

Abstract
Hepatorenal fibrocystic diseases (HRFCDs) are among the most common inherited human disorders. The discovery that proteins defective in the autosomal dominant and recessive polycystic kidney diseases (ADPKD and ARPKD) localize to the primary cilia and the recognition of the role these organelles play in the pathogenesis of HRFCDs led to the term “ciliopathies.” While ADPKD and ARPKD are the most common ciliopathies associated with both liver and kidney disease, variable degrees of renal and/or hepatic involvement occur in many other ciliopathies, including Joubert, Bardet–Biedl, Meckel–Gruber, and oral–facial–digital syndromes. The ductal plate malformation (DPM), a developmental abnormality of the portobiliary system, is the basis of the liver disease in ciliopathies that manifest congenital hepatic fibrosis (CHF), Caroli syndrome (CS), and polycystic liver disease (PLD). Hepatocellular function remains relatively preserved in ciliopathy-associated liver diseases. The major morbidity associated with CHF is portal hypertension (PH), often leading to esophageal varices and hypersplenism. In addition, CD predisposes to recurrent cholangitis. PLD is not typically associated with PH, but may result in complications due to mass effects. The kidney pathology in ciliopathies ranges from non-functional cystic dysplastic kidneys to an isolated urinary concentration defect; the disorders contributing to this pathology, in addition to ADPKD and ARPKD, include nephronophithisis (NPHP), glomerulocystic kidney disease and medullary sponge kidneys. Decreased urinary concentration ability, resulting in polyuria and polydypsia, is the first and most common renal symptom in ciliopathies. While the majority of ADPKD, ARPKD, and NPHP patients require renal transplantation, the frequency and rate of progression to renal failure varies considerably in other ciliopathies. This review focuses on the kidney and liver disease found in the different ciliopathies.
Keywords: ductal plate malformation, congenital hepatic fibrosis, Caroli syndrome, polycystic liver disease, portal hypertension, polycystic kidney disease, nephronophthisis, cystic dysplastic kidneys, medullary sponge kidney, multicystic dysplastic kidneys, primary cilia, ciliopathy
INTRODUCTION
Hepatorenal fibrocystic diseases (HRFCDs), characterized by developmental abnormalities of the portobiliary system in association with fibrocystic degeneration of the kidneys, are common and potentially lethal inherited disorders [Zerres et al., 1984; Johnson et al., 2003; Tahvanainen et al., 2005]. Polycystic kidney diseases (PKD) comprise the largest subset of HRFCDs, whose kidney pathology ranges from isolated urinary concentrating defects to cystic dysplastic kidneys with no function. Nephronophthisis (NPHP), a primarily fibrotic renal disease with secondary cyst formation, also contributes to the spectrum of kidney pathology observed in HRFCDs [Hildebrandt and Zhou, 2007]. Liver involvement includes congenital hepatic fibrosis (CHF), Caroli disease (CD), and polycystic liver disease (PLD).
The proteins defective in human and murine PKDs localize to the cell’s primary cilium and its basal body, pointing to the potential role these organelles play in the pathogenesis of HRFCDs [Fliegauf et al., 2007]. Primary (non-motile) cilia are micro-tubule-based antenna-like organelles that extend outward from the surface of many differentiated eukaryotic cells, including those of the renal and biliary epithelium. Primary cilia host critical signal transduction pathways that involve Hedgehog, Wnt, PDGFR-alpha, and integrin signaling. They also sense chemical, osmotic, and mechanical stimuli such as luminal fluid flow, and regulate important cellular functions including proliferation and maintenance of planar cell polarity and mitotic spindle orientation to ensure normal epithelial function and normal diameter of tubular structures such as renal and biliary ducts [Masyuk et al., 2008; Rodat-Despoix and Delmas, 2009]. HRFCDs belong to the larger group of disorders collectively referred to as “ciliopathies” [Veland et al., 2009]. More accurate gene-based classification of HRFCDs [Gunay-Aygun et al., 2009] and better understanding of their cellular pathology now allows targeted therapies for the liver and kidney disease of ciliopathies [Patel et al., 2009]. This review focuses on the clinical characteristics of the liver and kidney disease in various ciliopathies.
LIVER DISEASE IN CILIOPATHIES
Intact cilia-based signaling is required for normal development of the biliary and portal system in the liver [Masyuk et al., 2006]. The majority of diseases manifesting with hepatic fibrocystic pathology are caused by defective ciliary proteins; exceptions include autosomal dominant polycystic liver disease (ADPLD) and portal fibrosis associated with Congenital Disorder of Glycosylation (CDG) type Ib [Gunay-Aygun et al., 2008].
Hepatic fibrocystic pathologies can be grouped into three major descriptive categories: CHF; CD; and PLD [Kerr et al., 1961, 1978; Summerfield et al., 1986; Gunay-Aygun and Heller, 2008]. CHF is a histopathological diagnosis with three main components; that is, defective remodeling of the ductal plate (DPM) (Fig. 1); abnormal portal veins; and progressive fibrosis of the portal tracks [Desmet, 1992a,b, 1998]. The major morbidity associated with CHF is portal hypertension (PH) [Kerr et al., 1978]. CD refers to macroscopic saccular or fusiform dilations of the medium and large sized intrahepatic bile ducts on imaging of the biliary system [Caroli, 1968, 1973]. Caroli syndrome (CS) refers to CD in association with CHF [Caroli, 1968, 1973; Kerkar et al., 2006]. CD and CS probably represent different presentations of a continuum of pathology; CD and CS are reported to affect different members of the same family [Caroli, 1973]. The liver cysts in CS are non-obstructive dilatations of the intrahepatic biliary tree; hence, they are continuous with the biliary system [Caroli, 1973]. In contrast, the liver cysts in PLD are isolated, closed cysts that originate from biliary microhamartomas (von Meyenburg complexes) embedded in fibrous tissue; they are not in continuity with the intrahepatic biliary tree [Caroli, 1968, 1973; Tahvanainen et al., 2005; Morgan et al., 2006]. Imaging of the intrahepatic biliary system, preferably performed using magnetic resonance cholangiography because endoscopic pancreatico-cholangiography increases the risk for cholangitis, is useful in differential diagnosis [Morgan et al., 2006].
Figure 1.

Artist’s rendering of normal bile duct development in comparison with the ductal plate malformation and corresponding liver histopathology showing normal portal triad and congenital hepatic fibrosis. The ductal plate initially forms as a sleeve-like structure around the portal vein branches. Normal remodeling of the ductal plate involves resorption of parts of this structure and migration of the remodeled ducts centrally closer to the portal vein (left panel at bottom). The liver biopsy at 10× magnification shows a normal portal tract with sections of portal vein, bile duct, and hepatic artery. Defective remodeling, termed the ductal plate malformation, is characterized by retention of excessive numbers of bile duct remnants in their original peripheral interrupted ring-like position (right panel at bottom). The biopsy with CHF shows persistence of bile duct remnants (magnification = 40×). (Liver biopsies are from Potter’s pathology of the fetus, infant and child, 2nd edition, Ed: Gilbert-Barness E. Mosby Elsevier.)
DPM is the main pathology underlying the liver disease in ciliopathies [Desmet, 1992b, 1998] (Fig. 1). The severity of DPM and the level of the portobiliary tree affected by DPM vary within and among individual ciliopathies [Desmet, 1992b, 1998; Gunay-Aygun and Heller, 2008]. DPM results in a spectrum of abnormalities including CHF (microscopic bile ducts), CHF/CS (microscopic and medium size bile ducts), and CD (medium and large bile ducts). The isolated liver cysts in the PLD of ADPKD probably represent DPM affecting the most peripheral end of the biliary system [Desmet, 1992b, 1998].
