

Abstract
Our triazole-based histone deacetylase inhibitor (HDACI), octanedioic acid hydroxyamide[3-(1-phenyl-1H-[1,2,3]triazol-4-yl)phenyl]amide (4a), suppresses pancreatic cancer cell growth in vitro, with the lowest IC50 value of 20 nM against MiaPaca-2 cell. In this study, we continued our efforts to develop triazol-4-ylphenyl bearing hydroxamate analogs by embellishing the terminal phenyl ring of 4a with different substituents. The isoform inhibitory profile of these hydroxamate analogs was similar to those of 4a. All of these triazol-4-ylphenyl bearing hydroxamates are pan-HDACIs like SAHA. Moreover, compounds 4h and 11a were found to be very effective inhibitors of cancer cell growth in the HupT3 (IC50 = 50 nM) and MiaPaca-2 (IC50 = 40 nM) cancer cell lines, respectively. Compound 4a was found to re-activate the expression of CDK inhibitor proteins and to suppress pancreatic cancer cell growth in vivo. Taken together these data further support the value of the triazol-4-ylphenyl bearing hydroxamates in identifying potential pancreatic cancer therapies.
Introduction
Epigenetic alterations involve regulation of gene expression, and are critical to the pathogenesis of many diseases including cancer and various neurodegenerative diseases.1, 2 Histone modification is one of the molecular mechanisms that mediate epigenetic phenomena.3 Two types of enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs), control the acetylation of histones. In general, HATs function to acetylate lysine groups in nuclear histones, resulting in neutralization of the charges on the histones and a more open, transcriptionally active chromatin structure, while HDACs function to deacetylate and suppress transcription. A shift in the balance of acetylation on chromatin may 3 in changes in the regulation of patterns of gene expression.4 HDAC inhibitors (HDACIs) represent a class of molecularly targeted agents that can modulate epigenetic changes to histone proteins and thereby counteract aberrant gene expression. HDACIs can be classified into structural classes, including short chain fatty acids, small-molecule hydroxamates,5 cyclic peptides,2 benzamides,6 thiol-based compounds,7 ketones8 and other hybrid compounds (Figure 1). A number of HDACIs such as (E)-(1S,4S,10S,21S)-7-[(Z)-ethylidene]-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8.7.6] tricos-16-ene-3,6,9,19,22-pentone (FR901228)2 have now reached clinical trials, and suberoylanilide hydroxamic acid (SAHA, 1a) is marketed by Merck for use in cutaneous T-cell lymphoma (CTCL).9 Published work on the use of HDACIs in inflammatory disease,10 neurodegeneration,11 heart disease12 and protozoan infections13 reflect the growing recognition that HDACIs might serve as therapeutic interventions for diseases other than cancer.
Figure 1.

Classes of HDACIs
There are 11 HDAC isoforms identified that operate through zinc-dependent mechanisms.14 These include the class I HDACs 1, 2, 3, and 8, class II that includes 4, 5, 6, 7, 9, and 10, and class IV that contains HDAC11. Class III enzymes are HDACs in yeast and include the SIRTs (sirtuins) or Sir2-related proteins, which are NAD-dependent HDACs.15 Most class I HDACs are subunits of multiprotein nuclear complexes that are crucial for transcriptional repression and epigenetic landscaping. A variety of data suggest that HDAC1 plays an important role in tumorigenesis,16 and therefore class I inhibitors are being sought for use as anti-cancer drugs. Class II HDACs regulate cytoplasmic processes or function as signal transducers that shuttle between the cytoplasm and the nucleus. The class IV enzyme, HDAC11 is poorly understood compared to other HDAC isoforms, and recently it has been found to negatively regulate the expression of the gene encoding interleukin 10 (IL-10) in antigen-presenting cells.17 HDACs of class I are expressed in most cell types, whereas the expression pattern of class II HDACs is more tissue-restricted. For example, HDAC4 and 5 are highly expressed in cardiomyocytes and play a role in hypertrophy.18 HDACs 4, 8, and 9 are expressed to a greater extent in tumor tissues.19 High expression level of HDAC11 transcripts is limited to kidney, heart, brain, skeletal muscle, and testis while the class II enzyme, HDAC6, is expressed in most neurons.20 To understand precise roles of HDAC isoforms will expand our understanding of the connections between individual HDAC isoform and pathophysiology as well as guide the development of selective HDACIs as strategic therapeutic agents that elicit fewer undesirable side effects.
