

Abstract
Salmonid alphavirus (SAV) is an emerging virus in salmonid aquaculture, with SAV-3 being the only subtype found in Norway. Until now, there has been little focus on the alpha interferon (IFN-α)-induced antiviral responses during virus infection in vivo or in vitro in fish. The possible involvement of IFN-γ in the response to SAV-3 is also not known. In this study, the two IFNs were cloned and expressed as recombinant proteins (recombinant IFN-α [rIFN-α] and rIFN-γ) and used for in vitro studies. SAV-3 infection in a permissive salmon cell line (TO cells) results in IFN-α and IFN-stimulated gene (ISG) mRNA upregulation. Preinfection treatment (4 to 24 h prior to infection) with salmon rIFN-α induces an antiviral state that inhibits the replication of SAV-3 and protects the cells against virus-induced cytopathic effects (CPE). The antiviral state coincides with a strong expression of Mx and ISG15 mRNA and Mx protein expression. When rIFN-α is administered at the time of infection and up to 24 h postinfection, virus replication is not inhibited, and cells are not protected against virus-induced CPE. By 40 h postinfection, the alpha subunit of eukaryotic initiation factor 2 (eIF2α) is phosphorylated concomitant with the expression of the E2 protein as assessed by Western blotting. Postinfection treatment with rIFN-α results in a moderate reduction in E2 expression levels in accordance with a moderate downregulation of cellular protein synthesis, an approximately 65% reduction by 60 h postinfection. rIFN-γ has only a minor inhibitory effect on SAV-3 replication in vitro. SAV-3 is sensitive to the preinfection antiviral state induced by rIFN-α, while postinfection antiviral responses or postinfection treatment with rIFN-α is not able to limit viral replication.
Salmon pancreas disease virus (SPDV) is the causative agent of pancreas disease (PD) in Atlantic salmon and rainbow trout and is an emerging pathogen in Europe and North America (21). SPDV belongs to the genus Alphavirus within the family Togaviridae and is phylogenetically related to arthropod-borne alphavirus groups such as the Semliki Forest virus (SFV) group, the Sindbis virus (SINV) group, and Venezuelan equine encephalitis virus (VEEV)/Eastern equine encephalitis virus (EEEV) (24). SPDV was later termed salmonid alphavirus (SAV) and has been divided into subtypes (14), and six subtypes have now been recognized, SAV-1 to SAV-6 (9). SAV-1 was first isolated from farmed Atlantic salmon in Ireland and Scotland (37). Subsequently, sleeping disease virus (SDV) (and later SAV-2), which affects mainly rainbow trout, was discovered in the United Kingdom and France (35, 38). A third and new subtype, SAV-3, is found exclusively in Norway and affects both Atlantic salmon and rainbow trout (14, 33). Three additional subtypes of SAV from Scotland and Ireland have been described (9). Diseased fish are clinically characterized by inappetence, fecal casts, and emaciation, with main pathological changes found in pancreas, heart, and skeletal muscle (21, 25). Immunity to alphavirus infections in salmonids is poorly understood and has not been studied in any detail. In a previous study, interferon (IFN) responses were not detected following SAV-1 infection in salmon parr, possibly due to the low sensitivity of the detection method used (7). A more recent study showed Mx mRNA upregulation in response to SAV infection in vitro by using a macrophage cell line from salmon and suggested that Mx has an antiviral function against SAV (11). However, so far, no studies have been carried out to examine the functional aspects of type I IFN responses related to SAV infections of salmonids in vivo or in vitro.
Type I interferons (alpha/beta IFN [IFN-α/β]) and interferon-stimulated genes (ISGs) are known to play a pivotal role in innate immune responses against viral infections in all living organisms (18) and, therefore, also in fish (42). Mice devoid of type I IFN receptors are extremely susceptible to alphavirus infections, whereas there is no apparent role for IFN-γ (22). Studies of infections in higher vertebrates have shown that the IFN-α/β pathway represents the primary protective response against alphavirus infection involving the limitation of virus replication (1, 13). The antiviral effect of mammalian type I IFN is exerted through binding to the receptor of IFN-α/β, triggering the JAK-STAT pathway, which results in the expression of ISGs, some of which encode antiviral proteins, including Mx, ISG15, and double-stranded RNA (dsRNA)-dependent protein kinase R (PKR) (29). Numerous ISGs such as 2′,5′-oligoadenylate synthetase (OAS)/RNase L, zinc finger antiviral protein (ZAP), Mx, and ISG15 likely execute a direct or indirect inhibition of alphavirus replication in higher vertebrates (5, 16, 19), although the detailed mechanisms are not understood. The importance of ISGs in relation to alphavirus infections has not been studied for fish. Type I IFN and several of its stimulated genes have been cloned from Atlantic salmon, and their analogous antiviral functions are also partly characterized, which allows a detailed characterization of the responses at the transcript level. Furthermore, the antiviral nature of Atlantic salmon IFN was recently characterized by neutralizing IFN with specific antibodies in vitro (3, 23). Mx and ISG15 have been cloned from Atlantic salmon (26, 27), and their functional antiviral activity has been demonstrated against another salmon virus, infectious pancreatic necrosis virus (17). Atlantic salmon PKR protein has been cloned, but no functional studies have been performed. However, PKR from Japanese flounder (Paralichthys olivaceus) was recently characterized and shown to increase the phosphorylation of the alpha subunit of eukaryotic initiation factor 2 (eIF2α) and inhibit the replication of rhabdovirus in flounder embryonic cells (41). Atlantic salmon PKZ has a direct inhibitory effect on protein synthesis after transient expression in Chinook salmon embryo (CHSE) cells (4).
In this study we were interested in understanding IFN-α- and IFN-γ-induced responses to SAV-3 infection in vitro and the possible role of these cytokines in controlling virus replication during the early stages of infection. The two IFNs were cloned and expressed as recombinant proteins and used for in vitro studies using a salmon macrophage cell line (36) as a model for SAV infection (11). We show that in SAV-3-infected cell cultures, IFN-α and ISG mRNAs are upregulated and Mx protein expression is strongly induced. By 40 h postinfection, eIF2α is phosphorylated, which coincides with a strong expression of the E2 protein of SAV-3. There is no clear arrest of macromolecular synthesis at early times postinfection (first 24 h postinfection), while from 36 h, there is a moderate downregulation of protein synthesis. The treatment of cell cultures with recombinant IFN-α (rIFN-α) 4 to 24 h prior to infection (preinfection treatment) induces an antiviral response that markedly inhibits the replication of SAV-3 and protects against cytopathic effects (CPE). Treatment with rIFN-α at time of infection and up to 24 h postinfection has a time-dependent effect on limiting SAV-3 replication: the later the treatment, the less efficient, and later treatment will not protect against CPE. Preinfection treatment (24 h) with IFN-γ has only a minor inhibitory effect on the replication of SAV-3 in vitro.
MATERIALS AND METHODS
Virus isolation and cell culture.
Chinook salmon embryonic cells (CHSE-214; ATCC CRL-1681) were maintained at 20°C with L-15 medium (Invitrogen) supplemented with 5% fetal bovine serum (FBS), l-glutamine, and gentamicin. TO cells (macrophage cell line), originating from salmon head kidney leukocytes (36), were grown at 20°C in HMEM (Eagle's minimal essential medium [MEM] with Hanks' balanced salt solution [BSS]) supplemented with l-glutamine, MEM nonessential amino acids, gentamicin sulfate, and 10% FBS. The SAV-3 isolate (named H10) used in this study was isolated from heart of an Atlantic salmon with clinical symptoms of PD. The isolate was propagated by inoculating 80% confluent CHSE-214 cells maintained with growth medium supplemented with 2% FBS. The isolate was identified as a SAV-3 variant by sequencing (14) and was passaged nine times in cell culture before being used in this study.