Congenital Hepatic Fibrosis/Caroli Syndrome
Ultrasonography (USG) is the most informative imaging modality in diagnosing CHF/CS; in patients with CS, it reveals increased echogenicity of the liver with a coarse, non-homogeneous pattern, splenic enlargement as an evidence for PH, and macroscopic liver cysts [Akhan et al., 2007]. These findings, especially in the context of fibrocystic renal involvement, are diagnostic of CHF/CS; liver biopsy is not required in such cases. However, liver biopsy is often performed in HRFCD patients who present in late childhood or adulthood with splenomegaly or thrombocytopenia (caused by hypersplenism). Liver biopsy in CHF shows abnormal portal tracts with an excess number of abnormally shaped embryonic bile ducts retained in their primitive ductal plate configuration (incorrectly referred to as bile duct proliferation), an abnormal portal vein, and periportal fibrosis without inflammation [Kerr et al., 1978] (Fig. 1). Portal–portal bridging of the fibrotic bands is common in advanced CHF; absence of portal tract to central vein bridging and preserved hepatocellular function distinguishes CHF from cirrhosis.
Typically, CHF/CS occurs as part of a multisystem disorder usually associated with fibrocystic renal disease [Johnson et al., 2003; Gunay-Aygun and Heller, 2008] (Table I) CHF/CS rarely appears as an isolated finding, but no causative gene has been identified to date. CHF/CS most commonly presents as part of autosomal recessive polycystic kidney disease (ARPKD) [Zerres et al., 1996; Guay-Woodford and Desmond, 2003; Gunay-Aygun et al., 2006]. Although all patients with ARPKD have CHF on microscopic examination at birth, abnormal liver echogenicity and splenomegaly may not be detectable during early childhood because periportal fibrosis and PH are time-dependent pathologies that progress with age. The majority of patients with the CHF associated with ARPKD develop PH, which results in splenomegaly, esophageal varices and thrombocytopenia and leukopenia due to hypersplenism (Fig. 2A). The hepatocellular function of the liver is relatively well preserved, and liver enzymes are normal or mildly elevated [Kerr et al., 1978]. An increased risk for cholangiocarcinoma in adulthood and for cholangitis at any age are the major morbidities caused by Caroli syndrome [Bloustein, 1977; Yamato et al., 1998].
TABLE I.
Ciliary Disorders Associated With Congenital Hepatic Fibrosis/Caroli’s Syndrome
| Disease | Mode of inheritance | Gene(s) | Prevalence |
|---|---|---|---|
| Autosomal recessive polycystic kidney disease | AR | PKHD1 | ~1 in 20,000 |
| Meckel–Gruber syndrome | AR | MKS1, TMEM67, CEP290, RPGRIP1L, CC2D2A | ~1 in 140,000 |
| Joubert syndrome and related disorders including COACH syndrome | AR | AHI1, NPHP1, CEP290, TMEM67, RPGRIP1L, ARL13B, CC2D2A | ~1 in 100,000 |
| Bardet–Biedl syndrome | AR | BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, BBS9, BBS10, TRIM32, BBS12, MKS1, CEP290 | ~1 in 100,000 |
| Oral–facial–digital syndrome Type I | X-linked | OFD1 | ~1 in 100,000 |
| Renal–hepatic–pancreatic dysplasia | AR | NPHP3 | Rare |
| Jeune chondrodysplasia | AR | IFT80 | Rare |
| Cranioectodermal dysplasia | AR | Unknown | Rare |
| Ellis–Van Creveld syndrome | AR | EVC, EVC2 | Rare |
| Mainzer–Saldino syndrome | AR | Unknown | Rare |
| Glomerulocystic kidney disease | AR, AD | HNF-1b | Rare |
| Autosomal dominant polycystic kidney disease | AD | PKD1, PKD2 | ~1 in 1,000 |
| Nephronophthisis | AR | NPHP1, INVS, NPHP3, NPHP4, IQCB1, CEP290, GLIS2, RPGRIP1L, NEK8 | ~1 in 100,000 |
Figure 2.
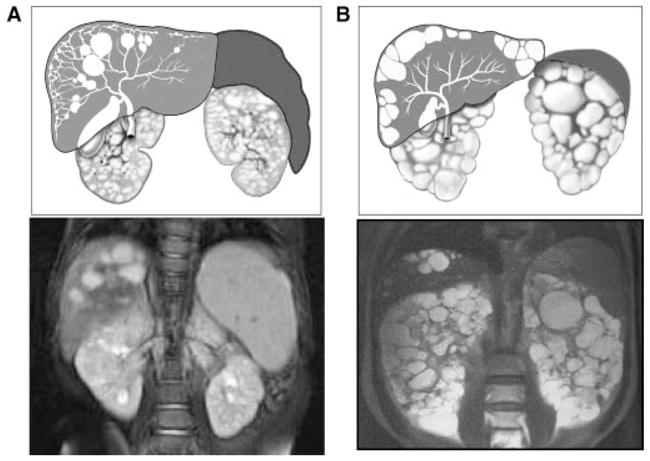
Artist’s rendering and MRI images of kidneys, liver, and spleen in ARPKD and ADPKD. A: ARPKD kidneys preserve their reniform contour. CHF in ARPKD is often complicated by portal hypertension evidenced by the enlarged spleen on the MRI. The majority of ARPKD patients also have dilatations of the intra- and extra-hepatic biliary system. B: In contrast, ADPKD kidneys often have distorted contours due to macrocysts. Portal hypertension is not typical in ADPKD and the spleen is not enlarged. Liver cysts in ADPKD are isolated cysts that are not contiguous with the biliary tree. (ADPKD MRI is from Heptinstall’s pathology of the kidney, Ed: Jennette J., Olson J., Schwartz M., Silva F.)
Other disorders in which CHF is a universal finding include Meckel–Gruber syndrome (MKS) [Fraser and Lytwyn, 1981], COACH syndrome (a subset of Joubert syndrome and related disorders (JSRD) with cerebellar vermis hypoplasia, oligophrenia, ataxia, coloboma, and hepatic fibrosis) [Satran et al., 1999; Uemura et al., 2005; Doherty et al., 2009], and renal–hepatic–pancreatic dysplasia (RHPD) [Bernstein et al., 1987; Torra et al., 1996; White et al., 2000] (Table I). Symptomatic CHF/CS, associated with PH or cholangitis, occurs in a subset of patients with various other ciliopathies including JSRDs [Lewis et al., 1994; Silverstein et al., 1997; Parisi et al., 2007], Bardet–Biedl (BBS) [Pagon et al., 1982; Nakamura et al., 1990], oral–facial–digital (OFD) syndromes [Thauvin-Robinet et al., 2006; Toprak et al., 2006; Gurrieri et al., 2007; Prattichizzo et al., 2008], and ciliary skeletal dysplasias such as Jeune asphyxiating thoracic dystrophy [Hudgins et al., 1992; Labrune et al., 1999; Yerian et al., 2003] (Table I). In most cases, the inheritance pattern of CHF/CS is autosomal recessive. Two rare exceptions are X-linked CHF/CS in association with oral–facial–digital syndrome type I (OFD1) [Prattichizzo et al., 2008] and autosomal dominant CHF/CS in rare families with ADPKD [Gunay-Aygun and Heller, 2008]. Whether the microscopic pathology of CHF is a universal finding in these syndromes remains to be determined. Asymptomatic or mild to moderate CHF/CS might be under-diagnosed in these disorders, because the pathologies of other systems often become the focus of attention and lifespan can be limited.
Polycystic Liver Disease
PLD refers to the presence of multiple macroscopic cysts in the liver. The cysts are isolated and are not part of the biliary tree. Most commonly, PLD exists as part of ADPKD (Fig. 2B). Otherwise, PLD occurs in patients with autosomal dominant polycystic liver disease (ADPLD), an entity that is genetically distinct from ADPKD and not typically associated with renal cysts [Drenth et al., 2005]. ADPLD is genetically heterogeneous; the PRKCSH and SEC63 genes, encoding the proteins hepatocystin and Sec63, account for less than half of ADPLD patients [Drenth et al., 2003; Li et al., 2003; Tahvanainen et al., 2003; Davila et al., 2004; Waanders et al., 2006]. Unlike other cystoproteins, hepatocystin and Sec63 are not ciliary proteins; they are involved in the ER processing of proteins [Drenth et al., 2005].