Classical HDACIs have three chemical domains, each with a specific function relating to the structure of the enzyme. Figure 2 shows the structure and functional domains of SAHA (1a), which includes a zinc binding group (ZBG), a linker region, and a surface recognition domain (cap). To understand the mechanism of HDAC is beneficial for a study of the interactions between HDACIs and the enzyme. The active site of class I, II, and IV HDACs is found within a highly conserved catalytic domain containing a divalent zinc cation that is coordinated to both histidine and aspartate residues. Deacetylation of the HDAC substrates occurs through attack by a water molecule that is activated through interaction with this zinc cation coupled with deprotonation through a histidine-aspartate charge-relay system. To date, x-ray co-crystallographic information is available for HDAC4, HDAC7 and 8, in complex with several different inhibitors including SAHA (1a), TSA (1b), and others.21 These crystal structures of the HDAC enzymes and the derived homology models provide valuable information in the design of selective HDACIs. We have been able to obtain varying degrees of isozyme selectivity for the HDACIs through chemical manipulations of the CAP region in concert with the ZBG. For example, we have reported that certain mercaptoacetamide-based HDACIs show inherent selectivity for HDAC6 versus HDAC1, and that some of these thiol-based agents are able to provide robust neuroprotection in the homocysteic acid model of oxidative stress.7, 22 Moreover, one of these thiol-based HDACIs was found to reduce neuronal cell loss in a rodent model of traumatic brain injury.23 We also have identified HDACIs containing a phenylisoxazole as the cap group that possess excellent selectivity and picomolar potency for HDAC6.24 In other studies, we have reported a series of trazolylphenyl-based HDACIs for which we demonstrated that modifications to the cap region were able to alter selectivity for HDAC1 versus HDAC6.25 Preliminary studies of the anticancer and antimalarial activity of these triazol-4-ylphenyl bearing hydroxamates (4a) suggested that these compounds were worth of further structural optimization (Figure 2). In this paper, we present additional details of the structure-activity relationship (SAR) studies of the parent compound 4a that focus on both steric and/or electronic effects on CAP region. The biological studies presented here include HDAC isoform profiling and cell-based anti-proliferative screening towards pancreatic cancer cell lines of all the newly synthesized triazolylphenyl-based HDACIs, together with cell-cycle dysregulation, induction of CDK inhibitor proteins and in vivo xenograft study of 4a.
Figure 2.

Functional domains of HDACIs
Results and Discussion
Chemistry
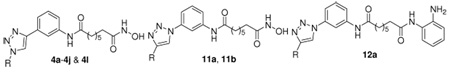
The copper-catalyzed [3+2]-cycloaddition of acetylene 1 with azides 2a–2l provided the 1,4-disubstituted triazoles 3a–3l, in analogy to work previously reported by us.26 The terminal ester groups of compounds 3a–3j and 3l were then treated with KOH/NH2OH in methanol to afford the final hydroxamates 4a–4j and 4l (Scheme 1).
Scheme 1. Synthesis of ligands 4a–4j and 4l.

Reagents and Conditions: (a) copper sulfate pentahydrate, sodium ascorbate, azide 2a–2j, rt; (b) H2, 10% Pd-C, rt, 4 h; (c) NH2OH, KOH, rt.