Protein synthesis shutdown.
TO cells were infected with SAV-3 at a multiplicity of infection (MOI) of 20. At different time points postinfection, the cells were washed three times with phosphate-buffered saline (PBS) and then incubated for 30 min in Dulbecco's modified Eagle's medium lacking methionine and supplemented with 0.1% FBS and 20 μCi [35S]methionine/ml. The cells were then harvested sequentially at 12, 24, 36, 48, 60, and 72 h and lysed by using CelLytic M reagent (Sigma). Protein was separated by SDS-PAGE and blotted onto a polyvinylidene difluoride (PVDF) membrane. The membrane was exposed in a PhosphorImager cassette and then scanned by using a Typhoon imager (GE Healthcare). The protein amount measured by densitometry was quantified with ImageJ software, and the value was expressed relative to a mock-infected control and corrected for the protein amount loaded into each lane.
RNA isolation and cDNA synthesis.
Total RNA was isolated by using the RNeasy Plus minikit (Qiagen), and the concentration of RNA was determined by spectrophotometry (Nanodrop ND1000). For each sample, 500 ng of total RNA was subjected to cDNA synthesis using a Transcriptor first-strand cDNA kit (Roche) in a total volume of 20 μl. The synthesized cDNA was stored at −20°C until further use.
Quantitative real-time PCR.
Quantitative PCR was performed by using LightCycler 480 SYBR green I master mix and the LightCycler 480 system (Roche). For each gene, 2 μl of cDNA was used as a template in a mixture of specific primers (10 μM) in a final volume of 20 μl. The mixtures were first incubated at 95°C for 10 min, followed by 40 amplification cycles (10 s at 95°C, 20 s at 60°C, and 8 s at 72°C). The sequences of primers used to assess the in vivo and in vitro expressions of Mx, ISG15, γIP10, and viral E2 are given in Table 1. For the viral E2 gene, the reaction mix contained 10 μl of LightCycler 480 Probes Master, 1 μl of primer-probe mix (final concentrations, 0.9 μM each primer and 0.25 μM probe), 2 μl of cDNA template, and 7 μl water and was incubated for 10 min at 95°C, followed by 45 amplification cycles (10 s at 95°C, 30 s at 60°C, and 1 s at 72°C). The specificity of the PCR products from each primer pair was confirmed by melting-curve analysis and subsequent agarose gel electrophoresis. The 2−ΔΔCT method was used to calculate the gene products as described elsewhere previously (20). 2−ΔΔCT is the relative mRNA expression representing the fold induction over the control group. All quantifications were normalized to β-actin (endogenous gene).
TABLE 1.
Primer and probe sequences for cloning and quantitative real-time PCRa
| Primer | Sequence | Use | GenBank accession no. |
|---|---|---|---|
| IFNα-F1 | CAGTATGCAGAGCGTGTGT | pGEMT cloning | AY216594 |
| IFNα-R1 | CGTAGCTTCTGAAATGAGTCTGG | ||
| pET-IFNα-F1 | GCGCATATGTGTGACTGGATCCGACAC | pET32 cloning | |
| pET-IFNα-R1 | GCGCTCGAGGTACATCTGTGCTGCAAG | ||
| pET-IFNα-R2 | GCGCTCGAGGTTCATTTTTCTCAGAAC | ||
| IFNα-F2 | TGGGAGGAGATATCACAAAGC | qPCR | |
| IFNα-R2 | TCCCAGGTGACAGATTTCAT | ||
| Mx-F | TGCAACCACAGAGGCTTTGAA | qPCR | U66475 |
| Mx-R | GGCTTGGTCAGGATGCCTAAT | ||
| pET14b-E2-F | GCGCATATGGCTGTGTCTGCGTCGCCT | pET14b cloning | |
| pET14b-E2-R | GCGCTCGAGTTACGCACGAGCCCCAGG | ||
| SAV-3 E2-F | CAGTGAAATTCGATAAGAAGTGCAA | qPCR | EF675594 |
| SAV-3 E2-R | TGGGAGTCGCTGGTAAAGGT | ||
| E2 probe | FAM-5′-AGCGCTGCCCAAGCGACCG-3′-MGB | ||
| IFNγ-F1 | AGGCGGTCTCGTTAAGTCAA | pGEMT cloning | AY795563 |
| IFNγ-R1 | TAAACTGACCCAAGATCAGC | ||
| pET32-IFNγ-F | GCGCATATGGCTCAGTACACATCAATT | pET32 cloning | |
| pET32-IFNγ-R1 | GCGCTCGAGCATGATGCTTGATTTGAG | ||
| pET32-IFNγ-R2 | GCGCTCGAGTTTCTCGTAGATGGTAAT | ||
| IFNγ-F4 | CTAAAGAAGGACAACCGCAG | qPCR | |
| IFNγ-R4 | CACCGTTAGAGGGAGAAATG | ||
| γIP10-F | TGCCAGAACATGGAGATCAT | qPCR | EF619047 |
| γIP10-R | TTTACTGCACACTCCTTTGGTT | ||
| ISG15-F | AAGTGATGGTGCTGATTACGG | qPCR | AY926456 |
| ISG15-R | CACCGTTAGAGGGAGAAATG | ||
| β-actin-F | CCAGTCCTGCTCACTGAGGC | qPCR | AF012125 |
| β-actin-R | GGTCTCAAACATGATCTGGGTCA |
Restriction sites are underlined. qPCR, quantitative PCR; FAM, 6-carboxyfluorescein; MGB, MGB-TaqMan probe.
Cloning, expression, and purification of recombinant IFN-α and IFN-γ.
Total RNA from the head kidney of Atlantic salmon was used as a template for cDNA synthesis by using a Transcriptor first-strand cDNA synthesis kit (Roche). For initial cloning, two pairs of primers, IFNα-F1 and IFNα-R1, and IFNγ-F1 and IFNγ-R1, were designed according to the Atlantic salmon salmo salar IFN-α1 (SasaIFN-α1) mRNA sequence (GenBank accession no. AY216594) and the Atlantic salmon SasaIFN-γ mRNA sequence (GenBank accession no. AY795563). For IFN-α, we amplified a region from 42 bp downstream of the start codon of the open reading frame (ORF) to 27 bp downstream of the stop codon. For IFN-γ, a region from 107 bp upstream of the start codon of the ORF to 75 bp downstream of the stop codon was amplified. The PCR products were purified by using the QIAquick gel extraction kit (Qiagen) and cloned into the pGEM-T Easy vector (Promega). The region coding for the predicted full-length forms without the signal peptide was subcloned from pGEM-T into the prokaryotic vector pET-32c (Novagen) by using primer sets pET-IFNα-F1 and pET-IFNα-R1, and pET-IFNγ-F1 and pET-IFNγ-R1, containing NdeI and XhoI restriction sites. Truncated forms of IFN-α (trIFN-α) and IFN-γ (trIFN-γ), with deletions of the C-terminal 28 amino acids (aa) and 29 aa, respectively, were also constructed by using different reverse primers (pET-IFNα-R2 and pET-IFNγ-R2). The vectors were designed to express a C-terminal 6×His fusion protein to facilitate the purification of recombinant proteins. Full-length and truncated forms of IFN-α and IFN-γ were designated rIFN-α and trIFN-α and rIFN-γ and trIFN-γ, respectively, which will be used throughout. The recombinant vectors pET-rIFNα and pET-trIFNα, and pET-rIFNγ and pET-tr IFNγ, were confirmed by DNA sequencing and were transformed into the bacterial host, BL21(DE3), for expression driven by the T7 polymerase. Induction was carried out at 37°C for 2 h with 1 mM isopropyl thiogalactopyranoside (IPTG). The purification of 6×His-tagged rIFN was performed under denaturing conditions by using a His-Bind purification kit (Novagen). Protein concentrations were determined by using the Quick Start Bradford protein assay kit (Bio-Rad) with bovine serum albumin (BSA) as a standard. The expression and the purity of recombinant IFN-α and IFN-γ were checked on a 12% Bis-Tris precast SDS-PAGE gel (Invitrogen Life Technologies) stained with PageBlue protein staining solution (Fermentas). Western blot analysis was performed to confirm the identity of the recombinant IFN-α by using a monoclonal anti-polyhistidine antibody (Sigma).