PLD as a Part of ADPKD
ADPKD represents the most common form of HRFCD, with a frequency of 1 in 500–1,000, and PLD is the most common extra-renal manifestation of ADPKD [Harris and Torres, 2008b]. Liver cysts in ADPKD increase in number and size as patients grow older; 58% of 15- to 24-yearolds and 94% of 35-to 46-year olds have liver cysts [Bae et al., 2006] (Fig. 2B). Although the prevalence of liver cysts is comparable in men (79%) and women (85%), cyst volume is greater in women, especially after multiple pregnancies and use of oral contraceptives [Everson and Taylor, 2005]. Enlarged cysts can cause chronic upper abdominal pain and distention, early satiety, nausea and dyspnea due to a mass effect, but hepatic function remains normal [Arnold and Harrison, 2005]. Cyst infection and hemorrhage can occur. CHF complicated by PH has been described in rare ADPKD families [Gunay-Aygun and Heller, 2008], and CS has also been reported [Shedda and Robertson, 2007]. PLD develops in ADPKD due to both PKD1 and PKD2 mutations [Harris and Torres, 2008b], but no genotype–phenotype correlation exists for PLD associated with ADPKD.
KIDNEY DISEASE IN CILIOPATHIES
In ciliopathies, the kidneys are the most commonly affected organs, displaying pathologies ranging from a urinary concentration defect in normal appearing kidneys to cystic dysplastic kidneys (Fig. 3). While ADPKD and ARPKD represent the most common ciliopathies, nephronophthisis, cystic dysplastic kidneys, medullary sponge kidney, and several overlap phenotypes contribute to the spectrum of kidney diseases observed in ciliopathies (Fig. 3).
Figure 3.
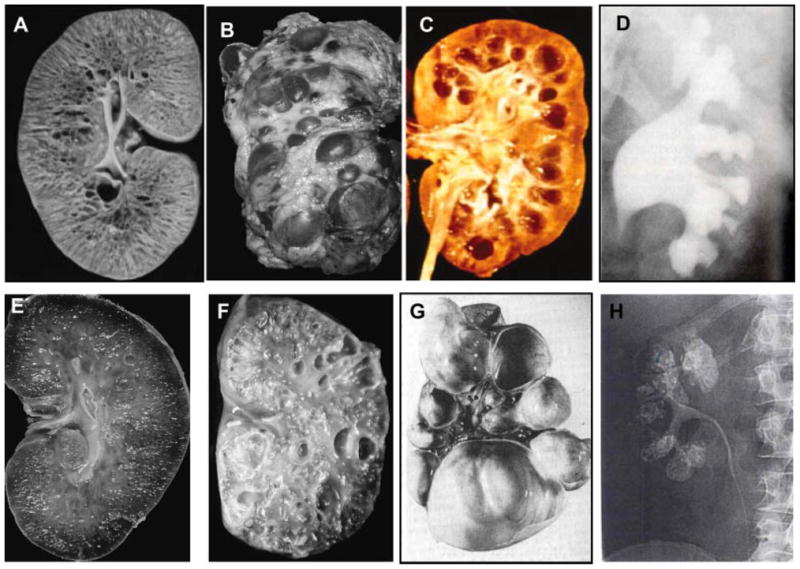
Kidney disease in ciliopathies. A: Kidneys of an infant with ARPKD that are enlarged with preserved contours due to uniform microcystic non-obstructive dilatation of collecting ducts. B: Massivelyenlarged kidneys of an adult with ADPKD that display large macrocysts distorting the kidney contour. C: Kidneys of an individual who has renal failure due nephronophithisis. D: Intraveneous pyelography of Bardet–Biedl syndrome kidneys displaying clubbing, blunting, and distortion of renal calyces in the absence of distal obstruction. E: Kidneys of an individual with glomerulocystic kidney disease displaying small cysts mostly located in the renal cortex. F: Cystic dysplastic kidneys in an infant with Meckel–Gruber syndrome. G: Multicystic dysplastic kidneys. H: Intraveneous pyelography displaying medullary sponge kidney. Sizes of the kidneys are not to scale. (A, B, E, and F are from Bisceglia et al., 2006, C is from Hildebrandt and Zhou, 2007, D is from Alton DJ and McDonald MB, Radiology, 109: 659–663, 1973. G is from Zerres et al., 1984. H is from Heptinstall’s Pathology of the kidney, Ed: Jennette J., Olson J., Schwartz M., Silva F.)
Bilateral, diffusely echogenic and/or cystic kidneys on prenatal or postnatal ultrasound are commonly the presenting findings in ciliopathy patients [Brun et al., 2004; de Bruyn and Marks, 2008]. In healthy infants, the renal cortex is more echogenic than the liver, but this physiological hyperechogenicity of the cortex disappears by about 3 months. In ciliopathies, the kidneys are termed “echogenic” “hyperechogenic” “hyperechoic” or “bright”, describing abnormally increased renal signal density on USG, that is, greater than that of the liver. In addition to ciliopathies, the broad differential diagnosis for hyperechoic kidneys includes congenital infections, renal vein thrombosis and congenital nephrotic syndrome. Determination of the presence or absence of congenital abnormalities other than hepatorenal disease, such as polydactyly, mid and hindbrain defects, iris or retinal colobomas, and retinal degeneration, helps to focus the diagnostic possibilities. The differential diagnosis for bilateral diffusely echogenic and/or cystic kidneys in a non-dysmorphic infant or child without extra-hepatorenal involvement includes ARPKD, ADPKD, glomerulocystic kidney disease (GCKD), and tuberous sclerosis [Brun et al., 2004; Avner and Sweeney, 2006]. Although renal cysts in tuberous sclerosis are typically associated with angiomyolipomas, cysts can occur without angiomyolipomas during the first years of life [Dell and Avner, 2008]. The appearance of kidneys on USG can be similar in infants with ARPKD, perinatal-onset ADPKD, glomerulocystic kidney disease, cystic dysplastic kidneys (as in classical Meckel–Gruber syndrome), BBS-related kidney disease, MKS-3-related kidney disease and some forms of NPHP [Brun et al., 2004; de Bruyn and Marks, 2008; Gunay-Aygun et al., 2009]. In all cases, the kidneys are enlarged and diffusely hyperechogenic with loss of corticomedullary differentiation, with or without macrocysts (visible discrete cysts).
Autosomal Dominant Polycystic Kidney Disease (ADPKD)
ADPKD is a systemic ciliopathy in which the kidneys and liver are primarily affected, but cysts can occur in the pancreas, arachnoid membrane, and seminal vesicles [Gabow, 1996; Harris and Torres, 2008, 2009]. Other manifestations include intracranial aneurysms, dilatation of the aortic root, dissection of the thoracic aorta, and mitral valve prolapse. The majority of ADPKD patients have a positive family history; 5–10% of patients have their disease due to de novo mutations [Harris and Torres, 2008, 2009]. ADPKD is genetically heterogeneous, with two causative genes identified. PKD1 encoding Polycystin-1 accounts for approximately 85% of affected individuals; PKD2 encoding Polycystin-2 is mutated in the remaining 15% of patients [Anon., 1994; Hughes et al., 1995; Mochizuki et al., 1996]. Although PKD1 and PKD2 sequencing is available, the diagnosis of ADPKD is made clinically, primarily based on imaging studies. The mutation detection rates for PKD1 and PKD2 are similar at approximately 85–90% (http://pkdb.mayo.edu/) [Harris and Torres, 2008]. PKD1 and PKD2 gene mutations result in similar extra-renal manifestations, including PLD and intracranial aneurysms [Rossetti et al., 2003a; Rossetti and Harris, 2007], but PKD1 mutations cause more severe kidney disease associated with a 20-year earlier onset of end stage renal disease (ESRD) compared with PKD2 mutations (54.3 years for PKD1; 74.0 years for PKD2) [Hateboer et al., 1999]. Mutations at the 5′ end of PKD1 are more likely to be associated with intracranial aneurysms and early renal failure than are 3′ mutations [Rossetti et al., 2002, 2003a].