As shown in Scheme 2, the synthesis of the reversed 1,4-disubstituted triazoles 11a and 11b started from the mono-Boc protected 1,3-phenyldiamine 5. Compound 6 was prepared by a coupling reaction between compound 5 and suberic acid monomethyl ester in the presence of POCl3 and pyridine, which was further converted to compound 7 by deprotection of the Boc group in trifluoroacetic acid. The diazotization of the resulting aniline 7 with NaN3 provided the phenylazide 8. The cycloaddition of compound 8 with phenylacetylene or cyclohexylacetylene provided the reversed triazoles 10a and 10b, respectively.26 Treating 10a and 10b with KOH/NH2OH in methanol gave the desired hydroxamates 11a and 11b (Scheme 2).
Scheme 2. Synthesis of ligands 11a and 11b.

Reagents and Conditions: (a) suberic acid monomethyl ester, POCl3, pyridine, 0 °C; (b) TFA, 0 °C; (c) HOAc, NaNO2, 0 °C; (d) NaN3, 0 °C; (e) copper sulfate pentahydrate, sodium ascorbate, phenylacetylene (9a) or cyclohexylacetylene (9b), rt; (f) NH2OH, KOH, rt.
HDAC isoform inhibition assay
The inhibitory effects of compounds 4b–4j, 4l, 11a–11b, and 12a on HDAC activity were determined by using a fluorescence-based assay as described before.7 The data are presented as IC50 values in Table 1. TSA (1b) was used as a positive control. The inhibitory data for SAHA (1a) are also presented for comparison.
Table 1.
HDAC isoform inhibitory activity (IC50, nM) of the triazole-based ligands
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | ClogP b | R | IC50 [nM] [a] | |||||
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC10 | HDAC6 | |||
| 1a | 1.44 | 96 | 282 | 17 | 2290 | 72 | 14 | |
| 1b | 2.23 | 4 | 14 | 2 | 1380 | 5 | 1 | |
| 4a | 2.39 | Ph | 18 ± 2 | 63.9 ± 12.5 | 7.2 ± 0.7 | 2780 ± 470 | 27.7 ± 1.9 | 9.6 ± 0.6 |
| 4b | 2.59 | 4-F-Ph | 31.9 ± 2.9 | 60 ± 10 | 18.1 ± 1.9 | 12000 ± 800 | 32.8 ± 5.0 | 8.4 ± 1.8 |
| 4c | 3.55 | 4-I-Ph | 8.0 ± 0.6 | 24.6 ± 0.8 | 3.0 ± 0.1 | > 30000 | 10.7 ± 0.7 | 2.2 ± 0.1 |
| 4d | 3.35 | 4-CF3-Ph | 8.7 ± 0.3 | 28.5 ± 1.0 | 3.4 ± 0.2 | 5220 ± 390 | 14.5 ± 0.6 | 2.9 ± 0.1 |
| 4e | 2.79 | 3,5-F,F-Ph | 5.3 | NA c | 1.4 | NA | NA | 1.7 |
| 4f | 1.47 | 4-OHCH2-Ph | 3.04 | NA | 1.66 | NA | NA | 7.6 |
| 4g | 0.96 | 3,5-OHCH2-, OHCH2-Ph |
5.7 ± 0.2 | 27.8 ± 0.7 | 3.3 ± 0.2 | 1320 ± 80 | 5.8 ± 0.1 | 1.9 ± 0.1 |
| 4h | 2.37 | 4-F-,3-CH3O- Ph |
5.06 | NA | 3.39 | NA | NA | 3.85 |
| 4j | 3.44 | Cyclohexyl | 8.6 ± 0.2 | 27.5 ± 1.0 | 3.7 ± 0.1 | 1190 ± 490 | 9.8 ± 0.1 | 3.3 ± 0.1 |
| 4l | 1.47 | 4-NH2-Ph | 1.01 | NA | 1.55 | NA | NA | 2.52 |
| 11a | 2.39 | Ph | 4.6 ± 0.1 | 25.1 ± 2.0 | 2.8 ± 0.2 | 4180 ± 390 | 6.8 ± 0.3 | 3.1 ± 0.2 |
| 11b | 3.44 | Cyclohexyl | 12.1 ± 0.4 | 48.2 ± 2.3 | 5.3 ± 0.5 | 2750 ± 120 | 16.6 ± 0.5 | 4.3 ± 0.5 |
| 12a | 3.85 | Ph | > 30000 | > 30000 | 122 ± 9 | > 30000 | > 30000 | > 30000 |
The isoform inhibition was tested at Nanosyn http://www.nanosyn.com;
CLogP (KOWWIN) were calculated from http://146.107.217.178/lab/alogps/start.html
NA = not assayed
The IC50 values of the newly synthesized hydroxamate HDACIs are similar to those of the lead compound 4a. Most of these analogs inhibit HDAC1, 3, 10, and 6 in the nanomolar range and are slightly less potent at HDAC2 while showing only micromolar activity against HDAC8. No significant isozyme selectivity was observed in this set of structures (4a–4j, 4l, and 11a–11b). Introduction of different substituents on the terminal phenyl ring of 4a increased its HDAC inhibitory activity although these modifications failed to influence the levels of HDAC isozyme selectivity. For example, compound 4d with an electron withdrawing