Cloning, expression, and purification of E2 and preparation of E2 antiserum.
Viral RNA was extracted from the cell supernatant by using a QIAamp viral RNA minikit (Qiagen) and used as a template for cDNA synthesis by using a Transcriptor first-strand cDNA synthesis kit (Roche). A pair of primers, pET14b-E2-F and pET14b-E2-R, was designed according to the E2 gene of the Norwegian salmonid alphavirus isolate (GenBank accession no. AY604236). The cloning, expression, and purification of recombinant E2 proteins were performed as described above. The purified recombinant E2 protein was sent to PickCell Laboratories BV (Netherlands) for rabbit immunization and subsequently characterized by Western blotting and immunofluorescence antibody test (IFAT) staining of SAV-3-infected cultures.
Induction of ISG expression by recombinant IFN-α and IFN-γ in vitro.
TO cells were seeded in 24-well plates and cultured until confluent. For IFN-α, the cells were stimulated with 2.5 μg/ml rIFN-α and trIFN-α, nontreated cells were included as negative control, and samples were harvested at 3, 6, 12, and 24 h poststimulation. The cells were also treated with 10-fold serial dilutions of rIFN-α with an initial starting concentration of 0.47 mg/ml; at 24 h, samples were taken, and RNA was extracted. The induction of ISGs by IFN-α was documented by studying the gene upregulation of Mx and ISG15 by using real-time PCR. For IFN-γ, purified rIFN-γ was serially diluted from 0.33 mg/ml in cell medium and incubated with TO cells for 24 h. The induction of ISGs by IFN-γ was documented by studying the gene upregulation of γIP10 (gamma-IP CXCL10-like chemokine) using real-time PCR. The data are expressed as the mean fold changes in gene expression ± standard errors of different dilutions of the interferon-treated group relative to the nontreated control group after normalization to β-actin.
Antiviral assays.
A CPE reduction assay was used to measure the protective effect of IFN-α against cytopathic effects in pretreated and infected cells (8). TO cells grown in 96-well plates were treated with a serial dilution of rIFN-α and trIFN-α, and rIFN-γ and trIFN-γ, for 24 h and subsequently infected with 1 MOI of SAV-3. Virus was left on the cells for 2 h to adsorb (15°C), after which the cells were washed three times with PBS. Untreated cells, infected and noninfected, were included as controls. The cell viability of the cell cultures subjected to the different treatments was assayed by using the CellTiter 96 AQueous One solution cell proliferation assay kit (Promega) at day 10 postinfection when strong CPE developed in untreated cells. The cell cultures were incubated at 15°C for 4 h, and the absorbance was measured at 490 nm by using a microplate reader (Tecan). For the virus yield reduction assay, culture supernatant and cell total RNA from infected cells in 24-well plates were collected. The titration of virus was done with CHSE-214 cells by the 50% tissue culture infective dose (TCID50) method as described previously by Kärber (15), and viral RNA was quantified by real-time PCR.
Detection of Mx and E2 expression by IFAT.
Cells seeded into 24-well culture plates were fixed with 4% paraformaldehyde for 30 min. After being washed with PBS, the cells were permeabilized with 0.1% Triton X-100 for 5 min on ice. The cells were washed once in PBS and blocked with 5% dry milk in PBS for 2 h before being incubated for 1 h with primary antibody. Anti-salmonid Mx (diluted 1:400; kindly provided by Jo-Ann Leong, Hawaii Institute of Marine Biology) and anti-E2 polyclonal antibody were used to detect Mx and E2 protein expressions, respectively. The cells were washed and incubated with Alexa 594 or Alexa 488 Fluor goat anti-rabbit IgG (Molecular Probes, Invitrogen) diluted 1:200 for 1 h. Finally, the cells were washed and examined by using a fluorescence microscope (Olympus). When nuclear counterstaining was used, it included Hoechst 33324 dye at 5 μg/ml.
Protein analysis of E2, Mx, and p-eIF2α in virus-infected cells.
TO cells were infected with SAV-3 (MOI of 1) or left uninfected. At days 1, 2, 3, and 4 postinfection, uninfected and infected cells were treated with a 103 dilution of IFN-α for 16 h or left untreated. The cells were then lysed by using CelLytic M reagent (Sigma) and scraped from the dish. Expressions of the E2 (antiserum prepared as described above), phosphorylated eIF2α (p-eIF2α) (Cell Signaling), Mx, and actin (Sigma-Aldrich) proteins were detected by Western blotting.
Statistical analysis.
All statistical analyses of gene expression results were performed with the help of GraphPad Prism 5.0 (GraphPad Software Inc.). Two-way analysis of variance (ANOVA) was used to calculate differences in the CPE protection assay at different concentrations. The significant level for rejection of hypothesis 0 (Ho) was set to a P value of <0.05.
RESULTS
Responses to SAV-3 infection in TO cells.
The initial studies were designed to characterize the expression of IFN-α and Mx mRNA in response to SAV-3 infection in TO cells. The kinetics of the virus infection were monitored by real-time reverse transcription (RT)-PCR analysis of the expression of the protein E2-encoding gene, which was detected as early as 6 h postinfection and increased 1,600-fold from 6 h to 24 h postinfection (Fig. 1 a). Over the same time period (6 h to 24 h), IFN-α transcript levels increased 4-fold, while Mx expression levels increased 10-fold (Fig. 1a). From 24 h postinfection to 4 days postinfection (dpi), there was a marked and parallel increase in levels of IFN-α, Mx, and E2 transcripts, all peaking at 4 dpi and declining at later times (Fig. 1a). Staining for the Mx protein at 8 days postinfection showed widespread expression in the infected cell culture (Fig. 1b). These findings provided a strong indication that SAV-3 infection will not result in a shutoff of interferon or interferon-induced responses.
FIG. 1.