Although all cells in an ADPKD kidney carry the germline mutation, cysts originate only from a small subset (~5%) of cells that acquire a second hit [Germino, 1997]. Most ADPKD patients are born with normal kidneys, and cysts develop and increase in number and size over a lifetime; glomerular function typically begins to decline in adulthood after massive cystic enlargement of the kidneys [Bae et al., 2006; Grantham et al., 2006]. Decreased urinary concentrating ability and hypertension occur before the start of the decline in glomerular function. ADPKD cysts originate from any part of the nephron and become isolated cysts early in the course of the disease [Osathanondh and Potter, 1964; Wilson, 2004]. Early in ADPKD, kidney USG reveals multiple cysts usually in association with normal parenchyma; in children cystic involvement may be unilateral. Rarely, ADPKD may present perinatally with enlarged hyperechogenic kidneys, indistinguishable from ARPKD kidneys on imaging. After the cysts form, they continue to grow into large macrocysts. Consequently, imaging in adults with ADPKD reveals massively enlarged kidneys full of macrocysts and often distorting the contour of the kidneys (Figs. 2B and 3B). Among individuals at 50% risk for ADPKD, diagnostic sensitivity of USG at age 30 years and younger is 95% for PKD1-related ADPKD and 67% for PKD2-related ADPKD [Nicolau et al., 1999]. A diagnosis of ADPKD cannot be excluded by a negative USG until the individual is 35 years old. Differing from ARPKD, ADPKD patients suffer from renal pain caused by nephrolithiasis, cyst hemorrhage or infection.
Autosomal Recessive Polycystic Kidney Disease (ARPKD)
ARPKD, the most common childhood-onset ciliopathy, occurs with a frequency of approximately 1 in 20,000 live births, making the carrier frequency approximately 1 in 70 [Zerres et al., 1996, 1998; Roy et al., 1997; Fonck et al., 2001; Capisonda et al., 2003; Guay-Woodford and Desmond, 2003]. ARPKD patients have non-obstructive fusiform dilatations of the renal collecting ducts, leading to progressive renal insufficiency, and DPM of the liver, resulting in CHF/CS [Jorgensen, 1977; Desmet, 1992a]. Most ARPKD patients present perinatally with enlarged kidneys, and approximately 30% die of pulmonary hypoplasia. CHF invariably accompanies ARPKD; although mild in early childhood, it eventually leads to PH, esophageal varices and hypersplenism in most cases [Kerr et al., 1978; Alvarez et al., 1981; Summerfield et al., 1986; Goilav et al., 2006]. Severe, early-onset systemic hypertension is seen in approximately 80% of ARPKD patients. It typically requires multi-agent antihypertensive treatment in infancy and becomes relatively easier to control later in childhood. Some ARPKD patients present late in childhood or adulthood, usually with PH associated with milder kidney disease. The majorityof ARPKD patients who become symptomatic perinatally require kidney transplantation in late childhood to adulthood. The diagnosis of ARPKD/CHF relies upon clinical, radiographic or biopsy evidence of typical renal pathology and CHF with autosomal recessive inheritance.
Differing from ADPKD, ARPKD kidneys are already diffusely affected at birth. They are symmetrically enlarged due to dilated collecting ducts, and retain their reniform configuration (Figs. 2A and 3A). Although most dilated collecting ducts in ARPKD continue to have urine flow in their lumen, some dilated ducts become closed cysts as children with ARPKD grow. This results in the appearance of macrocysts on the background of diffusely increased echogenicity on USG. As a consequence, imaging findings of ARPKD patients who present in childhood or adulthood can be difficult to distinguish from early ADPKD kidneys.
All patients with ARPKD thus far ascertained, including those who present in childhood or in adulthood with liver-predominant symptoms, have had mutations identified in the PKHD1 gene that encodes fibrocystin/polyductin [Onuchic et al., 2002; Ward et al., 2002; Bergmann et al., 2004; Sweeney and Avner, 2006]. Truncating PKHD1 mutations are associated with a more severe phenotype; typically, ARPKD patients who survive the neonatal period have at least one missense mutation [Rossetti et al., 2003b; Bergmann et al., 2005] (http://www.humgen.rwth-aachen.de).
Nephronophthisis (NPHP)
NPHP consists of a group of autosomal recessive tubulointerstitial disorders that initially present with a urinary concentrating defect and anemia and subsequently require renal transplant, usually in childhood [Hildebrandt and Zhou, 2007; Salomon et al., 2008]. Fibrotic tubulointerstitial pathology predominates in NPHP; renal cysts, typically located at the corticomedullary junction, form secondarily (Fig. 3C). The typical NPHP biopsy shows thickening and disruption of the tubular basement membrane, tubular atrophy, disproportionate tubulointerstitial fibrosis with minimal inflammation, and macrocysts at the corticomedullary junction [Omran, 2008]. Infantile, juvenile, and adolescent forms of NPHP result in ESRD at median ages of 1, 13, and 19 years, respectively. Juvenile NPHP, the most common form, generally presents between ages 4 and 6 years with polyuria, polydipsia, and anemia. Blood pressure is typically normal before the onset of renal failure. Renal USG shows normal size or small kidneys with increased echogenicity, with or without corticomedullary cysts. In infantile NPHP, the kidney size may be enlarged.
A subset of individuals with NPHP have mild retinal degeneration (tapetoretinal degeneration), which is most often asymptomatic [Omran, 2008]. Other extra-renal manifestations include CHF, structural cerebellar and midbrain abnormalities overlapping with JSRD, and severe retinal degeneration (Senior–Løken syndrome) [Hildebrandt and Zhou, 2007]. To date, nine genes (NPHP1–9) have been shown to cause NPHP [Hildebrandt and Zhou, 2007; Hildebrandt et al., 2009], but those nine genes account for only 30% of NPHP patients. NPHP1 mutations are the most common cause of NPHP (21%), and each of the other known genes is responsible for 3% or less of the disease. Clinical symptoms and renal histology are similar in all forms of NPHP except for NPHP2, which causes infantile NPHP. CHF is associated with NPHP2 and NPHP3 mutations [Boichis et al., 1973; Delaney et al., 1978; Olbrich et al., 2003]. It remains to be determined whether other NPHP genes are associated with CHF. MKS3 mutations can cause NPHP with CHF [Otto et al., 2009]. In addition, some patients with MKS3 mutations display an overlap phenotype with features of both NPHP and ARPKD [Gunay-Aygun et al., 2009].
The renal disease in Jeune asphyxiating thoracic dystrophy displays features most consistent with nephronophtisis [Shah, 1980; Ozcay et al., 2001].
Kidney Disease in Bardet–Biedl Syndrome
Renal disease is a major cause of morbidity and mortality in BBS [Hurley et al., 1975; Linne et al., 1986; Harnett et al., 1988; O’Dea et al., 1996]. Polyuria and polydypsia due to a urinary concentration defect occur frequently. On ultrasound imaging, BBS kidneys exhibit findings ranging from retained fetal lobulation to enlarged kidneys, resembling those in ARPKD, with diffuse hyperechogenicity and loss of corticomedullary differentiation, with or without macrocysts. On intravenous contrast imaging such as intravenous pyelography, BBS kidneys show renal calyceal clubbing, blunting, and distortion in the absence of distal obstruction (Fig. 3D) [Harnett et al., 1988]. Glomerular function declines at variable rates, requiring renal replacement therapy in some patients. Biopsy of BBS kidneys with declining glomerular function reveals pathology resembling NPHP tubulointerstitial disease [Linne et al., 1986].
Glomerulocystic Kidney Disease (GCKD)
Histopathologically, GCKD is characterized by a predominance of glomerular cysts without tubular dilatation [Avner and Sweeney, 2006; Bisceglia et al., 2006]. On USG, GCKD kidneys might be small, normal in size, or enlarged with small (<1 cm) cysts in the echogenic renal cortex but not in the medulla. Most GCKD is inherited in an autosomal dominant fashion, but it is not due to PKD1 or PKD2 mutations [Harris and Torres, 2008b ]. In autosomal dominant GCKD, kidneys are bilaterally enlarged and diffusely cystic (Fig. 3E). Infants with GCKD can have presentations that mimic ARPKD, and adults with GCKD can present with flank pain, hematuria, and hypertension.
