

Abstract
Paired immunoglobulin (Ig)-like type 2 receptor alpha (PILRα) and PILRβ are paired receptors that are highly homologous to each other. When engaged by ligand, PILRα is inhibitory whereas PILRβ is activating. PILRα is a newly identified herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) receptor and is associated with membrane fusion and entry activity of HSV-1. PILRα is a 303-amino-acid protein with an Ig-like V (variable)-type domain from amino acid 31 to 150, whereas PILRβ is a 217-amino-acid protein with an Ig-like V-type domain from amino acid 21 to 143. We report that PILRβ is not a receptor for HSV-1 and HSV-2. Domain swaps between PILRα and PILRβ reveal that the Ig-like V-type domain of PILRα, but not PILRβ, plays a critical role in cell membrane fusion activity and the binding of PILRα to gB. Individual replacement of 13 amino acids in PILRα showed that most of these mutations had no effect on cell fusion activity. However, mutation of the tryptophan residue at amino acid 139 significantly impaired cell fusion activity for HSV-1 and eliminated binding to gB.
Herpes simplex virus type 1 (HSV-1) is a member of the alphaherpesvirus subfamily and can cause recurrent mucocutaneous lesions on the mouth, face, or genitalia and potentially meningitis or encephalitis. Four membrane glycoproteins (gB, gD, gH, and gL) encoded by HSV mediate membrane fusion, a process required for entry of HSV into cells. Membrane fusion requires gD binding to a cellular entry receptor. To date, at least four gD receptors have been identified, including herpesvirus entry mediator (HVEM) (19), nectin-1 (2, 6, 17, 18, 29), nectin-2 (14, 43), and modified heparan sulfate (30, 31). HVEM is a member of the tumor necrosis factor receptor family and is expressed by cells of the immune system as well as other cell types, including epithelial, stromal, and endothelial cells (42). Nectin-1 and nectin-2 are cell adhesion molecules belonging to the immunoglobulin (Ig) superfamily and are widely expressed by a variety of cell types, including epithelial cells and neurons (38). Modified heparan sulfate generated by particular 3-O-sulfotransferases can serve as a gD-binding entry receptor (31). The binding of gD to a receptor is associated with a conformational change in gD that is thought to enable gD to interact with gB and/or the heterodimer gH-gL to trigger fusion (10, 24).
The paired immunoglobulin-like type 2 receptor alpha (PILRα) was identified as an entry receptor that binds to gB (26). The interaction of gB and PILRα can mediate viral entry and cell-cell fusion provided that gD also binds to one of its receptors (26). HSV-1 entry into CHO cells expressing PILRα was via virus-cell fusion at the cell surface, not endocytosis (1). Interestingly, human and murine PILRα were able to mediate entry of pseudorabies virus but not HSV-2 (1).
The PILR locus on chromosome 7 contains inhibitory PILRα and activating PILRβ (4, 20). The PILRβ gene is located 5.6 kb upstream of the PILRα gene (20). Both PILRα and PILRβ are conserved among mammals (44). PILRα was first identified using a yeast two-hybrid system with the immune regulator protein tyrosine phosphatase SHP-1 as bait (20). PILR gene products are considered novel regulators of the innate and adaptive immune systems (28). The inhibitory PILRα is a transmembrane protein with an Ig-like variable (V) extracellular domain and cytoplasmic immunoreceptor tyrosine-based inhibitory motif (ITIM) (20). The activating PILRβ has a smaller cytoplasmic tail. Similar to its inhibitory counterpart, PILRβ also has an Ig-like V-type domain; however, in place of ITIMs, it associates with DAP12 through an immunoreceptor tyrosine-based activation motif (ITAM) and delivers activating signals (28). Both PILRα and PILRβ are expressed on cells of the immune system, such as monocytes, dendritic cells, NK cells, B cells, macrophages, neutrophils, eosinophils, mast cells, and megakaryocyte/platelets (4, 11, 20, 21, 28, 39), as well as neurons (26). PILRβ is expressed by neurons throughout the brain as determined by in situ hybridization and protein blot analyses (37). CD99 is a natural ligand for both PILRα and PILRβ (28). The binding of either PILRα or PILRβ to CD99 depends on the presence of sialyated O-linked glycans on CD99 (41).
The aims of this study were (i) to identify whether PILRβ is a receptor for HSV-1 or HSV-2, (ii) to identify the specific regions and amino acids of PILRα that are important for HSV-1 entry, and (iii) to test the binding of PILRα and PILRβ to gB. We constructed chimeric proteins of PILRα and PILRβ and subsequently generated specific site-specific mutants to identify functional domains of PILRα and PILRβ. The mutants were tested for ability to mediate the cell-cell fusion activity of HSV-1. Our cell fusion experiment results indicate that PILRβ is not a receptor for HSV-1 and HSV-2. Our chimeric and point mutants identified the PILRα Ig-like V-type domain essential for cell fusion activity. A mutant with a single amino acid substitution at tryptophan 139, located in the V domain, showed that this residue is most critical for PILRα function. Finally, we found that PILRα, but not PILRβ, binds to gB.
MATERIALS AND METHODS
Cells.
Cell lines CHO-K1 (ATCC), 293 PEAK Rapid (Edge Biosystems), Hep-2 (ATCC), Arpe-19 (ATCC), CaSki (ATCC), HEL (ATCC), SK-N-SH (ATCC) and HDF-JM (kindly provided by Kathy Rundell, Northwestern University), and CHO-K1 hPILRα (expresses human PILRα; kindly provided by Hisashi Arase, Osaka University) and a CHO-K1 cell mutant selected for absence of PILRα ligands (26) were used in this study. The CHO-K1 cell line and derivatives were grown in Ham's F-12 medium supplemented with 10% fetal bovine serum (FBS), and Arpe-19 cells were grown in F-12 and DMEM ([Dulbecco modified Eagle medium] 1:1) with 10% FBS. CaSki cells were grown in RPMI with 10% FBS. SK-N-SH, HDF-JM, HEL, Hep-2, and 293 PEAK Rapid cells were grown in DMEM supplemented with 10% FBS.
Plasmids.
Plasmids expressing gB, gD, gH, and gL for HSV-1 and HSV-2 (22, 46), nectin-1 (pBG38) (6), HVEM (pBEG10) (19), human PILRα (pQF003) (3), and soluble human PILRα-IgG Fc hybrid protein (pME18S PILRα-Ig) (26) were used for this study. The plasmids generated for this study are shown in Table 1. A plasmid expressing human PILRβ (pQF020) was generated by subcloning the PILRβ open reading frame (ORF) from a commercially obtained cDNA clone (SC321885; OriGene) into pcDNA3, using primers 5′AATAAGCTTGCCGCCACCATGGGTCGGCCCCTGCTG3′ and 5′AATGGTACCCCACACTCTGTTGGTCAGAA3′ for PCR amplification followed by digestion with HindIII and Acc65I and ligation into prepared vector. Multiple cDNAs have been identified for PILRβ (44) and annotated by NCBI. For our studies we used NM_013440.3 (variant 1), which encodes the same product as NM_178238.1 (variant 3). The two cDNAs differ only in the number of 5′ noncoding exons. NM_175047.2 (variant 2) exhibits an alternative splicing event that results in an early frameshift mutation. Additional variants have also been described (44), but they are poorly characterized. FLAG-tagged PILRα (designated FLAG A in this paper) was generated by subcloning the human PILRα (without the native signal sequence from amino acid 1 to 19) from pQF003 using primer 5′ATAATGAATTCACAGCCTAGTGGCTCCACAGGA3′ and primer 5′AATGGTACCGGGCTGTCCATTGGTTAGGC3′ for PCR amplification followed by digestion with EcoRI and Acc65I and ligated into EcoRI- and Acc65I-digested pFLAG-myc-CMV-21 expression vector (E5776; Sigma). FLAG-tagged PILRβ (designated FLAG B in this paper) was generated by subcloning the human PILRβ (without the native signal sequence from amino acid 1 to 19) from pQF020 using primer 5′ATAATGAATTCACAGCCTGGTGGCTCCACAGGA3′ and primer 5′AATGGTACCCCACACTCTGTTGGTCAGAA3′ for PCR amplification followed by digestion with EcoRI and Acc65I and ligated into EcoRI- and Acc65I-digested pFLAG-myc-CMV-21. Chimeric forms of PILRα and PILRβ and other newly made plasmids (Table 1) used in this study were based on these constructs. All plasmids newly made for this study were sequenced by the Northwestern Genomic Core Facility.