(a) Expression levels of IFN-α, Mx, and viral E2 mRNA in TO cells infected with 1 MOI of SAV-3. The left y axis shows the fold increase of IFN-α and Mx mRNA relative to the nontreated control, and the right y axis shows the fold increase of viral E2 transcript levels relative to infected cells at 6 h. The results are expressed as means ± standard errors of the means (SEM) (n = 2). (b) Cytoplasmic Mx protein expression shown by indirect immunofluorescence in TO cells infected with SAV-3. Cells were fixed at 8 days postinfection. Nuclear staining was done with Hoechst 33324 dye.
rIFN-α induces expression of ISGs in salmon cell lines.
With the purpose to conduct functional studies with recombinant IFN-α using a macrophage cell line of Atlantic salmon (TO cells) (36), we first cloned and expressed full-length IFN-α (rIFN-α) and tested its functional activity. The obtained sequence was aligned with SasaIFN-α1 (GenBank accession no. AY216594) and SasaIFN-α2 (accession no. AY216595) sequences, and the amino acid similarities were 96.1% and 99.3%, respectively. As a negative control, we constructed a truncated form of IFN-α (trIFN-α), with a deletion of a 28-aa stretch at the C-terminal end (31). Both full-length and truncated IFN-α were expressed in Escherichia coli BL21(DE3) cells using the pET prokaryotic expression system. The protein yield in the cell soluble fraction was much lower than that of the inclusion bodies (data not shown); thus, the purification of proteins was performed under denaturing conditions. A 6×His tag added at the C-terminal end of the protein facilitated purification with a His-Bind column. The expression and purification of recombinant proteins were identified by SDS-PAGE and Western blot analysis using an anti-His monoclonal antibody (Fig. 2 a and b). The concentrations of purified full-length and truncated IFN-α were 0.47 mg/ml and 0.48 mg/ml, respectively. Dilutions of rIFN-α and trIFN-α refer to these initial concentrations in all experiments except when stated otherwise. A similar characterization was performed for IFN-γ by SDS-PAGE for the full-length and the truncated forms of purified samples (Fig. 2c). The protein concentration for purified full-length rIFN-γ was 0.33 mg/ml.
FIG. 2.

(a) Expression and purification of mature and truncated IFN-α (SDS-PAGE). Lanes: 1, protein marker; 2, full-length recombinant IFN-α (rIFN-α); 3, purified rIFN-α; 4, truncated IFN-α (trIFN-α); 5, purified trIFN-α. (b) Western blot analysis of rIFN-α (lane 1) and trIFN-α (lane 2) in bacterial lysates. The predicted molecular masses of full-length and truncated IFN-α are approximately 19.4 and 16.0 kDa, respectively (M, marker lane). (c) Expression and purification of recombinant and truncated IFN-γ. Lanes: 1, protein marker; 2, full-length IFN-γ (rIFN-γ); 3, purified rIFN-γ; 4, trIFN-γ; 5, purified trIFN-γ. The predicted molecular masses of full-length and truncated IFN-γ are approximately 19.7 and 16.3 kDa, respectively.
The biological function and activity of rIFN-α were assessed by measuring the expression of the interferon-induced Mx and ISG15 genes in TO cells using real-time RT-PCR after stimulation with rIFN-α. At 24 h poststimulation, Mx mRNA was found to be strongly expressed after rIFN-α treatment, while the truncated form was nonfunctional, and no expression of Mx was seen (Fig. 3 a). By serial dilution we found that 0.47 ng/ml of rIFN-α still induced the expression of Mx (2.5-fold upregulation) (106 dilution) (Fig. 3b). The expression of ISG15 mRNA by 12 h post-rIFN-α treatment was 1,000-fold upregulated, increasing to >1,500-fold by 24 h (Fig. 3c). The expression of the Mx protein was confirmed by indirect immunofluorescence 24 h after rIFN-α stimulation. TO cells were stained positive with an anti-Mx antibody (Fig. 4 a), while cells without treatment showed no staining (Fig. 4b).
FIG. 3.

Induction of Mx and ISG15 expression in TO cells after treatment with full-length recombinant and truncated IFN-α. (a and b) Time course expression levels of Mx (a) and dose-dependent induction of Mx mRNA by rIFN-α (b). Purified rIFN-α was diluted serially 10-fold from 0.47 mg/ml and incubated with TO cells for 24 h. The data are expressed as the mean fold changes in gene expression ± SEM of different dilutions for the rIFN-α-treated group relative to the nontreated control group after normalization to β-actin (n = 2). (c) ISG15 mRNA transcripts of TO cells stimulated with 2.5 μg/ml rIFN-α and trIFN-α. The data are expressed as the mean fold changes in gene expression ± standard errors for the IFN-α-treated group relative to the nontreated control group after normalization to β-actin (n = 2).
FIG. 4.

(a) Detection of Mx protein expression by indirect immunofluorescence in TO cells stimulated with 2.5 μg/ml rIFN-α for 24 h and then fixed and stained. (b) Nontreated control cells.
rIFN-α induced a preinfection antiviral state that inhibits SAV-3 replication.
We then went on to study the antiviral effects after pretreatment (24 h) of TO cells with rIFN-α. All studies were carried out using cell cultures originating from Atlantic salmon (Salmo salar L.), while we also included a limited study using CHSE cells (of Chinook salmon origin [Oncorhynchus tshawytscha]). We used the CPE reduction assay and the virus yield reduction assay (virus titer estimation) to measure the antiviral activity of rIFN-α on SAV-3 replication. TO cells were pretreated with a serial dilution of full-length and truncated IFN-α for 24 h and subsequently infected with 1 MOI of SAV-3. rIFN-α and trIFN-α were added to the cell cultures in culture medium, and the cultures were incubated for 24 h. At 0 h, medium was aspirated, virus was added, and the cultures were incubated for up to 10 days without changing the medium. Controls included infected cells not treated with rIFN-α or cells left uninfected. At 10 dpi when strong CPE occurred in the infected, non-IFN-α-treated cells, viability was assayed by using an MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] assay. Protection from CPE in rIFN-α-pretreated cells was dose dependent, with significant differences (P < 0.05) between nontreated, infected cells and cells pretreated with rIFN-α at 103-, 104-, 105-, and 106-fold dilutions (Fig. 5 a), corresponding to rIFN-α concentrations from 0.47 μg to 0.47 ng ml−1. Conversely, trIFN-α gave no protection against CPE (Fig. 5a). Antiviral activity was also demonstrated for CHSE cells, and significant protection (P < 0.05) was seen for cells treated with rIFN-α at 103-, 104-, and 105-fold dilutions (Fig. 5b).
FIG. 5.

Cytopathic effect (CPE) reduction assay expressed as percent protection from CPE in TO cells (a) and in CHSE cells (b). Cell cultures were stimulated with 10-fold serial dilutions of rIFN-α and trIFN-α 24 h prior to infection with 1 MOI of SAV-3. The lowest concentration with a biological effect (106 dilution) corresponds to 0.47 ng/ml, while the highest concentration used (103 dilution) was 0.47 μg/ml for IFN-α. For trIFN-α, the highest concentration used was 0.48 μg/ml (n = 3) (means ± SEM). Non-tr, nontreated cultures.
Morphologically, no CPE was observed for TO cells pretreated with rIFN-α at 103-, 104-, and 105-fold dilutions, while at a 106-fold dilution, CPE were observed (Fig. 6). Cultures pretreated with trIFN-α were not protected against CPE induced by SAV-3 infection even at the highest concentration used (Fig. 6).
FIG. 6.