p-trifluoro group on the terminal phenyl ring was 2- to 3-fold more potent than compound 4a against HDAC 1, 2, 3, 10 and 6 while it was 1.9-fold less potent than compound 4a in HDAC8. Comparison of inhibitors 4l and 4a shows that the addition of a para-amino group on the terminal phenyl ring of 4a also leads to an increase in inhibitory activity against HDAC1 (18-fold), HDAC3 (5-fold), and HDAC6 (4-fold). Replacement of the terminal phenyl ring of 4a with a cyclohexyl group as in 4j, led to a 2- to 3- fold increase in potency against all tested HDACs (HDAC 1, 2, 3, 8, 10, and 6). The reversed 1,4-disubstituted triazole 11a was more potent than compound 4a against HDACs 1, 2, 3, 10, and 6. The other reversed 1,4-disubstituted triazole 11b was slightly less potent than compound 4j in all tested HDACs. Compound 12a has previously been reported by us, and it represents a benzamide-based HDACI that selectively inhibits HDAC3 over the other tested HDAC isoforms.27
Triazole-based HDACIs re-activate expression of CDK inhibitors and suppress growth of pancreatic cancer cells
Pancreatic cancer is the fourth leading cause of cancer death in the United States, and essentially remains an incurable disease with the incidence nearly equalling the mortality. Altered HDAC activity is seen to be associated with many cancers, validating HDACs as promising targets for cancer therapy. In fact, treatment of pancreatic cancer cells with the broad spectrum class I/II HDACI, SAHA (1a), leads to up-regulation of p21, growth inhibition, and to an increase in gemcitabine-induction of apoptosis.28 Furthermore, SAHA (1a) was found to enhance the antiproliferative effect of a DNA methylation inhibitor, 5-aza-2’deoxycytidine, on pancreatic cancer cells.29
In continuation of our efforts to develop HDACIs as anti-pancreatic cancer therapeutics, we assayed these newly synthesized triazoles (4b–4j, 4l, 11a–11b and 12a) against a panel of pancreatic cancer cell lines, which consisted of BxPC-3, HupT3, MiaPaca-2, Pan04.03, and SU86.86 cell lines. The effect of the tested analogues on cancer cell growth was measured by MTT assay, and the IC50 values obtained against these cell lines are summarized in Table 2. The data for SAHA (1a) and compound 4a are provided for comparison purposes. As is apparent from the data in Table 2, the majority of our HDACIs (4a–4f, 4h–4j, 4l and 11a–11b) have IC50 values equal to or less than those of SAHA (1a) with the exception of compounds 4g and 12a. Compound 4a was the most active analog against the MiaPaca-2 cells line with an IC50 value of 20 nM. Generally, compound 4a was a more effective inhibitor of cancer cell growth than compounds 4c–4g, 4i–4l, 11b, and 12a. Moreover, compounds 4h and 11a were very effective inhibitors of cancer cell growth in the HupT3 (IC50 = 50 nM) and MiaPaca-2 (IC50 = 40 nM) cancer cell lines, respectively. On the other hand, compound 4g was able to only moderately inhibit cell growth in the five pancreatic cancer cell lines. The weak inhibitory activity of compound 4g against the different pancreatic cancer cell lines might be due to its polarity, as it has a lower ClogP value than those of the other triazole analogs. Correlative studies reporting aberrant expression and/or localization of HDAC3 in various tumors imply an important role of HDAC3 in carcinogenesis.30 HDAC3 is also reported to be a repressor of ULBPs expression in epithelial cancer cells31 and to be linked to the cell cycle machinery of MiaPaCa2 cells.32 While 12a exhibits HDAC3 selectivity, its poor growth inhibition may be due to its poorer HDAC inhibitory activity.