TABLE 1.
Plasmids generated for this study
| Construct | Protein | Commenta |
|---|---|---|
| pQF020 | hPILRβ | The PCR product of the PILRβ ORF from a cDNA clone of PILRβ (SC321885; OriGene) was cloned into pCDNA3 |
| pQF022 | Flag A | The PCR product of the human PILRα without the native signal peptide (aa 1-19) from pQF003 was cloned into the vector |
| pQF023 | Flag B | The PCR product of the human PILRβ without the native signal peptide (aa 1-19) from pQF020 was cloned into the vector |
| pQF052 | A194-B | The PCR product containing FLAG pepide-L194 of pQF22 and D187-F227 of pQF23 was cloned into the vector |
| pQF053 | B186-A | The PCR product containing FLAG pepide-L186 of pQF23 and E195-A303 of pQF22 was cloned into the vector |
| pQF026 | A196-B | The PCR product containing FLAG pepide-T196 of pQF22 and V192-F227 of pQF23 was cloned into the vector |
| pQF027 | B191-A | The PCR product containing FLAG pepide-R191 of pQF23 and A197-A303 of pQF22 was cloned into the vector |
| pQF050 | A150-B | The PCR product containing FLAG pepide-T150 of pQF22 and G144-F227 of pQF23 was cloned into the vector |
| pQF051 | B143-A | The PCR product containing FLAG pepide-K143 of pQF23 and Q151-A303 of pQF22 was cloned into the vector |
| pQF054 | A218-B | The PCR product containing FLAG pepide-L218 of pQF22 and W213-F227 of pQF23 was cloned into the vector |
| pQF055 | B212-A | The PCR product containing FLAG pepide-L212 of pQF23 and R219-A303 of pQF22 was cloned into the vector |
| pQF032 | A150-(B 144-191)-A | G151-T196 in pQF22 was replaced by G144-T191 from pQF23 by PCR |
| pQF037 | S22G | S22 in pQF22 was mutated to G22 |
| pQF038 | T63I | T63 in pQF22 was mutated to I63 |
| pQF039 | A64V | A64 in pQF22 was mutated to V64 |
| pQF040 | D66N | D66 in pQF22 was mutated to N66 |
| pQF028 | N100S | N100 in pQF22 was mutated to S100 |
| pQF041 | K106E | K106 in pQF22 was mutated to E106 |
| pQF042 | Q116R | Q116 in pQF22 was mutated to R116 |
| pQF043 | Q118E | Q118 in pQF22 was mutated to E118 |
| pQF044 | S133R | S133 in pQF22 was mutated to R133 |
| pQF045 | W139L | W139 in pQF22 was mutated to L139 |
| pQF046 | E143K | E143 in pQF22 was mutated to K143 |
| pQF047 | S148T | S148 in pQF22 was mutated to T148 |
| pQF031 | Δ151-195 | G151 to E195 from pQF22 were deleted |
| pQF029 | Y269F | Y269 in pQF22 was mutated to F269 |
| pQF030 | Y298F | Y298 in pQF22 was mutated to F298 |
| pQF035 | Y269FY298F | Y298 in pQF29 was mutated to F298 |
| pQF056 | B-L139W | L139 in pQF23 was mutated to W139 |
| pQF021 | hPILRβ-Ig | The PCR product of the PILRβ ectodomain from a cDNA clone was cloned into pDM19 |
| pQF62 | pME18S | pME18S PILRα-Ig was digested with XhoI and the larger XhoI fragment was religated |
| pQF59 | PILRβ-Ig | The PCR product of the PILRβ ectodomain of pQF20 was cloned into pQF62 |
| pQF58 | PILRα V-Ig | The PCR product of the human Ig-like V-type domain of pME18S PILRα-Ig was cloned into pQF62 |
| pQF57 | PILRα W-Ig | W139 pME18S PILRα-Ig was mutated to L |
| pQF61 | PILRα W-V-Ig | The PCR product of the human Ig-like V-type domain containing W139L of pQF45 was cloned into pQF62 |
The vector was the pFLAG-myc-CMV-21 expression vector (Sigma); mutation and deletion were made by QuikChange site-directed mutagenesis (Stratagene). aa, amino acids.
RT-PCR.
Reverse transcription-PCR (RT-PCR) for PILRα and PILRβ was performed using the RNAqueous kit (Ambion) and high-capacity cDNA reverse transcription kits (Applied Biosystems) according to the manufacturer protocols. RT-PCR primers were designed using the human PILRα sequence. The RT-PCR products were sequenced by the Northwestern University Genomic Core Facility and aligned against human PILRα and PILRβ sequences.
CELISA.
Cell-based enzyme-linked immunosorbent assay (CELISA) was used to test the cell surface expression of various proteins as described by Lin and Spear (13). Briefly, CHO-K1 cells expressing each of the PILR mutants, FLAG A, FLAG B, or empty vector were washed once with phosphate-buffered saline (PBS) at 24 h after transfection, and CELISA was performed using the monoclonal antibody anti-FLAG-M2 (F1804; Sigma). After incubation with antibody, the cells were washed, fixed, and incubated with biotinylated goat anti-mouse IgG (Sigma), followed by streptavidin-horseradish peroxidase (HRP) (GE Healthcare) and HRP substrate (BioFX). Absorbance readings were taken at 380 nm using a Wallac-Victor luminometer (Perkin-Elmer).
Cell fusion assay.
The cell fusion assay was done as previously described (22). Briefly, CHO-K1 and CHO-K1 PILRα ligand-negative cells were seeded in six-well plates at 1 day before transfection. The CHO-K1 cells (effector cells) were transfected with 400 ng each of plasmids expressing T7 RNA polymerase or HSV-1 or HSV-2 gD, gH, or gL; 800 ng of gB; and 5 μl of Lipofectamine 2000. The CHO-K1 PILRα ligand-negative cells or CHO-K1 cells (target cells) were transfected with 400 ng of a plasmid carrying the firefly luciferase gene under the control of the T7 promoter; 1.5 μg of empty vector or plasmid expressing either human FLAG A (pQF022), FLAG B (pQF023), PILR mutants, nectin-1, or HVEM; and 5 μl of Lipofectamine 2000. At 6 h after transfection, the cells were detached with EDTA and suspended in 1.5 ml of F-12 medium supplemented with 10% FBS. Effector and target cells were mixed in a 1:1 ratio and replated in 96-well plates for 18 h. Luciferase activity was quantitated by a luciferase reporter assay system (Promega) using a Wallac-Victor luminometer (Perkin-Elmer).
Western blotting.