Protection against CPE observed by morphology. The top row shows cells treated with rIFN-α, which showed dose-dependent protection against CPE at 103 and 105 dilutions, while initial CPE were seen at a 106 dilution. Conversely, trIFN-α did not show any protection against CPE (bottom row). Shown are representative findings for two independent experiments. NT, not treated; NI, not infected.
The purity of the cloned E2 protein (Fig. 7 a) and the specificity of the anti-E2 rabbit antiserum were documented by Western blotting (Fig. 7b) and IFAT staining (Fig. 7c) of SAV-3-infected cultures, and the inhibition of viral replication in IFN-α-treated cells was further demonstrated by an analysis of virus replication and viral protein (E2) synthesis. TO cells treated with 0.47 μg/ml rIFN-α totally inhibited the expression of the E2 protein at 2 days postinfection (Fig. 7e), while nontreated cells showed positive staining for E2 at the same time point (Fig. 7f). The antiviral activity of rIFN-α was confirmed by the virus yield reduction assay: the virus titer was reduced in TO cells treated with rIFN-α diluted 103-, 104-, and 105-fold (103- to 105-fold reduction) compared with nontreated control cells (Fig. 7d). These results showed that the pretreatment of cells with rIFN-α strongly inhibited SAV-3 replication and protected cells against virus-induced cell lysis (CPE).
FIG. 7.

(a) Purification of recombinant pET14b-E2 protein analyzed by SDS-PAGE. Lanes: 1, protein marker; 2, whole bacteria after induction; 3, soluble fraction after sonication; 4, insoluble fraction after sonication; 5, purified recombinant protein. (b) Characterization of polyclonal antibody against E2 by Western blotting. Lanes: 1, pET14b-E2 after induction; 2, pET14b-E2 without induction; 3, purified E2. (c) Reactivity of rabbit antibody against E2 by IFAT of CHSE cells infected with SAV-3 using a 1:400 dilution of the serum. (d) Virus yield reduction assay. TO cells in 24-well plates were stimulated with 10-fold serial dilutions of rIFN-α for 24 h and then infected with 1 MOI of SAV-3. Cell culture supernatants were collected when strong CPE were observed for the control infected cells. The virus titer of the supernatants at 10 days postinfection was determined by the TCID50 method (n = 2) (means ± SEM). (e) Inhibition of E2 protein synthesis in interferon-treated TO cells. TO cells were treated with 0.47 μg/ml IFN-α for 24 h and subsequently infected with 1 MOI of SAV-3. At 2 days postinfection, cells were stained with anti-E2 antibody, and no cells were found to express virus protein. (f) TO cells without IFN treatment infected as described in the text and stained by IFAT for E2, showing distinct cytoplasmic staining in infected cells.
Timing of rIFN-α treatment of SAV-3-infected cells.
We then explored the effect of the timing of rIFN-α treatment (0.47 μg/ml) relative to when the cells were infected, starting IFN-α treatment 24 h prior to infection and ending 24 h postinfection. Virus replication was assayed by examining the relative expression of E2 by real-time RT-PCR (Fig. 8), and at the same time, we evaluated the development of CPE in pre- and posttreated infected cell cultures. Treatment 24 h prior to infection inhibited SAV-3 replication 40,000-fold relative to nontreated controls (Fig. 8). Preinfection treatment (−4 h) gave close to a 2,000-fold reduction in E2 gene expression and protection against CPE in infected cultures (Fig. 8). Treatment closer to the time of infection (−2 h and 0 h) and postinfection did not protect against CPE; however, there was a marked reduction in E2 gene expression levels relative to those of nontreated cell cultures. Treatment 24 h postinfection gave a 3-fold reduction in replication levels and strong CPE. The results clearly show that the timing of the IFN-α treatment is critical for the induction of the antiviral state, and its magnitude is dependent on the dose (Fig. 5a) and time (Fig. 8). While an antiviral state can be established through preinfection treatment, concurrent or postinfection treatment with rIFN-α will not protect infected cells against CPE, indicating that the virus has developed a strategy to evade the IFN-induced antiviral response at later stages of the infection cycle.
FIG. 8.

Timing of IFN-α treatment of SAV-3-infected cells. Treatment 4 to 24 h prior to infection results in a marked reduction in virus replication (2,000- to 40,000-fold reduction) as measured by real-time PCR and protection against CPE. rIFN-α treatment at the time of infection or up to 24 h postinfection gives a reduction in virus replication but not protection against CPE. TO cells were infected with an MOI of 1, and collection was done at 4 days postinfection. Shown are representative data from two independent experiments, expressed relative to the nontreated control (NT). More details are given in Materials and Methods.
SAV-3 infection results in phosphorylation of eIF2α cells and moderate protein synthesis downregulation.
These findings led us to further characterize the expression of viral protein (E2) and Mx and the phosphorylation of eIF2α by Western blotting over a 96-h period postinfection. TO cells were infected at an MOI of 1 with SAV-3 (0 h), and one parallel culture was also treated with rIFN-α (0.47 μg/ml) at 24, 48, 72, and 96 h postinfection. Control treatments included noninfected/rIFN-α-treated and infected/non-rIFN-α-treated cells (Fig. 9). After rIFN-α treatment, the cells were left to grow for an additional 16 h (after each treatment time) to assess responses to treatment. For simplicity, we refer to time points when rIFN-α was added. E2 expression was seen at 24 h (faint staining) postinfection in cultures treated with rIFN-α and left untreated, while Mx was not detected at this time point (Fig. 9). Faint staining for p-eIF2α was also seen. From 48 h onwards, there was marked E2 and Mx expression and staining for p-eIF2α in the SAV-3-infected cells not treated with IFN-α. The combination of infection and rIFN-α treatment gave the same results as those for the SAV-3-infected-only cells although with a moderate reduction in levels of E2 expression at 48 and 72 h (Fig. 9). In noninfected cells treated with rIFN-α, there was marked Mx expression, while eIF2α was not found to be phosphorylated at any time point (Fig. 9). The finding that SAV-3 infection resulted in the phosphorylation of eIF2α prompted us to characterize the effect of virus infection on protein synthesis. We found a moderate reduction of protein synthesis in TO cells by 36 h (40% reduction), which was more prominent by 48 and 60 h postinfection, with 50% and 65% reductions, respectively (Fig. 10).
FIG. 9.

Protein analysis of E2, Mx, and p-eIF2α in virus-infected cells. TO cells were infected with SAV-3 (MOI of 1) or left uninfected, and at 24, 48, 72, and 96 h postinfection, uninfected (−) and infected (+) cells were treated with 0.47 μg/ml of rIFN-α for 16 h or left untreated. Cells were lysed by using CelLytic M reagent (Sigma) and scraped from the dish. The expression of the E2, Mx, p-eIF2α, and actin proteins was detected by Western blotting.
FIG. 10.