Table 2.
Triazole-based HDACIs suppress growth of pancreatic cancer cells
| inhibition of pancreatic cancer cell lines (IC50, µM) |
|||||
|---|---|---|---|---|---|
| Compd | BxPC-3 | Hup T3 | Mia Paca-2 | Panc 04.03 | SU 86.86 |
| SAHA (1a) | 5 | 0.8 | 1.1 | 1.2 | 1.3 |
| 4a | 0.4 | 0.2 | 0.02 | < 0.5 | 0.8 |
| 4b | 1 | 0.1 | 0.1 | 0.2 | 0.6 |
| 4c | 1.1 | 1.4 | 2.33 | 2.34 | 7.7 |
| 4d | 0.9 | 0.4 | 0.9 | 1.9 | 1.5 |
| 4e | 1.2 | 0.4 | 0.8 | 2.03 | 1.7 |
| 4f | 1 | 0.2 | 0.2 | 1 | 1 |
| 4g | 29 | 7 | 14 | 27 | 37 |
| 4h | 0.2 | 0.05 | 0.1 | 0.1 | 1 |
| 4i | 1.2 | 0.5 | 0.6 | 0.7 | 1.2 |
| 4j | 4 | 0.4 | 0.1 | 0.5 | 0.1 |
| 4l | 1.2 | 0.55 | 0.88 | 0.36 | 1.45 |
| 11a | 0.9 | 0.1 | 0.04 | 0.1 | 0.2 |
| 11b | 2 | 0.4 | 0.1 | 0.5 | 0.9 |
| 12a | > 50 | > 50 | > 50 | > 50 | 32 |
To directly examine the effect on proliferation, we performed cell cycle analysis on Panc04.03 cells, which were treated for 24 h with either vehicle alone or compound 4a, the best HDACI in this series. Compared to the vehicle-treated Panc04.03 cancer cells, which show a cell cycle profile consistent with rapidly proliferating cells, Panc04.03 cancer cells treated with compound 4a demonstrate a loss of S-phase cells and an increase in the percentage of cells in G1 (Figure 3). The increase in G2/M cells in the cells treated with compound 4a may reflect additional effects on microtubule stabilization, which is frequently associated with increases in acetylated α-tubulin, which is deacetylated by HDAC6.
Figure 3.

Effect of compound 4a on cell cycles. Panc04.03 cells were treated with vehicle (DMSO) or compound 4a (5 µM) for 24 h. Cells were subsequently harvested, stained with propidium iodine and cell cycle was analyzed using flow cytometry. The percentage of cells in G1, S, and G2M are shown.