Western blotting was performed to test the expression of PILR constructs. CHO-K1 cells seeded in six-well plates were transfected with 1.5 μg of empty vector or a plasmid expressing FLAG A, FLAG B, or each of the PILR mutants and 5 μl of Lipofectamine 2000. After 24 h of incubation, the cells were detached using EDTA, washed with PBS, and lysed with 200 μl of lysis buffer (25 mM Tris-HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA, 10 mM NaF, 1 mM Na3VO3, 1% Nonidet P-40) containing a protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN). Proteins were separated by SDS-PAGE on 4 to 20% gels after boiling for 5 min under reducing conditions. Western blot analyses were performed by using the rabbit anti-FLAG (F7425; Sigma) at a 1:1,000 dilution for 1 h at room temperature, anti-rabbit secondary antibodies coupled to horseradish peroxidase (HRP), and ECL Western blotting detection reagents (GE Healthcare).
Preparation of PILR-Ig supernatants.
Supernatants from media of pME18S PILRα-Ig-, PILRα V-Ig (pQF58)-, PILRα W-Ig (pQF57)-, PILRα W-V-Ig (pQF61)-, and PILRβ-Ig (pQF59)-transfected cells were prepared as described by Fan et al (3). Plates of 293 PEAK Rapid cells were transfected with PILR-Ig plasmids using 293 Fectin (Invitrogen). The concentration of PILRα-Ig in the culture supernatant was measured using a human IgG ELISA kit (Immunology Consultants Laboratory, Inc.) (3). The supernatants of PILRβ-Ig, PILRα V-Ig, PILRα W-Ig, and PILRα W-V-Ig were normalized according to concentration of PILRα-Ig determined by Western blotting. The supernatants were used to test the binding of PILR to gB by Western blotting or CELISA.
Assays for binding of PILRα-Ig and PILRβ-Ig to gB.
CELISA and the “monolayer binding assay” were used to determine whether PILRα-Ig or PILRβ-Ig binds to gB. For CELISA, we first tried this assay using CHO-K1 cells, but they gave us higher background than 293 cells so we used 293 cells for this purpose. 293 PEAK Rapid cells seeded in 96-well plates were transfected with 60 ng of empty vector or a plasmid expressing gB and 0.15 μl of Lipofectamine 2000, both diluted in Opti-MEM. The cells were washed once with PBS at 24 h after transfection, and CELISA was performed as described previously (13) using PILRα-Ig and PILRβ-Ig supernatants (about 70 ng/ml) and biotinylated goat anti-human Ig(H+L) (2010-08; Southern Biotech) at a 1:500 dilution.
A “monolayer binding assay” was used to further assess the abilities of PILRα-Ig and PILRβ-Ig to bind to cell surface-expressed gB (32). CHO-K1 cells seeded in six-well plates were transfected with 1.5 μg of empty vector or a plasmid expressing wild-type (WT) gB and 5 μl of Lipofectamine 2000. After 24 h of incubation, the cells were washed twice with cold PBS, incubated with normalized PILR-Ig at about 70 ng/ml for 1 h at 4°C, washed with cold PBS four times, and lysed with 200 μl of lysis buffer (25 mM Tris-HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA, 10 mM NaF, 1 mM Na3VO3, 1% Nonidet P-40) containing a protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN). Proteins were separated by SDS-PAGE on 4 to 20% gels after boiling for 5 min under reducing conditions. Western blot analyses were performed by using the rabbit polyclonal anti-gB (R74) at a 1:10,000 dilution and anti-human IgG(H&L) (HRP) (ab6759; Abcam) at a 1:2,000 dilution.
RESULTS
Selected human cell lines express PILRβ but not PILRα.
To investigate the expression of PILRα and PILRβ, we tested human cell lines for the expression of PILRα using RT-PCR. CHO-K1 cells expressing human PILRα were used as a control. The tested cell lines included Hep-2 (derived from an epidermoid carcinoma of the larynx), Arpe-19 (derived from normal retinal pigmented epithelium), CaSki (derived from a human cervix carcinoma), HEL (derived from human embryonic lung), SK-N-SH (derived from a human neuroblastoma), and HDF-JM (primary human dermal fibroblasts from foreskin).
RT-PCR bands from the seven human cell lines tested in this study were smaller than the product from the PILRα-expressing CHO-K1 cell line (Fig. 1, upper panel). The PILRα amplified product is predicted to be 332 bp, whereas the PILRβ product, if amplified using the PILRα primers, would be 307 bp. To investigate the differences in the observed sizes of the bands, the resolved bands were purified and sequenced. The sequencing results indicated that the RT-PCR product from the CHO-K1 cells expressing PILRα was human PILRα; however, the products from the human cell lines were human PILRβ. Our results indicate that PILRβ mRNA expression is more readily detected than PILRα expression in the cell lines we examined, which is compatible with the observation that PILRβ is detected in a wider variety of tissues than PILRα (44).
FIG. 1.

Selected human cell lines express PILRβ, and PILRβ is not a fusion receptor for HSV-1 and HSV-2. Upper panel, RT-PCR of selected cell lines. RNAs extracted from CHO-K1 cells expressing human PILRα and various human cell lines were used for RT-PCR. Lower panel, cell fusion activities of selected receptors for HSV-1 and HSV-2. CHO-K1 cells were transfected with empty vector (pcDNA3), PILRα (pQF003), PILRβ (pQF020), HVEM (pBEC10), or nectin-1 (pBG38) along with a reporter plasmid expressing luciferase under the control of the T7 promoter. The cells were replated with CHO-K1 cells transfected with plasmids expressing T7 polymerase, gB, gD, gH, and gL from HSV-1 or HSV-2. Luciferase activity was quantified as a measure of cell fusion. Each bar shows the mean and standard deviation of three independent determinations, with the results expressed as a percentage of wild-type PILRα activity. The values from the vector control were not subtracted in order to show the background level of fusion.
PILRβ does not mediate HSV-1 or HSV-2 cell fusion.
Since PILRβ appears to be expressed broadly in human cell lines, we further investigated whether PILRβ functions in fusion for HSV-1 or HSV-2 using a cell-cell fusion assay (Fig. 1, lower panel). For fusion mediated by either HSV-1 or HSV-2 glycoproteins, the level of fusion observed using PILRα-expressing target cells was set to 100%. In order to show the relative activity of PILRβ with PILRα, the values of vector controls were not subtracted. Transfection of PILRα increased the level of fusion compared to the vector control, but the level of fusion was much lower than that observed with HVEM- or nectin-1-expressing target cells (3) when HSV-1 gD, gB, and gH/gL were expressed in the effector cells. The levels of HSV-1 glycoprotein-mediated fusion with the PILRβ target cells were nearly identical to that with the vector-transfected cells, indicating that PILRβ likely does not function as an HSV-1 receptor. For HSV-2, the level of fusion with PILRα and PILRβ target cells was not significantly different than that with vector-transfected cells, indicating that PILRβ expression does not enhance HSV-2 fusion in this assay. We therefore conclude that the cell-cell fusion experiment did not detect cell fusion activity for either HSV-1 or HSV-2 mediated by PILRβ. The absence of PILRα function for HSV-2 fusion has previously been reported (1). Similar to the case for HSV-1, HSV-2 glycoprotein-mediated fusion with HVEM- and nectin-1-expressing cells was substantially higher than that observed with PILRα-expressing cells.
Construction of PILRα and PILRβ chimeras, PILRα-Ig and PILRβ-Ig, and site-specific mutations of PILRα and PILRβ.