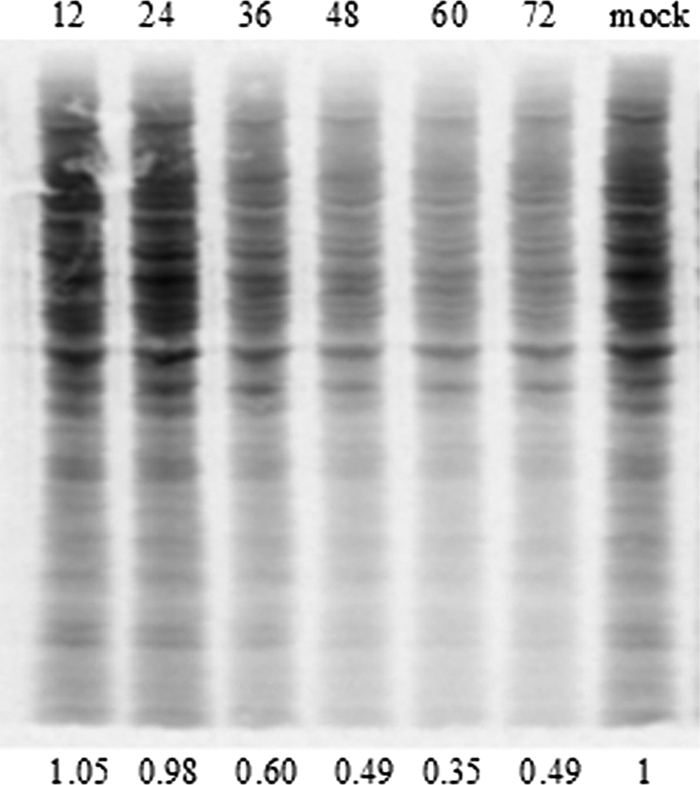
Residual protein synthesis in TO cells infected with SAV-3. Confluent TO cells were grown in a six-well plate and infected as described in Materials and Methods. The membrane was exposed in a PhosphorImager cassette and then scanned by using a Typhoon imager. Numbers above the figure represent hours postpulsing. The protein amount was quantified by densitometry with ImageJ software as described in Materials and Methods. The results show 40, 51, and 65% reduction by 36, 48, and 60 h postinfection, respectively. Mock infection is shown at the far right.
IFN-γ has a limited inhibiting effect on SAV-3 replication in vitro.
On the basis that it was previously shown that rIFN-γ has a limited effect on alphavirus replication in mice (21), we wanted to understand any antiviral effects induced by rIFN-γ in fish cells. The functional activity of rIFN-γ was documented by its ability to induce the expression of γIP10 (Fig. 11 a). The CPE reduction assay showed that rIFN-γ has no ability to rescue pretreated cells from CPE caused by SAV-3 infection (Fig. 11b), while there was an approximately 10-fold reduction of virus titers at the highest rIFN-γ concentration, determined by real-time PCR (1:200 dilution, corresponding to 1.65 μg/ml) (Fig. 11c).
FIG. 11.

(a) Induction of ISG mRNA by rIFN-γ in TO cells. Purified rIFN-γ was serially diluted from 0.33 mg/ml in cell medium and incubated with TO cells for 24 h. The data are expressed as the mean fold changes in gene expression ± standard errors of different dilutions of the IFN-γ-treated group relative to the nontreated control group after normalization to β-actin (n = 2) (±SEM). (b) Cytopathic effect reduction assay of IFN-γ-treated TO cells infected with SAV-3. TO cells were stimulated with serial dilutions of rIFN-γ and trIFN-γ for 24 h and subsequently infected with 1 MOI of SAV-3. There is no significant difference in protection against CPE between cell cultures pretreated with rIFN-γ and trIFN-γ (n = 4) (±SEM). Non-tr, nontreated cells; Non-inf, noninfected; OD490, optical density at 490 nm. (c) Real-time PCR quantification of virus in cell supernatants from TO cells stimulated with serial dilutions of mature IFN-γ for 24 h and subsequently infected with 1 MOI of SAV-3. Cell supernatants were collected when a strong CPE was observed in the control infected cells (8 days postinfection). The data are expressed as the mean crossing-point values ± standard errors using real-time PCR of different dilutions of an IFN-γ-treated group relative to the nontreated infected control group (n = 2) (±SEM).
DISCUSSION
This is the first report showing that IFN-α has a strong inhibitory effect on SAV-3 replication in vitro in permissive salmonid cell lines. This finding is in conformity with what was shown previously for other alphavirus species in higher vertebrates, where the stimulation of IFN-α/β pathways represents the primary protective response and plays a key role in protection against infection through the limitation of virus replication (1, 13, 32, 40). IFN-γ limits virus replication of SAV-3 marginally, again in concordance with what was found previously for higher vertebrates (22). These findings have implications for the understanding of host-pathogen interactions in SAV infections of salmonid fish and possibly provide new avenues for disease prevention.
The tools available to study the in vivo effects of type I IFN in infected fish at a functional level are very limited. In vitro methods using permanent cell lines permissive for virus infection are thus a good option for functional studies when this can be combined with treatment with functionally active cytokines. SAV-3 infection of TO cell was characterized by a rapid increase in viral E2 gene copy numbers from 12 to 24 h postinfection, representing a more-than-2-log10 increase over a 12-h period (Fig. 1). Over the same period, there was a strong upregulation of IFN-α and Mx transcripts, and these findings are in conformity with data from a previous study (11) using a different subtype of SAV (SAV-1). To what extent the cell line used in this study reflects a true in vivo situation is difficult to determine, but TO cells have morphological and functional traits of salmon macrophages (35) and support the replication of a number of salmonid viruses, like salmon anemia virus (36) and infectious pancreatic necrosis virus (3). Furthermore, the treatment of TO cells with low concentrations of rIFN-α induced strong expression levels of IFN-induced genes, Mx and ISG15, and also a strong expression of Mx at the protein level (shown by IFAT), indicating that a functionally active cell line was used. The induction of an antiviral state in the pretreated cells was dependent on dose and time relative to infection, and protection against CPE was obtained when rIFN-α was added to the cells earlier than 4 h prior to infection and at a dose higher than 0.47 μg/ml. Treatment at the time of infection (or 2 h prior to infection) and postinfection gave no protection against CPE, although there was a reduction in viral copy numbers.
In a recent study by Gahlawat et al. (11), it was proposed that the suppression of Mx responses correlates with SAV replication; however, those studies did not include a functional assessment of the responses involved. Those researchers showed a difference in mRNA IFN-α and Mx expression levels in two cell lines of salmon origin, and they proposed a possible interference with IFN signaling and the suppression of Mx responses (11). Their observations were based solely on the observation that a cell line with low susceptibility had high Mx responses and vice versa. From our studies we have no indication that SAV-3-infected cells have impaired Mx responses (Fig. 1 and 9), and the treatment of TO cells with rIFN-α followed by SAV-3 infection did not influence the expression of Mx as assessed by Western blotting. Furthermore, a strong expression of Mx postinfection seems to have little influence on virus replication. On the other hand, the timing of the antiviral responses relative to infection is obviously of major importance. This is concordant with a recent study where it was shown that mRNA of the Old World alphaviruses encoded by subgenomic RNA is resistant to eIF2α phosphorylation, while genomic RNA replication is highly sensitive to eIF2α phosphorylation (34). It was also found that the replication of genomic RNA ceased with the onset of eIF2α phosphorylation, which again correlated with the appearance of structural proteins. We also find that the synthesis of the spike protein (E2) coincided with the phosphorylation of eIF2α in SAV-3-infected and rIFN-α-treated/SAV-3-infected cells (Fig. 9), with some reduction of staining intensity of E2 with increasing p-eIF2α staining, but still, there was a successful production of virus progeny (Fig. 1). The antiviral state induced through preinfection treatment results in strong ISG induction, a marked inhibition of virus replication, and no E2 staining (Fig. 7 and 8). Thus, in line with what was described previously for other alphaviruses (28), the exact antiviral mechanisms controlling viral replication are not known; it seems that protein translation from genomic viral RNA (incoming positive strand) is sensitive to IFN-α-induced responses. At the same time, it cannot be ruled out that the synthesis of spike mRNA driven by the 26S promoter is resistant to IFN-α-induced cellular responses, but further studies are needed to understand the details of the mechanisms involved.