Autosomal dominantly inherited familial hypoplastic GCKD is a distinct form of the disease characterized by small kidneys and medullocalyceal abnormalities; affected individuals have heterozygous mutations in the hepatocyte nuclear factor-1-beta gene (HNF1β or TCF2) [Bingham et al., 2001]. Heterozygous mutations in HNF1β also result in maturity-onset diabetes of the young, type V (MODY V, also referred to as renal cysts and diabetes syndrome), which can display a spectrum of renal anomalies ranging from glomerular cysts to renal agenesis [Horikawa et al., 1997]. Familial and sporadic GCKD may be associated with renal medullary dysplasia and ductal plate malformation of the liver [Bisceglia et al., 2006].
The term glomerulocystic kidney (GCK), which differs from GCKD, refers to kidneys in the context of malformation syndromes such as oral–facial–digital syndrome—type I and trisomies 13 and 18; in these disorders, glomerular cysts are part of the kidney pathology [Bisceglia et al., 2006].
Cystic Dysplastic Kidneys (CDK)
Cystic dysplastic kidneys (Fig. 3F), which should not be confused with multicystic dysplastic kidneys (MCDK; Fig. 3G), are characterized by poorly differentiated and disorganized nephron segments with primitive elements such as cartilage [Limwongse and Cassidy, 1999; Watkins and Avner, 1999; Bisceglia et al., 2006]. CDKs are better differentiated than multicystic dysplastic kidneys, and have some normal nephrons. Similarly, the collecting system of cystic dysplastic kidneys can be hypoplastic or malformed, but is generally better developed than that of MCDKs. USG of cystic dysplastic kidneys shows echogenic kidneys that are often large, without any demarcation between medulla and cortex. Ciliopathies associated with cystic dysplastic kidneys include Meckel–Gruber and Dekaban-Arima syndromes and renal–hepatic–pancreatic dysplasia [Fraser and Lytwyn, 1981; Bernstein et al., 1987; Satran et al., 1999].
Multicystic Dysplastic Kidneys (MCDK)
In MCDK, the kidneys are non-reniform and so grotesquely cystic that they are described as “a bunch of grapes”(Fig. 3G) [Watkins and Avner, 1999; Bisceglia et al., 2006]. The calyceal system, ureter, and renal vasculature are usually atretic. There is no differentiated renal tissue; the non-cystic tissue shows nests of cartilage and mesenchymal mantles surrounding primitive tubules. Thus, MCDKs are always non-functional and usually unilateral, since bilateral MCDK is incompatible with life. USG reveals no identifiable renal parenchyma, but rather several small single cysts or a large mass of cysts of varying sizes surrounding a dominant large cyst. MCDK can occur in MODY 5, a disorder caused by heterozygous mutations in HNF1B; this transcription factor regulates expression of the ARPKD gene PKHD1 [Horikawa et al., 1997]. MCDK can rarely be familial.
Medullary Sponge Kidney (MSK)
Most cases of MSK are sporadic; autosomal dominant inheritance is suggested in some families [Zerres et al., 1984]. MSK is typically diagnosed based on intravenous urography imaging that shows characteristic radial linear streaking in the renal papillae due to collection of contrast medium in the ectatic papillary-collecting ducts; this appearance is often referred to as a “bouquet of flowers” (Fig. 3H) [Bisceglia et al., 2006]. In approximately 50% of the patients, MSK may be complicated by nephrocalcinosis. MSK is usually bilateral; kidneys are normal size and glomerular function is preserved. Patients usually remain asymptomatic in childhood and come to medical attention in adulthood due to renal lithiasis.
CONCLUSION
Although the study of ciliopathies is still in its infancy, we can draw a few general conclusions regarding the associated liver and kidney disease. First, the current monikers attached to specific ciliopathies do not strictly distinguish each disorder; there exists significant overlap that continues to expand as the phenotype becomes better defined. Second, new ciliopathies will be identified, and their causative genes are likely to encode proteins that interact with currently known ciliary proteins. Finally, detailed descriptions of the clinical characteristics of ciliopathy patients will lay the groundwork for diagnosis, prognosis, and therapeutic interventions. Many intracellular abnormalities downstream to defective cilia-based signaling (e.g., altered calcium fluxes, and increased cAMP levels) are shared among various ciliopathies and are common to both renal and biliary epithelia, making novel targeted therapies a real possibility. Many therapeutic agents are currently under phase II or III clinical trials [Harris and Torres, 2008a]. While some of these agents slow down the disease progression in both kidneys and the liver (c-Src inhibitors, octreotide), others (vasopressin 2 receptor antagonists) are affective only in kidneys.
ONGOING RESEARCH
An intramural NIH protocol evaluating the characteristics of liver and kidney disease in various ciliopathies, is continuing to enroll patients (www.clinicaltrials.gov, trial NCT00068224).
Biography
Meral Gunay-Aygun is a Pediatrician and Biochemical Geneticist at the National Human Genome Research Institute (NHGRI) at the National Institutes of Health (NIH), and an Associate Professor of Pediatrics at Johns Hopkins University School of Medicine. She received her medical degree from Hacettepe University School of Medicine in Ankara, Turkey and completed her residency and genetics fellowship at Case Western Reserve University. Her research interests include ciliopathies and congenital bleeding disorders associated with platelet organelle formation defects.
References
- Akhan O, Karaosmanoglu AD, Ergen B. Imaging findings in congenital hepatic fibrosis. Eur J Radiol. 2007;61:18–124. doi: 10.1016/j.ejrad.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Alvarez F, Bernard O, Brunelle F, Hadchouel M, Leblanc A, Odievre M, Alagille D. Congenital hepatic fibrosis in children. J Pediatr. 1981;99:370–375. doi: 10.1016/s0022-3476(81)80320-4. [DOI] [PubMed] [Google Scholar]
- Anonymous. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell. 1994;78:725. [PubMed] [Google Scholar]
- Arnold HL, Harrison SA. New advances in evaluation and management of patients with polycystic liver disease. Am J Gastroenterol. 2005;100:2569–2582. doi: 10.1111/j.1572-0241.2005.00263.x. [DOI] [PubMed] [Google Scholar]
- Avner ED, Sweeney WE., Jr Renal cystic disease: New insights for the clinician. Pediatr Clin North Am. 2006;53:889–909. ix. doi: 10.1016/j.pcl.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Bae KT, Zhu F, Chapman AB, Torres VE, Grantham JJ, Guay-Woodford LM, Baumgarten DA, King BF, Jr, Wetzel LH, Kenney PJ, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1:64–69. doi: 10.2215/CJN.00080605. [DOI] [PubMed] [Google Scholar]
- Bergmann C, Senderek J, Kupper F, Schneider F, Dornia C, Windelen E, Eggermann T, Rudnik-Schoneborn S, Kirfel J, Furu L, et al. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD) Hum Mutat. 2004;23:453–463. doi: 10.1002/humu.20029. [DOI] [PubMed] [Google Scholar]