PILRα and PILRβ are highly homologous proteins, having 81% identity in protein sequence. In order to investigate why PILRα but not PILRβ functions in cell-cell fusion, chimeric proteins were constructed by swapping domains between PILRα and PILRβ (Fig. 2A). To further investigate the roles of single amino acids and specific domains of PILRα, single-amino-acid substitution and deletion mutants were made by site-specific mutagenesis. Secreted variants of PILRα and PILRβ fused with the Fc segment of human IgG1 at the C terminus were also made, including selected chimeric and site-specific mutants (Fig. 2B). The constructs are listed in Table 1 and schematically shown in Fig. 2. For each of the constructs, the epitope for the FLAG monoclonal antibody was added at the amino terminus.
FIG. 2.

Schematic diagrams of domains swapped between PILRα and PILRβ (A) and amino acid substitutions and deletions of PILRα (B). All constructs were based on the FLAG-tagged PILRα (FLAG A) and FLAG-tagged PILR β (FLAG B), as described in Table 1. The amino acid sequence numbers of PILRα and PILRβ are designated according to sequences obtained from OriGene (Rockville, MD). Locations of the domains contained in PILRα and PILRβ are shown at the top of panel A. The Ig-like V-type domains for PILRα and PILRβ map between amino acids Y31 and T150 and between amino acids P21 and K143, respectively. The transmembrane (TM) regions for PILRα and PILRβ map to amino acids A197 to L218 and R191 to L212, respectively.
Expression and fusion activities of the PILRα and PILRβ chimeras.
First, we verified cell surface expression of the various PILRα and PILRβ chimeras. CELISAs were performed on CHO-K1 cells transfected with empty vector, PILRα (FLAG A), PILRβ (FLAG B), or each of the chimeric constructs, and a monoclonal antibody that recognizes the FLAG epitope was used to detect cell surface expression of the respective proteins. CELISA results are presented in Fig. 3 (upper panel), along with cell fusion activity. The data are represented as the percentage of wild-type PILRα (FLAG A). All of the chimeric constructs expressed at least 70% of the WT PILRα level, with the exception of B143-A, which expressed only 12% of the level of wild-type PILRα. Compatible with the CELISA expression of each of the constructs, Western blots (Fig. 3, lower panel) indicated that all the constructs were expressed well in whole-cell lysates. The expression of the A150-B protein was slightly reduced, but this was not a consistent result, since other blots testing the expression of the various proteins had similar levels of expression of this mutant with the other constructs.
FIG. 3.

Cell surface expression, cell fusion activity, and Western blotting of the PILR chimeric proteins and mutants in comparison with wild-type PILRα. Upper panel, CELISA and cell fusion activity for the chimeric constructs. For CELISA, CHO-K1 cells were transfected in 96-well plates with the various PILR chimeric plasmids. The cells were washed, incubated with an anti-FLAG M2 antibody, and washed extensively prior to fixation and incubation with a mouse secondary antibody and an HRP detection system. For cell fusion activity, PILRα ligand-negative CHO-K1 cells were transfected with pFLAG-CMV-myc-21 (empty vector), FLAG A, FLAG B, or the various PILR chimeric constructs along with a reporter plasmid expressing luciferase under the control of the T7 promoter. The transfected cells were replated with CHO-K1 cells transfected with plasmids expressing T7 polymerase, gB, gD, gH, and gL. Each bar shows the mean and standard deviation of three independent determinations, with the results expressed as a percentage of wild-type PILRα activity. Lower panel, Western blot analyzing the expression of the various PILR constructs. CHO-K1 cells expressing wild-type PILRα, PILRβ, or each of the chimeric constructs were lysed, resolved by SDS-PAGE, and blotted with rabbit anti-FLAG and goat anti-rabbit secondary antibody.
The cell fusion activity of the chimeric constructs was reduced significantly compared to that of the wild-type PILRα, with the exception of the chimera A150(B144-191)-A, a construct in which PILRα amino acids G151 to T196 were replaced by PILRβ amino acids G144 to T191 (Fig. 3, upper panel). This suggests that PILRα amino acids 151 to 196 do not play a critical role in cell fusion activity whereas the PILRα Ig-like V-type domain and the transmembrane domain and cytoplasmic tail of PILRα may play an important role. Supportive of an important role of the PILRα ectodomain in fusion function, most constructs containing the PILRα ectodomain (except for A196-B) had some fusion activity compared to PILRβ. In addition, the constructs containing the PILRβ ectodomain were all at near-background levels for fusion function. Chimeras A194-B, A196-B, A150-(B144-191)-A, A150-B, and A218-B all contain the PILRα Ig-like V-type domain. Chimera A150-B showed high levels of cell fusion activity (32% of the WT PILRα activity) among the chimeric proteins that contained just the PILRα Ig-like V-type domain with the remaining portion from PILRβ. Overall, these results indicate that the Ig-like V-type domain in PILRα plays an important role in cell fusion activity; however, the reduced cell fusion activity compared to that of wild-type PILRα when this domain is transferred to PILRβ indicates that other domains contained in the transmembrane domain and cytoplasmic tail of PILRα may also play important roles in fusion activity.
Effects of PILRα site-specific mutations on expression and cell fusion activity.
Since the PILRα Ig-like V-type domain was critical for cell fusion activity, we investigated whether any single amino acid in this region is responsible for the activity. Amino acid alignment indicated only 12 amino acid differences in the Ig-like V-type domains when PILRα was compared to PILRβ. Twelve single-amino-acid mutations in the PILRα Ig-like V-type domain were constructed by replacing the corresponding amino acids in PILRβ. A potential glycosylation site (N100 to T102) also was mutated (N100S). In addition, G151 to E195 from PILRα were deleted to test the importance of this region in fusion function.
We were also interested in whether signaling through PILRα is important for fusion activity. PILRα transduces inhibitory signals via the ITIM within its cytoplasmic domain. ITIMs play a pivotal role in regulating cells of the immune system (12). PILRα has three potential tyrosine phosphorylation sites, and Y269 and Y298 are important for tyrosine phosphorylation, the recruitment of cellular proteins, and cell signaling (20). We made the single-amino-acid substitution mutants Y269F and Y298F and a double mutant, Y269F Y298F, to determine if these tyrosines are important for cell fusion activity.
Figure 4 (upper panel) shows cell surface expression and cell fusion activity of the mutants relative to those of wild-type PILRα. Most of the mutants were expressed on the cell surface at levels similar to that of wild-type PILRα. Only N100S and Δ151-195 were expressed poorly (37% and 21% of wild-type PILRα expression, respectively). Western blots (Fig. 4, lower panel) showed that all mutants were expressed with the expected size except the N100S and Δ151-195 mutants. Both the N100S and Δ151-195 mutants appeared to have immature and not fully processed forms of PILRα, suggesting that these mutants were not appropriately processed and transported to the cell surface. Cell-cell fusion results showed that most mutants had fusion activity similar to that of WT PILRα. As might be expected, the two mutants with reduced cell surface expression, N100S and Δ151-195, had a complete abrogation of cell fusion activity. The T63I and K106E mutants had a reduction in cell fusion activity, to 57% and 56% of the WT PILRα activity, even though both were expressed well on the cell surface. Most interesting was the W139L mutant, which despite normal cell surface expression compared to wild-type PILRα was negative for cell fusion, indicating a key role for this amino acid in cell fusion.
FIG. 4.