One of the key features of HDACIs is that they have the capability to re-activate tumor suppressor genes in cancer cells that can inhibit proliferation and survival of the cancer cells. The CDK inhibitor protein p21 is frequently epigenetically silenced in tumor cells, and reactivation of this protein can cause cell cycle arrest and induce senescence and apoptosis in cancer cells. We therefore examined whether treatment of the BXPC-3 cell line with compound 4a would lead to the expression of p21. Indeed, although p21 protein levels are nearly undetectable in the BXPC-3 cell line treated with vehicle (DMSO) alone, the addition of compound 4a resulted in an increased expression of p21.
As shown in Figure 5, expression of CDK inhibitors p15, p16, p21, p27 and p57 was re-activated in mouse xenograft tumors upon treatment with compound 4a.
Figure 5. CDK inhibitors are re-expressed in SU86.86 and Panc04.03 mouse xenograft tumors upon treatment with HDACI 4a.

(A) Female athymic nude mice (8–10 weeks old) were inoculated subcutaneously with 3 × 106 Su86.86 (left flank) and Panc04.03 (right flank) pancreatic cancer cells mixed with Matrigel (BD Biosciences). Two weeks after injection, tumors were size matched and mice were randomized into three treatment groups: (a) control DMSO, 4 mice; (b) compound 4a (10 mg/kg), 4 mice; (c) compound 4a (100 mg/kg), 4 mice. Established SU86.86 and Panc04.03 xenografts (tumor volume 150–200 mm3) were treated by i.p. injections with diluent (50 µL DMSO) or compound 4a (10 and 100 mg/kg; every 12 h for 48 h). (B) Tumor proteins were extracted from fresh tumor tissues taken from mouse, separated by SDS-PAGE (50 µg/well), transferred to polyvinylidene difluoride membrane, and probed with the indicated antibodies.
Conclusion
A series of substituted 1-aryl-1H-[1,2,3]triazolylphenyl-based analogs of compound 4a was designed, synthesized, and profiled for their HDAC isoform selectivity and anti-proliferative activities against pancreatic cell lines. The preliminary in vivo evaluation of compound 4a on established pancreatic tumor cell line xenografts were also determined. We found that compound 4a could re-activate expression of CDK inhibitor proteins as well as suppresses pancreatic cancer cell growth in vivo. The fluorinated analogs 4b, 4e, 4h of compound 4a may have potential in PET imaging studies. Taken together, our current findings strongly suggest that compound 4a and its related compounds may ultimately be useful in various combination treatment strategies for pancreatic cancer.
Experimental Section
Chemistry
1H NMR and 13C NMR spectra were recorded on Bruker spectrometer at 300/400 MHz and 75/100 MHz respectively with TMS as an internal standard. Standard abbreviation indicating multiplicity was used as follows: s = singlet, d = doublet, t = triplet, q = quartet, quin = quintuplet, m = multiplet and br = broad. HRMS experiment was performed on Q-TOF-2TM (Micromass). TLC was performed with Merck 250-mm 60F254 silica gel plates. Preparative TLC was performed with Analtech 1000-mm silica gel GF plates. Column chromatography was performed using Merck silica gel (40–60 mesh). HPLC was carried out on Agilent 1100 HPLC system with a Synergi 4 µ Hydro-RP 80A column, with detection at 254 on a variable wavelength detector G1314A. Method 1: flow rate = 1.4 mL/min; gradient eluation over 20 minutes, from 30% CH3OH–H2O to 100% CH3OH with 0.05% TFA. Method 2: flow rate = 1.4 mL/min; gradient eluation over 20 minutes, from 10% CH3OH–H2O to 100% CH3OH with 0.05% TFA. Method 3: flow rate = 1.4 mL/min; gradient eluation over 20 minutes, from 100% H2O to 100% CH3OH with 0.05% TFA.