- Bergmann C, Kupper F, Dornia C, Schneider F, Senderek J, Zerres K. Algorithm for efficient PKHD1 mutation screening in autosomal recessive polycystic kidney disease (ARPKD) Hum Mutat. 2005;25:225–231. doi: 10.1002/humu.20145. [DOI] [PubMed] [Google Scholar]
- Bernstein J, Chandra M, Creswell J, Kahn E, Malouf NN, McVicar M, Weinberg AG, Wybel RE. Renal-hepatic-pancreatic dysplasia: A syndrome reconsidered. Am J Med Genet. 1987;26:391–403. doi: 10.1002/ajmg.1320260218. [DOI] [PubMed] [Google Scholar]
- Bingham C, Bulman MP, Ellard S, Allen LI, Lipkin GW, Hoff WG, Woolf AS, Rizzoni G, Novelli G, Nicholls AJ, Hattersley AT. Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet. 2001;68:219–224. doi: 10.1086/316945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: A review. Adv Anat Pathol. 2006;13:26–56. doi: 10.1097/01.pap.0000201831.77472.d3. [DOI] [PubMed] [Google Scholar]
- Bloustein PA. Association of carcinoma with congenital cystic conditions of the liver and bile ducts. Am J Gastroenterol. 1977;67:40–46. [PubMed] [Google Scholar]
- Boichis H, Passwell J, David R, Miller H. Congenital hepatic fibrosis and nephronophthisis. A family study. Q J Med. 1973;42:221–233. [PubMed] [Google Scholar]
- Brun M, Maugey-Laulom B, Eurin D, Didier F, Avni EF. Prenatal sonographic patterns in autosomal dominant polycystic kidney disease: A multicenter study. Ultrasound Obstet Gynecol. 2004;24:55–61. doi: 10.1002/uog.1098. [DOI] [PubMed] [Google Scholar]
- Capisonda R, Phan V, Traubuci J, Daneman A, Balfe JW, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: Outcomes from a single-center experience. Pediatr Nephrol. 2003;18:119–126. doi: 10.1007/s00467-002-1021-0. [DOI] [PubMed] [Google Scholar]
- Caroli J. Diseases of intrahepatic bile ducts. Isr J Med Sci. 1968;4:21–35. [PubMed] [Google Scholar]
- Caroli J. Diseases of the intrahepatic biliary tree. Clin Gastroenterol. 1973;2:147–161. [PubMed] [Google Scholar]
- Davila S, Furu L, Gharavi AG, Tian X, Onoe T, Qian Q, Li A, Cai Y, Kamath PS, King BF, Azurmendi PJ, Tahvanainen P, Kääriäinen H, Höckerstedt K, Devuyst O, Pirson Y, Martin RS, Lifton RP, Tahvanainen E, Torres VE, Somlo S. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–577. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- de Bruyn R, Marks SD. Postnatal investigation of fetal renal disease. Semin Fetal Neonatal Med. 2008;13:133–141. doi: 10.1016/j.siny.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Delaney V, Mullaney J, Bourke E. Juvenile nephronophthisis, congenital hepatic fibrosis and retinal hypoplasia in twins. Q J Med. 1978;47:281–290. [PubMed] [Google Scholar]
- Dell KM, Avner ED. GeneReviews at GeneTests: Medical Genetics Information Resource (database online) Copyright. University of Washington; Seattle: 2008. Autosomal recessive polycystic kidney disease; pp. 1997–2008. Available at http://wwwgenetestsorg. [Google Scholar]
- Desmet VJ. Congenital diseases of intra-hepatic bile ducts: Variations on the theme “ductal plate malformation”. Hepatology. 1992a;16:1069–1083. doi: 10.1002/hep.1840160434. [DOI] [PubMed] [Google Scholar]
- Desmet VJ. What is congenital hepatic fibrosis? Histopathology. 1992b;20:465–477. doi: 10.1111/j.1365-2559.1992.tb01031.x. [DOI] [PubMed] [Google Scholar]
- Desmet VJ. Ludwig symposium on biliary disorders—Part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–89. doi: 10.4065/73.1.80. [DOI] [PubMed] [Google Scholar]
- Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, Clericuzio C, Demir H, Dorschner M, van Essen A, Gahl WA, Gentile M, Gorden NT, Hikida A, Knutzen D, Ozyurek H, Phelps I, Rosenthal P, Verloes A, Weigand H, Chance PF, Dobyns WB, Glass IA. Mutations in 3 genes (MKS3, CC2D2A and RPGRIPIL) cause coach syndrome (Joubert syndrome with congenital hepatic fibrosis) J Med Genet. 2009 doi: 10.1136/jmg.2009.067249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33:345–347. doi: 10.1038/ng1104. [DOI] [PubMed] [Google Scholar]
- Drenth JP, Martina JA, van de Kerkhof R, Bonifacino JS, Jansen JB. Polycystic liver disease is a disorder of cotranslational protein processing. Trends Mol Med. 2005;11:37–42. doi: 10.1016/j.molmed.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Everson GT, Taylor MR. Management of polycystic liver disease. Curr Gastroenterol Rep. 2005;7:19–25. doi: 10.1007/s11894-005-0061-6. [DOI] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H. When cilia go bad: Cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- Fonck C, Chauveau D, Gagnadoux MF, Pirson Y, Grunfeld JP. Autosomal recessive polycystic kidney disease in adulthood. Nephrol Dial Transplant. 2001;16:1648–1652. doi: 10.1093/ndt/16.8.1648. [DOI] [PubMed] [Google Scholar]
- Fraser FC, Lytwyn A. Spectrum of anomalies in the Meckel syndrome, or: “Maybe there is a malformation syndrome with at least one constant anomaly”. Am J Med Genet. 1981;9:67–73. doi: 10.1002/ajmg.1320090112. [DOI] [PubMed] [Google Scholar]
- Gabow P. Definition and natural history of autosomal dominant polycystic kidney disease. In: Watson ML, Torres VE, editors. Polycystic kidney disease. Oxford: Oxford University Press; 1996. pp. 333–355. [Google Scholar]
- Germino GG. Autosomal dominant polycystic kidney disease: A two-hit model. Hosp Pract (Minneap) 1997;32:81–82. 85–88, 91–92. doi: 10.1080/21548331.1997.11443444. passim. [DOI] [PubMed] [Google Scholar]
- Goilav B, Norton KI, Satlin LM, Guay-Woodford L, Chen F, Magid MS, Emre S, Shneider BL. Predominant extrahepatic biliary disease in autosomal recessive polycystic kidney disease: A new association. Pediatr Transplant. 2006;10:294–298. doi: 10.1111/j.1399-3046.2005.00456.x. [DOI] [PubMed] [Google Scholar]
- Grantham JJ, Chapman AB, Torres VE. Volume progression in autosomal dominant polycystic kidney disease: The major factor determining clinical outcomes. Clin J Am Soc Nephrol. 2006;1:148–157. doi: 10.2215/CJN.00330705. [DOI] [PubMed] [Google Scholar]
- Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: The clinical experience in North America. Pediatrics. 2003;111:1072–1080. doi: 10.1542/peds.111.5.1072. [DOI] [PubMed] [Google Scholar]
- Gunay-Aygun M, Gahl WA, Heller T. GeneReviews at GeneTests: Medical Genetics Information Resource (database online) Copyright. University of Washington; Seattle: 2008. Congenital hepatic fibrosis overview; pp. 1997–2008. Available at http://wwwgenetestsorg. [PubMed] [Google Scholar]
- Gunay-Aygun M, Avner ED, Bacallao RL, Choyke PL, Flynn JT, Germino GG, Guay-Woodford L, Harris P, Heller T, Ingelfinger J, Kaskel F, Kleta R, LaRusso NF, Mohan P, Pazour GJ, Shneider BL, Torres VE, Wilson P, Zak C, Zhou J, Gahl WA. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: Summary statement of a first National Institutes of Health/Office of Rare Diseases conference. J Pediatr. 2006;149:159–164. doi: 10.1016/j.jpeds.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunay-Aygun M, Parisi M, Doherty D, Tuchman M, Tsilou E, Kleiner DE, Huizing M, Turkbey B, Choyke P, Guay-Woodford L, Heller T, Szymanska K, Johnson CA, Glass I, Gahl WA. MKS3-related Ciliopathy with Features of Autosomal Recessive Polycystic Kidney Disease, Nephronophthisis and Joubert Syndrome. J Pediatr. 2009;155:386–392. doi: 10.1016/j.jpeds.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurrieri F, Franco B, Toriello H, Neri G. Oral-facial-digital syndromes: Review and diagnostic guidelines. Am J Med Genet Part A. 2007;143A:3314–3323. doi: 10.1002/ajmg.a.32032. [DOI] [PubMed] [Google Scholar]