Cell surface expression, cell fusion activity, and Western blotting of amino acid substitution and deletion mutants of PILRα in comparison with wild-type PILRα. Upper panel, CELISA and cell fusion activity for the amino acid substitution and deletion mutants. For CELISA, CHO-K1 cells were transfected in 96-well plates with the various amino acid substitution and deletion mutants. The cells were washed, incubated with an anti-FLAG M2 antibody, and washed extensively prior to fixation and incubation with a mouse secondary antibody and an HRP detection system. For cell fusion activity, PILRα ligand-negative CHO-K1 cells were transfected with pFLAG-CMV-myc-21 (empty vector), FLAG A, FLAG B, or the various PILR amino acid substitution and deletion mutants along with a reporter plasmid expressing luciferase under the control of the T7 promoter. The transfected cells were replated with CHO-K1 cells transfected with plasmids expressing T7 polymerase, gB, gD, gH, and gL. Each bar shows the mean and standard deviation of three independent determinations, with the results expressed as a percentage of wild-type PILRα activity. Lower panel, Western blot analyzing the expression of the various amino acid substitution and deletion mutants. CHO-K1 cells expressing wild-type PILRα, PILRβ, or each of the amino acid substitution and deletion mutants were lysed, were resolved by SDS-PAGE, and blotted with rabbit anti-FLAG and goat anti-rabbit secondary antibody.
Mutation of leucine 139 to tryptophan in PILRβ does not confer cell fusion activity on PILRβ.
We were particularly interested in tryptophan 139 of PILRα, since mutation of this amino acid to leucine, the homologous residue in PILRβ, completely abrogated fusion activity. Replacement of the other divergent PILRα residues had no effect or only modest effects on cell fusion activity, implying that this tryptophan may be solely responsible for the difference in function between PILRα and PILRβ. To investigate this possibility, an L139W substitution was added to PILRβ. This mutated form of PILRβ was expressed well on the cell surface (Fig. 5, upper panel) and in cell lysates by Western blotting (Fig. 5. lower panel). Despite good expression, however, no significant increase in cell fusion activity was detected when the PILRβ L139R mutant was compared to the WT PILRβ. These results suggest that L139W is not sufficient to restore cell fusion activity of PILRβ. Other domains or specific amino acids of PILRα are likely required for full fusion function, as was suggested by the studies with the chimeric proteins.
FIG. 5.
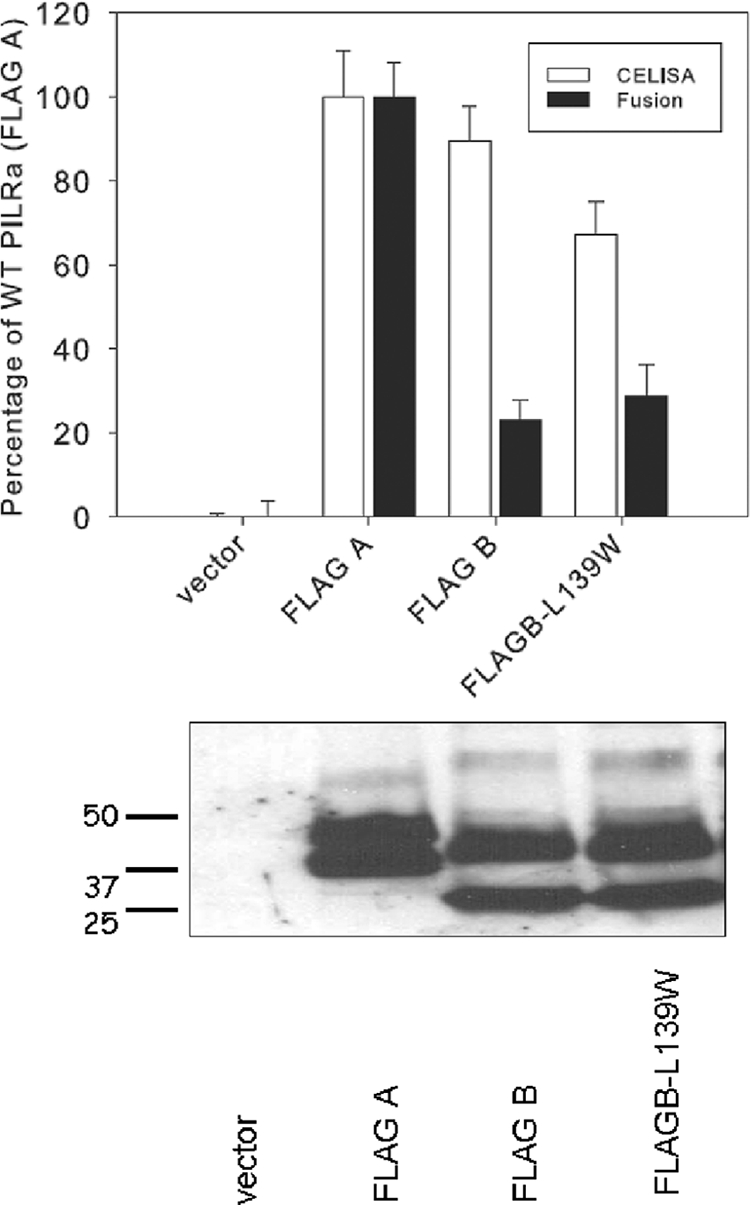
PILRβ L139W is defective in cell fusion activity compared to PILRα. Upper panel, CELISA and cell fusion activity for the PILRβ L139W. For CELISA, CHO-K1 cells were transfected in 96-well plates with PILRβ L139W and controls. The cells were washed, incubated with an anti-FLAG M2 antibody, and washed extensively prior to fixation and incubation with a mouse secondary antibody and an HRP detection system. For cell fusion activity, PILRα ligand-negative CHO-K1 cells were transfected with pFLAG-CMV-myc-21 (empty vector), FLAG A, FLAG B, or PILRβ L139W along with a reporter plasmid expressing luciferase under the control of the T7 promoter. The transfected cells were replated with CHO-K1 cells transfected with plasmids expressing T7 polymerase, gB, gD, gH, and gL. Each bar shows the mean and standard deviation of three independent determinations, with the results expressed as a percentage of wild-type PILRα activity. Lower panel, Western blot analyzing the expression of PILRβ L139W. CHO-K1 cells expressing wild-type PILRα, PILRβ, or PILRβ L139W were lysed, were resolved by SDS-PAGE, and blotted with rabbit anti-FLAG and goat anti-rabbit secondary antibody.
Binding of PILRα, PILRβ, the PILRα Ig-like V-type domains, and tryptophan mutants to gB.
Previous studies have shown that when the PILRα ectodomain is fused to IgG Fc, the resulting fusion protein (PILRα-Ig) can bind to gB on cell surfaces (3, 26) and to its natural ligand, CD99 (41). To assess whether wild-type PILRβ or mutant PILRα could bind to gB expressed on the cell surface of CHO-K1 cells, we made several chimeric constructs. First, we cloned the ectodomain of PILRβ into pDM19 containing the rabbit Fc portion of rabbit Ig, generating pQF21, and performed a CELISA and an immunoprecipitation to determine if PILRβ could bind to gB. Results from these experiments indicated that PILRβ did not bind to gB (data not shown). Since pME18S PILRα-Ig has a human IgG, we next made soluble proteins using the same vector (pQF62) for consistency in our analysis. The human PILRβ-Ig was generated by fusing the ectodomain of PILRβ with the human IgG Fc domain like the PILRα-Ig expression construct. Similarly, the N-terminal portion of PILRα containing the Ig-like V-type domain was fused with the human IgG Fc domain (PILRα V-Ig). The W139L mutation was added to both the PILRα-Ig construct (PILRα W-Ig) and the PILRα V-Ig construct containing the V domain only (PILRα W-V-Ig).
Using CELISA, we investigated the binding of these PILR-Ig proteins to gB. 293 PEAK Rapid cells were transfected with the PILR-Ig constructs, and the secreted protein levels in the supernatants were normalized. Binding of PILR-Ig from these supernatants to 293 cells expressing gB was analyzed by CELISA. As expected, we found that PILRα-Ig bound well to cells expressing gB (Fig. 6, upper panel). PILRα V-Ig also bound to cells expressing gB, consistent with the fusion results suggesting that the V domain is the critical domain for fusion. Also consistent with the cell fusion data, PILRβ-Ig, PILRα-Ig (W139L), and PILRα V-Ig (W139L) did not bind to cells expressing gB.