A known strategy by which alphaviruses evade the IFN response is through global shutoff of host gene expression (28). The transcriptional shutoff caused by Old World alphaviruses depends on a nonstructural protein (nsP2), and in addition, nsP2 of Sindbis virus has an anti-interferon effect, likely through decreased IFN production, and mutations in nsP2 can attenuate Sindbis virus cytopathogenicity (10, 12). There is no protein shutoff in SAV-3-infected cells, but there is a downregulation of protein synthesis. This is more in line with New World alphaviruses that result in a more moderate downregulation of protein synthesis (2, 10, 12), where the capsid protein possibly plays a role in the arrest of macromolecular synthesis during early stages of infection, which will limit the formation of antiviral proteins in infected cells (2, 12). The New World alphavirus VEEV is also able to antagonize the IFN response using distinct mechanisms independent of host shutoff by inhibiting the phosphorylation of cytoplasmic signal transducer and activator of transcription 1 (STAT-1), which prevents the nuclear translocation of phosphorylated STAT-1/STAT-2 heterodimers (30). The result is a reduced formation of antiviral effector molecules like Mx and ISG15. More-recent studies have added complexity to the picture described above and have shown that VEEV nonstructural proteins arrested cellular translation (39). To what extent SAV-3 employs specific mechanisms to counteract the IFN response by targeting certain signaling proteins involved in the IFN pathway or IFN signaling pathway needs to be investigated. A better understanding of the induction of IFN-α and related responses and possible antiviral effects and regulation of host gene expression imposed by nonstructural and/or structural proteins of SAV-3 is strongly needed, and it is our understanding that this can best be studied by using reverse genetics methods.
The strong upregulation of IFN-α mRNA in response to SAV-3 infection is in contrast to what has been seen for SINV infection of permissive cell lines, while it is in conformity with what was observed for VEEV infection (in the same cell lines), where an upregulation of IFN-α mRNA transcripts postinfection was observed (39). The aquatic alphaviruses are defined as a separate clade within the genus Alphavirus, and they are phylogenetically different from Old World and New World alphaviruses (24). Their identification has raised interesting questions as to the origin of different variants of alphaviruses in general. While the Old World and New World alphaviruses have been classified into arthrogenic and neurotropic variants, the salmonid alphaviruses do not seem to fall into either of these categories, and they are not transferred by vectors (21). However, in a recent study (6), histopathological changes were found in the central nervous system following infection of Atlantic salmon with the SAV-3 subtype, while no such changes were found for SAV-1 variants. It remains to be decided if this is a general phenomenon of SAV-3 infections, but this observation combined with the similarities in cell responses to infection raises an interesting possibility that SAV-3 is functionally closer to the VEEV variants than the Old World viruses like SINV and Semliki Forest virus.
Acknowledgments
This study was carried out with financial support from the Norwegian Research Council, project no. 182035, Immunization Strategies against Viral Pathogens of Atlantic Salmon.
We are also indebted to Jo-Ann Leong, School of Ocean, Earth Science Technology, Hawaii Institute of Marine Biology, for providing the antibody to the Mx protein.
Footnotes
Published ahead of print on 23 June 2010.
REFERENCES
- 1.Aguilar, P. V., S. Paessler, A. S. Carrara, S. Baron, J. Poast, E. Wang, A. C. Moncayo, M. Anishchenko, D. Watts, R. B. Tesh, and S. C. Weaver. 2005. Variation in interferon sensitivity and induction among strains of eastern equine encephalitis virus. J. Virol. 79:11300-11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguilar, P. C., S. C. Weaver, and C. F. Basler. 2007. Capsid protein of eastern encephalitis virus inhibits host cell gene expression. J. Virol. 81:3866-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berg, T. S. K., B. Sun, and B. Robertsen. 2009. An antiserum against Atlantic salmon IFNa1 detects IFN and neutralizes antiviral activity produced by poly I:C stimulated cells. Dev. Comp. Immunol. 33:638-645. [DOI] [PubMed] [Google Scholar]
- 4.Bergan, V., R. Jagus, S. Lauksund, O. Kileng, and B. Robertsen. 2008. The Atlantic salmon Z-DNA binding protein kinase phosphorylates translation initiation factor 2 alpha and constitutes a unique orthologue to the mammalian dsRNA-activated protein kinase R. FEBS J. 275:184-197. [DOI] [PubMed] [Google Scholar]
- 5.Brehin, A. C., I. Casademont, M. P. Frenkiel, C. Julier, A. Sakuntabhai, and P. Despres. 2009. The large form of human 2′,5′-oligoadenylate synthetase (OAS3) exerts antiviral effect against Chikungunya virus. Virology 384:216-222. [DOI] [PubMed] [Google Scholar]
- 6.Christie, K. E., D. A. Graham, M. F. McLoughlin, S. Villoing, D. Todd, and D. Knappskog. 2007. Experimental infection of Atlantic salmon Salmo salar pre-smolts by i.p. injection with new Irish and Norwegian salmonid alphavirus (SAV) isolates: a comparative study. Dis. Aquat. Organ. 75:13-22. [DOI] [PubMed] [Google Scholar]
- 7.Desvignes, L., C. Quentel, F. Lamour, and A. Le Ven. 2002. Pathogenesis and immune response in Atlantic salmon (Salmo salar L.) parr experimentally infected with salmon pancreas disease virus (SPDV). Fish Shellfish Immunol. 12:77-95. [DOI] [PubMed] [Google Scholar]
- 8.Ferreira, P. C., M. L. Peixoto, M. A. Silva, and R. R. Golgher. 1979. Assay of human interferon in Vero cells by several methods. J. Clin. Microbiol. 9:471-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fringuelli, E., H. M. Rowley, J. C. Wilson, R. Hunter, H. Rodger, D. A. Graham. 2008. Phylogenetic analyses and molecular epidemiology of European salmonid alphaviruses (SAV) based on partial E2 and nsP3 gene nucleotide sequences. J. Fish Dis. 31:811-823. [DOI] [PubMed] [Google Scholar]
- 10.Frovola, E. I., R. R. Fayzulin, S. H. Cook, D. E. Griffin, C. M. Rice, and I. Frolov. 2002. Roles of nonstructural protein nsp2 and alpha/beta interferon in determining the outcome of Sindbis virus infection. J. Virol. 76:11254-22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gahlawat, S. K., A. E. Ellis, and B. Collet. 2009. Expression of interferon and interferon-induced genes in Atlantic salmon Salmo salar cell lines SHK-1 and TO following infection with salmon alphavirus SAV. Fish Shellfish Immunol. 26:672-675. [DOI] [PubMed] [Google Scholar]
- 12.Garmashova, N., R. Gorchakov, E. Volkova, S. Paessler, E. Frolova, and I. Frolov. 2007. The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol. 81:2472-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grieder, F. B., and S. N. Vogel. 1999. Role of interferon and interferon regulatory factors in early protection against Venezuelan equine encephalitis virus infection. Virology 257:106-118. [DOI] [PubMed] [Google Scholar]