- Harnett JD, Green JS, Cramer BC, Johnson G, Chafe L, McManamon P, Farid NR, Pryse-Phillips W, Parfrey PS. The spectrum of renal disease in Laurence-Moon-Biedl syndrome. N Engl J Med. 1988;319:615–618. doi: 10.1056/NEJM198809083191005. [DOI] [PubMed] [Google Scholar]
- Harris PC, Torres VE. GeneReviews at GeneTests: Medical Genetics Information Resource (database online) Copyright. University of Washington; Seattle: 2008. Polycystic Kidney Disease, Autosomal Dominant; pp. 1997–2008. Available at http://wwwgenetestsorg. [Google Scholar]
- Harris PC, Torres VE. Polycystic kidney disease. Annual Rev Med. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999;353:103–107. doi: 10.1016/s0140-6736(98)03495-3. [DOI] [PubMed] [Google Scholar]
- Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18:1855–1871. doi: 10.1681/ASN.2006121344. [DOI] [PubMed] [Google Scholar]
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: Disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20:23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, Lindner T, Yamagata K, Ogata M, Tomonaga O, Kuroki H, Kasahara T, Iwamoto Y, Bell GI. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17:384–385. doi: 10.1038/ng1297-384. [DOI] [PubMed] [Google Scholar]
- Hudgins L, Rosengren S, Treem W, Hyams J. Early cirrhosis in survivors with Jeune thoracic dystrophy. J Pediatr. 1992;120:754–756. doi: 10.1016/s0022-3476(05)80241-0. [DOI] [PubMed] [Google Scholar]
- Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–160. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- Hurley RM, Dery P, Norady MB, Drummond KN. The renal lesion of the Laurence-Moon-Biedl syndrome. J Pediatr. 1975;87:206–209. doi: 10.1016/s0022-3476(75)80580-4. [DOI] [PubMed] [Google Scholar]
- Johnson CA, Gissen P, Sergi C. Molecular pathology and genetics of congenital hepatorenal fibrocystic syndromes. J Med Genet. 2003;40:311–319. doi: 10.1136/jmg.40.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen MJ. The ductal plate malformation. Acta Pathol Microbiol Scand Suppl. 1977;257:1–87. [PubMed] [Google Scholar]
- Kerkar N, Norton K, Suchy FJ. The hepatic fibrocystic diseases. Clin Liver Dis. 2006;10:55–71. v–vi. doi: 10.1016/j.cld.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Kerr DN, Harrison CV, Sherlock S, Walker RM. Congenital hepatic fibrosis. Q J Med. 1961;30:91–117. [PubMed] [Google Scholar]
- Kerr DN, Okonkwo S, Choa RG. Congenital hepatic fibrosis: The long-term prognosis. Gut. 1978;19:514–520. doi: 10.1136/gut.19.6.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrune P, Fabre M, Trioche P, Estournet-Mathiaud B, Grangeponte MC, Rambaud C, Maurage C, Bernard O. Jeune syndrome and liver disease: Report of three cases treated with ursodeoxycholic acid. Am J Med Genet. 1999;87:324–328. doi: 10.1002/(sici)1096-8628(19991203)87:4<324::aid-ajmg8>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Lewis SM, Roberts EA, Marcon MA, Harvey E, Phillips MJ, Chuang SA, Buncic JR, Clarke JT. Joubert syndrome with congenital hepatic fibrosis: An entity in the spectrum of oculo-encephalo-hepato-renal disorders. Am J Med Genet. 1994;52:419–426. doi: 10.1002/ajmg.1320520406. [DOI] [PubMed] [Google Scholar]
- Li A, Davila S, Furu L, Qian Q, Tian X, Kamath PS, King BF, Torres VE, Somlo S. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72:691–703. doi: 10.1086/368295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limwongse CCS, Cassidy SB. Syndromes and malformations of the urinary tract. In: Barratt TM, Avner ED, Harmon WE, editors. Pediatric nephrology. Baltimore: Lippincott Williams & Wilkins; 1999. pp. 427–452. [Google Scholar]
- Linne T, Wikstad I, Zetterstrom R. Renal involvement in the Laurence-Moon-Biedl syndrome. Functional and radiological studies. Acta Paediatr Scand. 1986;75:240–244. doi: 10.1111/j.1651-2227.1986.tb10192.x. [DOI] [PubMed] [Google Scholar]
- Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–920. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masyuk AI, Masyuk TV, LaRusso NF. Cholangiocyte primary cilia in liver health and disease. Dev Dyn. 2008;237:2007–2012. doi: 10.1002/dvdy.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- Morgan DE, Lockhart ME, Canon CL, Holcombe MP, Bynon JS. Polycystic liver disease: Multimodality imaging for complications and transplant evaluation. Radiographics. 2006;26:1655–1668. doi: 10.1148/rg.266065013. quiz 1655. [DOI] [PubMed] [Google Scholar]
- Nakamura F, Sasaki H, Kajihara H, Yamanoue M. Laurence-Moon-Biedl syndrome accompanied by congenital hepatic fibrosis. J Gastroenterol Hepatol. 1990;5:206–210. doi: 10.1111/j.1440-1746.1990.tb01826.x. [DOI] [PubMed] [Google Scholar]
- Nicolau CTR, Badenas C, Vilana R, Bianchi L, Gilabert R, Darnell A, Bru C. Autosomal dominant polycystic kidney disease types 1 and 2: Assessment of US sensitivity for diagnosis. Radiology. 1999;213:273–276. doi: 10.1148/radiology.213.1.r99oc05273. [DOI] [PubMed] [Google Scholar]
- O’Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J. The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kidney Dis. 1996;27:776–783. doi: 10.1016/s0272-6386(96)90513-2. [DOI] [PubMed] [Google Scholar]
- Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT, Sasmaz G, Trauer U, Reinhardt R, Sudbrak R, Antignac C, Gretz N, Walz G, Schermer B, Benzing T, Hildebrandt F, Omran H. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet. 2003;34:455–459. doi: 10.1038/ng1216. [DOI] [PubMed] [Google Scholar]
- Omran HaE-OB. Nephronophithisis and medullary cystic kidney disease. In: Geary DF, Schaefer F, editors. Comprehensive pediatric nephrology. Elsiever; 2008. pp. 143–154. [Google Scholar]
- Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schöneborn S, Mrug M, Sweeney W, Avner ED, Zerres K, Guay-Woodford LM, Somlo S, Germino GG. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osathanondh V, Potter EL. Pathogenesis of polycystic kidneys. Historical survey. Arch Pathol. 1964;77:459–465. [PubMed] [Google Scholar]
- Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, Wise EL, Wolf MTF, Utsch B, Becker C, Nürnberg G, Nürnberg P, Nayir A, Saunier S, Antignac C, Hildebrandt F. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11) Journal of Medical Genetics. 2009;46:663–670. doi: 10.1136/jmg.2009.066613. [DOI] [PubMed] [Google Scholar]
- Ozcay F, Derbent M, Demirhan B, Tokel K, Saatci U. A family with Jeune syndrome. Pediatr Nephrol. 2001;16:623–626. doi: 10.1007/s004670100627. [DOI] [PubMed] [Google Scholar]
- Pagon RA, Haas JE, Bunt AH, Rodaway KA. Hepatic involvement in the Bardet-Biedl syndrome. Am J Med Genet. 1982;13:373–381. doi: 10.1002/ajmg.1320130405. [DOI] [PubMed] [Google Scholar]
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300) Eur J Hum Genet. 2007;15:511–521. doi: 10.1038/sj.ejhg.5201648. [DOI] [PubMed] [Google Scholar]