FIG. 6.
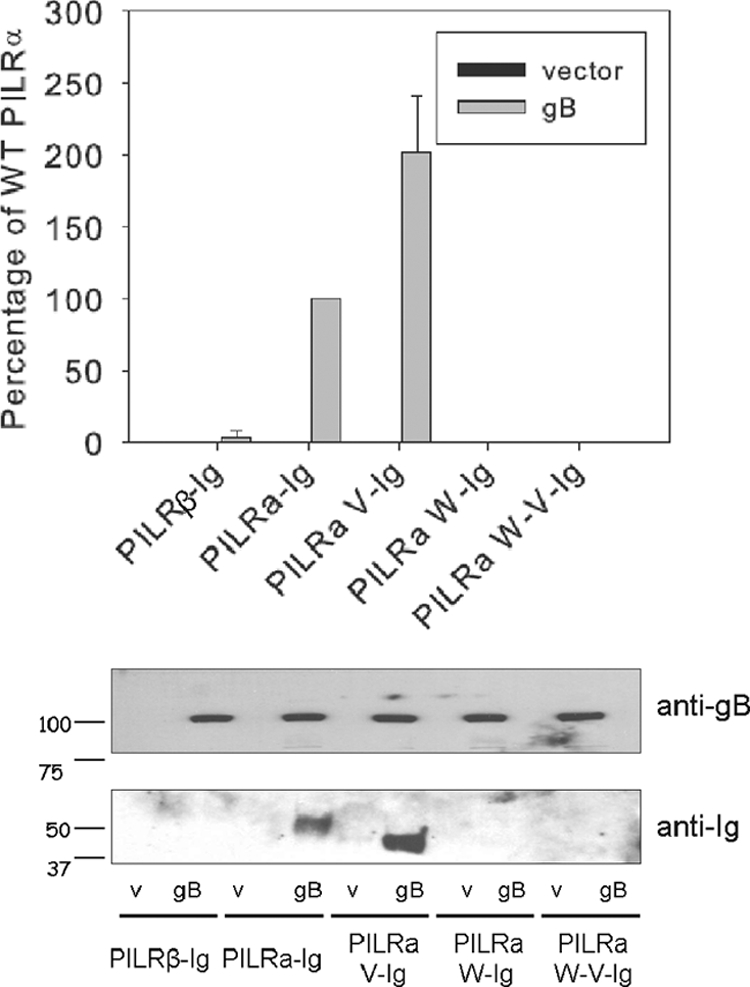
CELISA and Western blot demonstrating that PILRα, but not PILRβ, binds to gB. Upper panel, CELISA, 293 PEAK Rapid cells transfected with empty vector and gB in 96-well plates were incubated with PILR-Ig supernatants, fixed, and then incubated with biotinylated goat anti-Human Ig(H+L) and an HRP detection system. Lower panel, Western blot. CHO-K1 cells expressing empty vector and gB in six-well plates were incubated with PILR-Ig supernatants and washed, and intact cells were lysed and resolved for SDS-PAGE.
Since CHO-K1 cells were used in the cell-cell fusion experiments, we further tested the binding of the secreted PILR-Ig proteins to gB expressed on CHO-K1 cells. CHO-K1 cells were transfected with gB or empty vector. The cells were incubated with supernatants normalized for PILR-Ig protein content as described above, washed thoroughly, and subsequently lysed for SDS-PAGE and Western blot analysis. The Western blots were probed with an anti-gB serum or anti-human IgG separately. As expected, only PILRα-Ig and PILRα V-Ig bound to cells expressing gB. PILRβ and other forms of PILR-Ig did not bind to gB (Fig. 6, lower panel) even though gB expression was readily detected in the transfected cells (Fig. 6, middle panel). Interestingly, PILRα V-Ig showed stronger binding to gB than PILRα-Ig did to gB, as shown in Fig. 6 (upper panel).
DISCUSSION
PILRα not only acts as an HSV-1 gB receptor (26) but also mediates entry of pseudorabies virus, but not HSV-2 (1). In addition to the HSV-1 gB receptor PILRα, myelin-associated glycoprotein (MAG) was recently identified as a gB receptor for varicella-zoster virus (VZV) (36). MAG is also associated with HSV-1 gB and enhanced HSV-1 infection of promyelocytes (36). Since we determined that a variety of human cell lines expressed PILRβ but not PILRα (Fig. 1, upper panel), we investigated the functional roles of PILRα and PILRβ in HSV-induced membrane fusion. Similar to results of previous studies (1), we found that PILRα does not function in HSV-2-induced membrane fusion despite a high level of conservation between HSV-1 and HSV-2 gB (89% amino acid identity). To our surprise, despite good conservation between PILRα and PILRβ, we found that PILRβ does not function in HSV-1 or HSV-2 membrane fusion. To investigate this result, we made chimeric proteins and site-specific mutants of PILRα and PILRβ to determine which domains of PILRα are important for function in fusion. By exchanging only the Ig-like V-type domain between PILRα and PILRβ, we found that this domain was a key determinant of PILRα function. All chimeras except A196-B with the Ig-like V domain of PILRα generally had some fusion activity characteristic of human PILRα, whereas chimeras with the Ig-like V domain of PILRβ had no fusion activity.
In comparing the current studies with previous studies analyzing the interaction of gD with the cellular receptors nectin-1 and nectin-2, several interesting differences become apparent. Nectin-1 and nectin-2 share a similar ectodomain structure consisting of an N-terminal Ig-like V domain and two Ig-like constant (C) domains. Previous studies demonstrated that the N-terminal Ig-like V domains of nectin-1 and nectin-2 were important for HSV entry (9, 15, 16, 33, 34), whereas the Ig-like C domains of nectin-1 and nectin-2 were important not for entry but rather for expression and oligomerization. In addition, all chimeras with the Ig-like V domain of human nectin-2 functioned similarly to the wild type regardless of the derivation of the rest of the ectodomain, transmembrane domain, or cytoplasmic tail. Similar results were obtained for nectin-1 chimeras, which showed that only chimeras and mutants containing the entire Ig-like V domain conferred cell fusion and entry activity (35). In contrast, our results indicate that domains outside the PILRα Ig-like V-type domain may also be important for fusion function, since chimeras replacing just the Ig-like V-type domain of PILRβ with the Ig-like V-type domain of PILRα had a marked reduction in cell fusion activity (Fig. 3) compared to wild-type PILRα, indicating that the transmembrane domain and/or the cytoplasmic tail of PILRα may play a role in membrane fusion. Interestingly, previous studies also showed that the ability of chimeric nectin-1-related receptors to bind gD is not necessarily sufficient for viral entry (5), similar to our results that the Ig-type V-type domain of PILRα can confer wild-type-like binding to gB while the binding does not confer wild-type-like cell fusion activity. Since previous studies have shown that tyrosine phosphorylation of host cellular proteins is triggered by HSV entry (23), we also investigated if phosphorylation of Y269 and Y298, located in the cytoplasmic ITIM of PILRα, was important for HSV-1-induced membrane fusion. These two tyrosine residues provide binding sites for the amino-terminal and carboxyl-terminal SH2 domains of SHP-1. Y269 in particular plays an important role in recruitment of SHP-1 to PILRα (20). Mutations in the ITIM (Y269 and Y298) did not alter PILRα expression and cell fusion activity (Fig. 4). Further investigation will be required to determine if these tyrosines have a role in the tyrosine phosphorylation observed when HSV-1 infects cells and if this phosphorylation is important for any aspect of HSV-1 infection. N-linked glycans play an important role in recycling membrane proteins within the endoplasmic reticulum (ER) until they are folded properly (7). The mutant with a mutation at PILRα N100 had reduced cell surface expression and completely abrogated cell fusion activity (Fig. 4). The failure of the N100 mutant to mediate HSV fusion probably resulted from its failure to be transported to a membrane. The deletion mutant Δ151-195, which also was poorly expressed in CELISA, may also have a folding defect resulting in retention in the ER.