- 14.Hodneland, K., A. Bratland, K. E. Christie, C. Endresen, and A. Nylund. 2005. New subtype of salmonid alphavirus (SAV), Togaviridae, from Atlantic salmon Salmo salar and rainbow trout Oncorhynchus mykiss in Norway. Dis. Aquat. Organ. 66:113-120. [DOI] [PubMed] [Google Scholar]
- 15.Kärber, G. 1931. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch. Exp. Pathol. Pharmakol. 162:480-483. [Google Scholar]
- 16.Landis, H., A. Simon-Jodicke, A. Kloti, C. Di, J. J. Schnorr, S. Schneider-Schaulies, H. P. Hefti, and J. Pavlovic. 1998. Human MxA protein confers resistance to Semliki Forest virus and inhibits the amplification of a Semliki Forest virus-based replicon in the absence of viral structural proteins. J. Virol. 72:1516-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larsen, R., T. P. Røkenes, and B. Robertsen. 2004. Inhibition of infectious pancreatic necrosis virus replication by Atlantic salmon Mx1 protein. J. Virol. 78:7938-7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lenschow, D. J., N. V. Giannakopoulos, L. J. Gunn, C. Johnston, A. K. O'Guin, R. E. Schmidt, B. Levine, and H. W. Virgin IV. 2005. Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo. J. Virol. 79:13974-13983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lenschow, D. J., C. Lai, N. Frias-Staheli, N. V. Giannakopoulos, A. Lutz, T. Wolff, A. Osiak, B. Levine, R. E. Schmidt, A. Garcia-Sastre, D. A. Leib, A. Pekosz, K. P. Knobeloch, I. Horak, and H. W. Virgin IV. 2007. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. U. S. A. 104:1371-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 21.McLoughlin, M. F., and D. A. Graham. 2007. Alphavirus infections in salmonids—a review. J. Fish Dis. 30:511-531. [DOI] [PubMed] [Google Scholar]
- 22.Müller, U., U. Steinhoff, L. F. Reis, S. Hemmi, J. Pavlovic, R. M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918-1921. [DOI] [PubMed] [Google Scholar]
- 23.Ooi, E. L., N. Verjan, I. Haraguchi, T. Oshima, H. Kondo, I. Hirono, T. Aoki, H. Kiyono, and Y. Yuki. 2008. Innate immunomodulation with recombinant interferon-alpha enhances resistance of rainbow trout (Oncorhynchus mykiss) to infectious hematopoietic necrosis virus. Dev. Comp. Immunol. 32:1211-1220. [DOI] [PubMed] [Google Scholar]
- 24.Powers, A. M., A. C. Brault, Y. Shirako, E. G. Strauss, W. Kang, J. H. Strauss, and S. C. Weaver. 2001. Evolutionary relationships and systematics of the alphaviruses. J. Virol. 75:10118-10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raynard, R., and G. Houghton. 1993. Development towards an experimental protocol for the transmission of pancreas disease of Atlantic salmon Salmo salar. Dis. Aquat. Organ. 15:123-128. [Google Scholar]
- 26.Robertsen, B., G. Trobridge, and J. A. Leong. 1997. Molecular cloning of double-stranded RNA inducible MX genes from Atlantic salmon (Salmo salar L.). Dev. Comp. Immunol. 21:397-412. [DOI] [PubMed] [Google Scholar]
- 27.Rokenes, T. P., R. Larsen, and B. Robertsen. 2007. Atlantic salmon ISG15: expression and conjugation to cellular proteins in response to interferon, double-stranded RNA and virus infections. Mol. Immunol. 44:950-959. [DOI] [PubMed] [Google Scholar]
- 28.Ryman, K., and W. B. Klimstra. 2008. Host responses to alphavirus infection. Immunol. Rev. 225:27-45. [DOI] [PubMed] [Google Scholar]
- 29.Samuel, C. E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14:778-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simmons, J. D., L. J. White, T. E. Morrison, S. A. Montgomery, A. C. Whitmore, R. E. Johnston, and H. T. Heise. 2009. Venezuelan equine encephalitis virus disrupts STAT1 signaling by distinct mechanisms independent of host shutoff. J. Virol. 83:10571-10581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slutzkia, M., D. A. Jaitin, T. B. Yehezkela, and G. Schreiber. 2006. Variations in the unstructured C-terminal tail of interferons contribute to differential receptor binding and biological activity. J. Mol. Biol. 360:1019-1030. [DOI] [PubMed] [Google Scholar]
- 32.Stanton, G. J., R. E. Lloyd, M. Sarzotti, and J. E. Blalock. 1989. Protection of mice from Semliki Forest virus infection by lymphocytes treated with low levels of interferon. Mol. Biother. 1:305-310. [PubMed] [Google Scholar]
- 33.Taksdal, T., A. B. Olsen, I. Bjerkas, M. J. Hjortaas, B. H. Dannevig, D. A. Graham, and M. F. McLoughlin. 2007. Pancreas disease in farmed Atlantic salmon, Salmo salar L., and rainbow trout, Oncorhynchus mykiss (Walbaum), in Norway. J. Fish. Dis. 30:545-558. [DOI] [PubMed] [Google Scholar]
- 34.Ventoso, I., M. A. Sanz, S. Molina, J. J. Berlanga, L. Carrasco, and M. Esteban. 2006. Translational resistance of late alphavirus mRNA to eIF2alpha phosphorylation: a strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 20:87-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villoing, S., M. Bearzotti, S. Chilmonczyk, J. Castric, and M. Bremont. 2000. Rainbow trout sleeping disease virus is an atypical alphavirus. J. Virol. 74:173-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wergeland, H. I., and R. A. Jakobsen. 2001. A salmonid cell line (TO) for production of infectious salmon anaemia virus (ISAV). Dis. Aquat. Organ. 44:183-190. [DOI] [PubMed] [Google Scholar]
- 37.Weston, J. H., M. D. Welsh, M. F. McLoughlin, and D. Todd. 1999. Salmon pancreas disease virus, an alphavirus infecting farmed Atlantic salmon, Salmo salar L. Virology 256:188-195. [DOI] [PubMed] [Google Scholar]
- 38.Weston, J., S. Villoing, M. Bremont, J. Castric, M. Pfeffer, V. Jewhurst, M. McLoughlin, O. Rodseth, K. E. Christie, J. Koumans, and D. Todd. 2002. Comparison of two aquatic alphaviruses, salmon pancreas disease virus and sleeping disease virus, by using genome sequence analysis, monoclonal reactivity, and cross-infection. J. Virol. 76:6155-6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin, J., C. L. Gardner, C. W. Burke, K. D. Ryman, and W. B. Klimstra. 2009. Similarities and differences in antagonism of neuron alpha/beta interferon responses by Venezuelan equine encephalitis and Sindbis alphaviruses. J. Virol. 83:10036-10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang, Y., C. W. Burke, K. D. Ryman, and W. B. Klimstra. 2007. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol. 81:11246-11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu, R., Y. B. Zhang, Q. Y. Zhang, and J. F. Gui. 2008. Functional domains and the antiviral effect of the double-stranded RNA-dependent protein kinase PKR from Paralichthys olivaceus. J. Virol. 82:6889-6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zou, J., C. Tafalla, J. Truckle, and C. J. Secombes. 2007. Identification of a second group of type I IFNs in fish sheds light on IFN evolution in vertebrates. J. Immunol. 179:3859-3871. [DOI] [PubMed] [Google Scholar]