- Patel V, Chowdhury R, Igarashi P. Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens. 2009;18:99–106. doi: 10.1097/MNH.0b013e3283262ab0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prattichizzo C, Macca M, Novelli V, Giorgio G, Barra A, Franco B. Mutational spectrum of the oral-facial-digital type I syndrome: A study on a large collection of patients. Hum Mutat. 2008;29:1237–1246. doi: 10.1002/humu.20792. [DOI] [PubMed] [Google Scholar]
- Rodat-Despoix L, Delmas P. Ciliar functions in the nephron. Pflugers Arch. 2009;458:179–187. doi: 10.1007/s00424-008-0632-0. [DOI] [PubMed] [Google Scholar]
- Rossetti S, Harris PC. Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol. 2007;18:1374–1380. doi: 10.1681/ASN.2007010125. [DOI] [PubMed] [Google Scholar]
- Rossetti S, Burton S, Strmecki L, Pond GR, San Millan JL, Zerres K, Barratt TM, Ozen S, Torres VE, Bergstralh EJ, Winearls CG, Harris PC. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol. 2002;13:1230–1237. doi: 10.1097/01.asn.0000013300.11876.37. [DOI] [PubMed] [Google Scholar]
- Rossetti S, Chauveau D, Kubly V, Slezak JM, Saggar-Malik AK, Pei Y, Ong AC, Stewart F, Watson ML, Bergstralh EJ, Winearls CG, Torres VE, Harris PC. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet. 2003a;361:2196–2201. doi: 10.1016/S0140-6736(03)13773-7. [DOI] [PubMed] [Google Scholar]
- Rossetti S, Torra R, Coto E, Consugar M, Kubly V, Malaga S, Navarro M, El-Youssef M, Torres VE, Harris PC. A complete mutation screen of PKHD1 in autosomal-recessive polycystic kidney disease (ARPKD) pedigrees. Kidney Int. 2003b;64:391–403. doi: 10.1046/j.1523-1755.2003.00111.x. [DOI] [PubMed] [Google Scholar]
- Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: Long-term outcome of neonatal survivors. Pediatr Nephrol. 1997;11:302–306. doi: 10.1007/s004670050281. [DOI] [PubMed] [Google Scholar]
- Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatr Nephrol. 2008 July 8; doi: 10.1007/s00467-008-0840-z. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satran D, Pierpont ME, Dobyns WB. Cerebello-oculo-renal syndromes including Arima, Senior-Loken and COACH syndromes: More than just variants of Joubert syndrome. Am J Med Genet. 1999;86:459–469. [PubMed] [Google Scholar]
- Shah KJ. Renal lesion in Jeune’s syndrome. Br J Radiol. 1980;53:432–436. doi: 10.1259/0007-1285-53-629-432. [DOI] [PubMed] [Google Scholar]
- Shedda S, Robertson A. Caroli’s syndrome and adult polycystic kidney disease. ANZ J Surg. 2007;77:292–294. doi: 10.1111/j.1445-2197.2006.03659.x. [DOI] [PubMed] [Google Scholar]
- Silverstein DM, Zacharowicz L, Edelman M, Lee SC, Greifer I, Rapin I. Joubert syndrome associated with multicystic kidney disease and hepatic fibrosis. Pediatr Nephrol. 1997;11:746–749. doi: 10.1007/s004670050381. [DOI] [PubMed] [Google Scholar]
- Summerfield JANY, Sherlock S, Canafalch J, Scheuer PJ. Hepatobiliary fibropolycystic disease: A clinical and histological review of 51 patients. J Hepatol. 1986;2:141–156. doi: 10.1016/s0168-8278(86)80073-3. [DOI] [PubMed] [Google Scholar]
- Sweeney WE, Jr, Avner ED. Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPKD) Cell Tissue Res. 2006;326:671–685. doi: 10.1007/s00441-006-0226-0. [DOI] [PubMed] [Google Scholar]
- Tahvanainen P, Tahvanainen E, Reijonen H, Halme L, Kaariainen H, Hockerstedt K. Polycystic liver disease is genetically heterogeneous: Clinical and linkage studies in eight Finnish families. J Hepatol. 2003;38:39–43. doi: 10.1016/s0168-8278(02)00348-3. [DOI] [PubMed] [Google Scholar]
- Tahvanainen E, Tahvanainen P, Kaariainen H, Hockerstedt K. Polycystic liver and kidney diseases. Ann Med. 2005;37:546–555. doi: 10.1080/07853890500389181. [DOI] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Cossee M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, Bieth E, Layet V, Parent P, David A, Goldenberg A, Mortier G, Héron D, Sagot P, Bouvier AM, Huet F, Cusin V, Donzel A, Devys D, Teyssier JR, Faivre L. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: A French and Belgian collaborative study. J Med Genet. 2006;43:54–61. doi: 10.1136/jmg.2004.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toprak O, Uzum A, Cirit M, Esi E, Inci A, Ersoy R, Tanrisev M, Ok E, Franco B. Oral-facial-digital syndrome type 1, Caroli’s disease and cystic renal disease. Nephrol Dial Transplant. 2006;21:1705–1709. doi: 10.1093/ndt/gfk013. [DOI] [PubMed] [Google Scholar]
- Torra R, Alos L, Ramos J, Estivill X. Renal-hepatic-pancreatic dysplasia: An autosomal recessive malformation. J Med Genet. 1996;33:409–412. doi: 10.1136/jmg.33.5.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Sanchez EQ, Ikegami T, Watkins D, Narasimhan G, McKenna GJ, Chinnakotla S, Dawson SIII, Randall HB, Levy MF, Goldstein RM, Klintmalm GB. Successful combined liver and kidney transplant for COACH syndrome and 5-yr follow-up. Clin Transplant. 2005;19:717–720. doi: 10.1111/j.1399-0012.2005.00409.x. [DOI] [PubMed] [Google Scholar]
- Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 2009;111:39–53. doi: 10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waanders E, te Morsche RH, de Man RA, Jansen JB, Drenth JP. Extensive mutational analysis of PRKCSH and SEC63 broadens the spectrum of polycystic liver disease. Hum Mutat. 2006;27:830. doi: 10.1002/humu.9441. [DOI] [PubMed] [Google Scholar]
- Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- Watkins SLMR, Avner ED. Renal dysplasia, hypoplasia and miscellaneous cystic disorders. In: Barratt TM, Avner ED, Harmon WE, editors. Pediatric nephrology. Baltimore: Lippincott Williams & Wilkins; 1999. pp. 415–425. [Google Scholar]
- White SM, Hurst JA, Hamoda H, Chamberlain P, Bowker CM. Renal-hepatic-pancreatic dysplasia: A broad entity. Am J Med Genet. 2000;95:399–400. doi: 10.1002/1096-8628(20001211)95:4<399::aid-ajmg19>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Wilson PD. Polycystic kidney disease. N Engl J Med. 2004;350:151–164. doi: 10.1056/NEJMra022161. [DOI] [PubMed] [Google Scholar]
- Yamato T, Sasaki M, Hoso M, Sakai J, Ohta H, Watanabe Y, Nakanuma Y. Intra-hepatic cholangiocarcinoma arising in congenital hepatic fibrosis: Report of an autopsy case. J Hepatol. 1998;28:717–722. doi: 10.1016/s0168-8278(98)80297-3. [DOI] [PubMed] [Google Scholar]
- Yerian LM, Brady L, Hart J. Hepatic manifestations of Jeune syndrome (asphyxiating thoracic dystrophy) Semin Liver Dis. 2003;23:195–200. doi: 10.1055/s-2003-39950. [DOI] [PubMed] [Google Scholar]
- Zerres K, Volpel MC, Weiss H. Cystic kidneys. Genetics, pathologic anatomy, clinical picture, and prenatal diagnosis. Hum Genet. 1984;68:104–135. doi: 10.1007/BF00279301. [DOI] [PubMed] [Google Scholar]
- Zerres K, Rudnik-Schoneborn S, Deget F, Holtkamp U, Brodehl J, Geisert J, Scharer K. Autosomal recessive polycystic kidney disease in 115 children: Clinical presentation, course and influence of gender. Arbeitsgemeinschaft fur Padiatrische, Nephrologie. Acta Paediatr. 1996;85:437–445. doi: 10.1111/j.1651-2227.1996.tb14056.x. [DOI] [PubMed] [Google Scholar]
- Zerres K, Mucher G, Becker J, Steinkamm C, Rudnik-Schoneborn S, Heikkila P, Rapola J, Salonen R, Germino GG, Onuchic L, Somlo S, Avner ED, Harman LA, Stockwin JM, Guay-Woodford LM. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): Molecular genetics, clinical experience, and fetal morphology. Am J Med Genet. 1998;76:137–144. [PubMed] [Google Scholar]