The most interesting PILRα mutant that we obtained is the W139L mutant, with a mutation located in the Ig-like V-type domain. It was expressed well on the cell surface and did not mediate cell fusion activity, suggesting this tryptophan plays a key role in cell fusion activity (Fig. 4) and binding of PILRα binding to gB (Fig. 6). Interestingly, the converse mutation of L139W in PILRβ did not confer the ability to mediate cell-cell fusion on PILRβ (Fig. 5), further supporting the idea that there are other amino acids that play a role in the binding and fusion activity of PILRα with gB. Tryptophans, with a large hydrophobic surface, have a unique role in many proteins, including the protein-folding process (8), assembly of membrane proteins (27), and overall tertiary structure (25). Replacement of the hydrophobic bulky tryptophan with similarly hydrophobic but less bulky leucine suggests that a local change in protein structure rather than a global change in structure may be responsible for loss of PILRα function, since leucine is found in PILRβ and it would be expected that PILRα and PILRβ would be structurally similar due to a high degree of sequence homology. The near-normal expression of this mutant is compatible with this interpretation. Tryptophan residues are associated with protein-protein interaction. For example, tryptophan residues in the portal protein of HSV-1 play critical roles in interaction with scaffold proteins and affect the incorporation of the portal into capsids (45). We tried to localize PILRα W139 to a particular structural determinant of solved structures of Ig V domains but failed because the Ig-like V-type domain of PILRα has very low amino acid identity to other forms of the Ig V domain (<30%). The role of W139 of PILRα in HSV cell membrane fusion needs to be further investigated.
Previous results have shown that PILRα binds to gB and CD99 (3, 26, 40). The binding of either PILRα or PILRβ to CD99 depends on the presence of sialyated O-linked glycans on CD99 (41). We investigated the binding properties of PILRα, PILRβ, and our PILRα-PILRβ chimeric proteins by making Fc fusions. From these studies, we were able to demonstrate that PILRα and the PILRα Ig-like V-type domain are able to bind to HSV-1 gB. Thus, the Ig-like V-type domain is sufficient to bind to gB. PILRβ-Ig did not bind to gB (Fig. 6). Our results also indicated that PILRβ-Ig did not bind to HSV-2 gB (data not shown). As might be expected from the cell fusion results, tryptophan 139 in PILRα was important for gB binding, providing evidence that this tryptophan plays a key role in protein-protein interactions required for both binding and the induction of cell fusion.
Overall, our results further define the domains and key amino acids of PILRα that are critical for fusion and provide a basis for further study of the role of PILRα in HSV fusion to and entry into target cells. Future studies will need to carefully address the relative importance of PILRα and other HSV receptors for the binding and entry of HSV and how domains outside the Ig-like V-type domain contribute to fusion function of PILRα. As the modes of entry are determined for different cell types in vivo and the contributions of the different HSV receptors to the pathogenesis of HSV in natural infections are understood, therapeutics that effectively target the HSV entry process may be developed.
Acknowledgments
We thank Hisashi Arase for providing us the H2KB and human PILRα cell lines. We thank N. Susmarski for timely and excellent technical assistance and members of the Longnecker Laboratory for their help with these studies, particularly Sarah Connolly and Cindy Rowe for reading the manuscript. Traditional sequencing services were performed at the Northwestern University Genomics Core Facility.
This work was supported by NIH grants CA021776 and AI067048 to R.L.
Footnotes
Published ahead of print on 23 June 2010.
REFERENCES
- 1.Arii, J., M. Uema, T. Morimoto, H. Sagara, H. Akashi, E. Ono, H. Arase, and Y. Kawaguchi. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor alpha. J. Virol. 83:4520-4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cocchi, F., L. Menotti, P. Mirandola, M. Lopez, and G. Campadelli-Fiume. 1998. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 72:9992-10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan, Q., E. Lin, T. Satoh, H. Arase, and P. G. Spear. 2009. Differential effects on cell fusion activity of mutations in herpes simplex virus 1 glycoprotein B (gB) dependent on whether a gD receptor or a gB receptor is overexpressed. J. Virol. 83:7384-7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fournier, N., L. Chalus, I. Durand, E. Garcia, J. J. Pin, T. Churakova, S. Patel, C. Zlot, D. Gorman, S. Zurawski, J. Abrams, E. E. Bates, and P. Garrone. 2000. FDF03, a novel inhibitory receptor of the immunoglobulin superfamily, is expressed by human dendritic and myeloid cells. J. Immunol. 165:1197-1209. [DOI] [PubMed] [Google Scholar]
- 5.Geraghty, R. J., A. Fridberg, C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 2001. Use of chimeric nectin-1(HveC)-related receptors to demonstrate that ability to bind alphaherpesvirus gD is not necessarily sufficient for viral entry. Virology 285:366-375. [DOI] [PubMed] [Google Scholar]
- 6.Geraghty, R. J., C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618-1620. [DOI] [PubMed] [Google Scholar]
- 7.Helenius, A., and M. Aebi. 2004. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73:1019-1049. [DOI] [PubMed] [Google Scholar]
- 8.Jonasson, P., G. Aronsson, U. Carlsson, and B. H. Jonsson. 1997. Tertiary structure formation at specific tryptophan side chains in the refolding of human carbonic anhydrase II. Biochemistry 36:5142-5148. [DOI] [PubMed] [Google Scholar]
- 9.Krummenacher, C., A. H. Rux, J. C. Whitbeck, M. Ponce de Leon, H. Lou, I. Baribaud, W. Hou, C. Zou, R. J. Geraghty, P. G. Spear, R. J. Eisenberg, and G. H. Cohen. 1999. The first immunoglobulin-like domain of HveC is sufficient to bind herpes simplex virus gD with full affinity, while the third domain is involved in oligomerization of HveC. J. Virol. 73:8127-8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krummenacher, C., V. M. Supekar, J. C. Whitbeck, E. Lazear, S. A. Connolly, R. J. Eisenberg, G. H. Cohen, D. C. Wiley, and A. Carfi. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 24:4144-4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubagawa, H., C. C. Chen, L. H. Ho, T. S. Shimada, L. Gartland, C. Mashburn, T. Uehara, J. V. Ravetch, and M. D. Cooper. 1999. Biochemical nature and cellular distribution of the paired immunoglobulin-like receptors, PIR-A and PIR-B. J. Exp. Med. 189:309-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanier, L. L. 1998. NK cell receptors. Annu. Rev. Immunol. 16:359-393. [DOI] [PubMed] [Google Scholar]
