and J. A. McCullers. 2007. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2:240-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCullers, J. A. 2006. Insights into the interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev. 19:571-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minor, P. D. 2010. Vaccines against seasonal and pandemic influenza and the implications of changes in substrates for virus production. Clin. Infect. Dis. 50:560-565. [DOI] [PubMed] [Google Scholar]
- 31.Müller, S., and J. L. Imler. 2007. Dicing with viruses: microRNAs as antiviral factors. Immunity 27:1-3. [DOI] [PubMed] [Google Scholar]
- 32.Otsuka, M., Q. Jing, P. Georgel, L. New, J. Chen, J. Mols, Y. J. Kang, Z. Jiang, X. Du, R. Cook, S. C. Das, A. K. Pattnaik, B. Beutler, and J. Han. 2007. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity 27:123-134. [DOI] [PubMed] [Google Scholar]
- 33.Rajewsky, N. 2006. microRNA target predictions in animals. Nat. Genet. 38(Suppl.):S8-S13. [DOI] [PubMed] [Google Scholar]
- 34.Rehwinkel, J., I. Behm-Ansmant, D. Gatfield, and E. Izaurralde. 2005. A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA 11:1640-1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russell, C. J., and R. G. Webster. 2005. The genesis of a pandemic influenza virus. Cell 123:368-371. [DOI] [PubMed] [Google Scholar]
- 36.Scaria, V., M. Hariharan, S. Maiti, B. Pillai, and S. K. Brahmachari. 2006. Host-virus interaction: a new role for microRNAs. Retrovirology 3:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinhauer, D. A., and J. J. Skehel. 2002. Genetics of influenza viruses. Annu. Rev. Genet. 36:305-332. [DOI] [PubMed] [Google Scholar]
- 38.Sullivan, C. S., A. T. Grundhoff, S. Tevethia, J. M. Pipas, and D. Ganem. 2005. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 435:682-686. [DOI] [PubMed] [Google Scholar]
- 39.Tomari, Y., and P. D. Zamore. 2005. Perspective: machines for RNAi. Genes Dev. 19:517-529. [DOI] [PubMed] [Google Scholar]
- 40.Toyoda, T., M. Kobayashi, S. Nakada, and A. Ishihama. 1996. Molecular dissection of influenza virus RNA polymerase: PB1 subunit alone is able to catalyze RNA synthesis. Virus Genes 12:155-163. [DOI] [PubMed] [Google Scholar]
- 41.Wienholds, E., and R. H. Plasterk. 2005. MicroRNA function in animal development. FEBS Lett. 579:5911-5922. [DOI] [PubMed] [Google Scholar]
- 42.Yekta, S., I. H. Shih, and D. P. Bartel. 2004. MicroRNA-directed cleavage of HOXB8 mRNA. Science 304:594-596. [DOI] [PubMed] [Google Scholar]
- 43.Yeung, M. L., Y. Bennasser, T. G. Myers, G. Jiang, M. Benkirane, and K. T. Jeang. 2005. Changes in microRNA expression profiles in HIV-1-transfected human cells. Retrovirology 2:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoon, S., and G. De Micheli. 2006. Computational identification of microRNAs and their targets. Birth Defects Res. C 78:118-128. [DOI] [PubMed] [Google Scholar]
- 45.Zamarin, D., A. Garcia-Sastre, X. Xiao, R. Wang, and P. Palese. 2005. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zamarin, D., M. B. Ortigoza, and P. Palese. 2006. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J. Virol. 80:7976-7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeng, Y., and B. R. Cullen. 2003. Sequence requirements for micro RNA processing and function in human cells. RNA 9:112-123. [DOI] [PMC free article] [PubMed] [Google Scholar]