- 13.Lin, E., and P. G. Spear. 2007. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 104:13140-13145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez, M., F. Cocchi, L. Menotti, E. Avitabile, P. Dubreuil, and G. Campadelli-Fiume. 2000. Nectin2α (PRR2α or HveB) and nectin2δ are low-efficiency mediators for entry of herpes simplex virus mutants carrying the Leu25Pro substitution in glycoprotein D. J. Virol. 74:1267-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez, W. M., and P. G. Spear. 2002. Amino acid substitutions in the V domain of nectin-1 (HveC) that impair entry activity for herpes simplex viruses 1 and 2 but not for pseudorabies virus or bovine herpesvirus 1. J. Virol. 76:7255-7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez, W. M., and P. G. Spear. 2001. Structural features of nectin-2 (HveB) required for herpes simplex virus entry. J. Virol. 75:11185-11195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menotti, L., E. Avitabile, P. Dubreuil, M. Lopez, and G. Campadelli-Fiume. 2001. Comparison of murine and human nectin-1 binding to herpes simplex virus glycprotein D (gD) reveals a weak interaction of murine nectin-1 to gD and a gD-dependent pathway of entry. Virology 282:256-266. [DOI] [PubMed] [Google Scholar]
- 18.Menotti, L., M. Lopez, E. Avitabile, A. Stefan, F. Cocchi, J. Adelaide, E. Lecocq, P. Dubreuil, and G. Campadelli-Fiume. 2000. The murine homolog of human Nectin1δ serves as a species nonspecific mediator for entry of human and animal αherpesviruses in a pathway independent of a detectable binding to gD. Proc. Nat. Acad. Sci.U. S. A. 97:4867-4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427-436. [DOI] [PubMed] [Google Scholar]
- 20.Mousseau, D. D., D. Banville, D. L'Abbe, P. Bouchard, and S.-H. Shen. 2000. PILRα, a novel immunoreceptor tyrosine-based inhibitory motif-bearing protein, recruits SHP-1 upon tyrosine phosphorylation and is paired with the truncated counterpart PILRβ. J. Biol. Chem. 275:4467-4474. [DOI] [PubMed] [Google Scholar]
- 21.Munitz, A., M. L. McBride, J. S. Bernstein, and M. E. Rothenberg. 2008. A dual activation and inhibition role for the paired immunoglobulin-like receptor B in eosinophils. Blood 111:5694-5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pertel, P., A. Fridberg, M. L. Parish, and P. G. Spear. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313-324. [DOI] [PubMed] [Google Scholar]
- 23.Qie, L., D. Marcellino, and B. C. Herold. 1999. Herpes simplex virus entry is associated with tyrosine phosphorylation of cellular proteins. Virology 256:220-227. [DOI] [PubMed] [Google Scholar]
- 24.Rey, F. A. 2006. Molecular gymnastics at the herpesvirus surface. EMBO Rep. 7:1000-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samanta, U., D. Pal, and P. Chakrabarti. 2000. Environment of tryptophan side chains in proteins. Proteins 38:288-300. [PubMed] [Google Scholar]
- 26.Satoh, T., J. Arii, T. Suenaga, J. Wang, A. Kogure, J. Uehori, N. Arase, I. Shiratori, S. Tanaka, Y. Kawaguchi, P. G. Spear, L. L. Lanier, and H. Arase. 2008. PILRa is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schiffer, M., C. H. Chang, and F. J. Stevens. 1992. The functions of tryptophan residues in membrane proteins. Protein Eng. 5:213-214. [DOI] [PubMed] [Google Scholar]
- 28.Shiratori, I., K. Ogasawara, T. Saito, L. L. Lanier, and H. Arase. 2004. Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J. Exp. Med. 199:525-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shukla, D., M. Dal Canto, C. L. Rowe, and P. G. Spear. 2000. Striking similarity of murine nectin-1α to human nectin-1α (HveC) in sequence and activity as a gD receptor for alphaherpesvirus entry. J. Virol. 74:11773-11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shukla, D., J. Liu, P. Blaiklock, N. W. Shworak, X. Bai, J. D. Esko, G. H. Cohen, R. J. Eisenberg, R. D. Rosenberg, and P. G. Spear. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13-22. [DOI] [PubMed] [Google Scholar]
- 31.Shukla, D., and P. G. Spear. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 108:503-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sorem, J., T. S. Jardetzky, and R. Longnecker. 2009. Cleavage and secretion of Epstein-Barr virus glycoprotein 42 promote membrane fusion with B lymphocytes. J. Virol. 83:6664-6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Struyf, F., W. M. Martinez, and P. G. Spear. 2002. Mutations in the N-terminal domains of nectin-1 and nectin-2 reveal differences in requirements for entry of various alphaherpesviruses and for nectin-nectin interactions. J. Virol. 76:12940-12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Struyf, F., A. E. Plate, and P. G. Spear. 2005. Deletion of the second immunoglobulin-like domain of nectin-1 alters its intracellular processing and localization and ability to mediate entry of herpes simplex virus. J. Virol. 79:3841-3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramanian, R. P., J. E. Dunn, and R. J. Geraghty. 2005. The nectin-1α transmembrane domain, but not the cytoplasmic tail, influences cell fusion induced by HSV-1 glycoproteins. Virology 339:176-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suenaga, T., T. Satoh, P. Somboonthum, Y. Kawaguchi, Y. Mori, and H. Arase. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. U. S. A. 107:866-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Syken, J., T. Grandpre, P. O. Kanold, and C. J. Shatz. 2006. PirB restricts ocular-dominance plasticity in visual cortex. Science 313:1795-1800. [DOI] [PubMed] [Google Scholar]
- 38.Takai, Y., J. Miyoshi, W. Ikeda, and H. Ogita. 2008. Nectins and nectin-like moleecules: roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 9:603-615. [DOI] [PubMed] [Google Scholar]
- 39.Uehara, T., M. Blery, D. W. Kang, C. C. Chen, L. H. Ho, G. L. Gartland, F. T. Liu, E. Vivier, M. D. Cooper, and H. Kubagawa. 2001. Inhibition of IgE-mediated mast cell activation by the paired Ig-like receptor PIR-B. J. Clin. Invest. 108:1041-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang, J., Q. Fan, T. Satoh, J. Arii, L. L. Lanier, P. G. Spear, Y. Kawaguchi, and H. Arase. 2009. Binding of herpes simplex virus glycoprotein B (gB) to paired immunoglobulin-like type 2 receptor alpha depends on specific sialylated O-linked glycans on gB. J. Virol. 83:13042-13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang, J., I. Shiratori, T. Satoh, L. L. Lanier, and H. Arase. 2008. An essential role of sialylated O-linked sugar chains in the recognition of mouse CD99 by paired Ig-like type 2 receptor (PILR). J. Immunol. 180:1686-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ware, C. F. 2008. Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol. Rev. 223:186-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warner, M. S., R. J. Geraghty, W. M. Martinez, R. I. Montgomery, J. C. Whitbeck, R. Xu, R. J. Eisenberg, G. H. Cohen, and P. G. Spear. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2 and pseudorabies virus. Virology 246:179-189. [DOI] [PubMed] [Google Scholar]
- 44.Wilson, M. D., J. Cheung, D. W. Martindale, S. W. Scherer, and B. F. Koop. 2006. Comparative analysis of the paired immunoglobulin-like-receptor (PILR) locus in six mammalian genomes: duplication, conversion, and the birth of new genes. Physiol. Genomics 27:201-218. [DOI] [PubMed] [Google Scholar]
- 45.Yang, K., and J. D. Baines. 2009. Tryptophan residues in the portal protein of herpes simplex virus 1 critical to the interaction with scaffold proteins and incorporation of the portal into capsids. J. Virol. 83:11726-11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zago, A., and P. G. Spear. 2003. Differences in the N termini of herpes simplex virus type 1 and 2 gDs that influence functional interactions with the human entry receptor nectin-2 and an entry receptor expressed in Chinese hamster ovary cells. J. Virol. 77:9695-9699. [DOI] [PMC free article] [PubMed] [Google Scholar]