7-{3-[1-(4-Fluoro-phenyl)-1H-[1,2,3]triazol-4-yl]phenylcarbamoyl}heptanoic acid methyl ester (yc-5–198, 3b)
In a mixture of 1 (100 mg, 0.34 mmol) and 1-azido-4-fluorobenzene (2b, 143 mg, 1.04 mmol) in water and ethyl alcohol (v/v = 1:1, 10 mL), sodium ascorbate (28 mg, 0.14 mmol, dissolved in 1 mL of water) was added, followed by the addition of copper 33 sulfate pentahydrate (17 mg, 0.07 mmol, dissolved in 1 mL of water). The heterogeneous mixture was stirred vigorously overnight at room temperature. The reaction mixture was diluted with EtOAc and washed thoroughly with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (hexanes/EtOAc 1:1) to afford 77 mg (52%) of 3b. Rf = 0.47 (1:1 Hexane:EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ = 1.30 (br s, 4H), 1.53 (t, J = 8.0 Hz, 2H), 1.58 (t, J = 8.0 Hz, 2H), 2.28–2.34 (m, 4H), 3.57 (s, 3H), 7.40 (t, J = 8.0 Hz, 1H), 7.49 (t, J = 8.0 Hz, 2H), 7.55 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 8.01–8.04 (m, 2H), 8.26 (s, 1H), 9.24 (s, 1H), 10.02 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.7, 25.3, 28.6, 28.7, 33.6, 36.7, 51.6, 116.3, 117.0, 117.3, 119.3, 120.3, 120.6, 122.7, 122.8, 129.7, 131.0, 133.6, 140.3, 147.7, 171.8, 173.7.
7-{3-[1-(4-Iodophenyl)-1H-[1,2,3]triazol-4-yl]phenylcarbamoyl}heptanoic acid methyl ester (yc-5–176, 3c)
Compound 3c (yield 38%) was prepared from 1 and 1-azido-4-iodobenzene (2c) according to the methodology described for the preparation of compound 3b. Rf = 0.71 (1:1 Hexane:EtOAc); 1H NMR (DMSO-d6, 300 MHz): δ = 1.28 (br s, 4H), 1.51 (t, J = 6.8 Hz, 2H), 1.58 (t, J = 6.8 Hz, 2H), 2.25–2.33 (m, 4H), 3.55 (s, 3H), 7.38 (t, J = 7.9 Hz, 1H), 7.53 (d, J = 7.5 Hz, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.97 (d, J = 8.2 Hz, 2H), 8.25 (s, 1H), 9.27 (s, 1H), 10.01 (s, 1H); 13C NMR (DMSO-d6, 75 MHz): δ = 25.1, 25.7, 29.0, 29.1, 34.0, 37.1, 51.9, 116.6, 119.7, 120.3, 121.0, 122.6, 130.1, 131.2, 137.0, 139.4, 140.7, 148.2, 172.1.
7-{3-[1-(4-Trifluoromethylphenyl)-1H-[1,2,3]triazol-4-yl]phenylcarbamoyl}heptanoic acid methyl ester (yc-5–201, 3d)
Compound 3d (yield 51%) was prepared from 1 and 1-azido-4-trifluoro-methylbenzene (2d) according to the methodology described for the preparation of compound 3b.Rf = 0.54 (1:1 Hexane:EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ = 1.31(br s, 4H), 1.55 (t, J = 8.0 Hz, 2H), 1.61 (t, J = 8.0 Hz, 2H), 2.35-2.28 (m, 4H), 3.57 (s, 3H), 7.42 (t, J = 8.0 Hz, 1H), 7.58 (t, J = 8.0 Hz, 2H), 8.03 (d, J = 8.0 Hz, 2H), 8.24 (d, J = 8.0 Hz, 2H), 8.30 (s, 1H), 9.42 (s, 1H), 10.03 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.7, 25.3, 28.6, 28.7, 33.6, 36.7, 51.6, 116.3, 119.5, 120.2, 120.7, 120.8, 125.6, 127.6, 127.7, 128.9, 129.2, 129.8, 130.7, 139.8, 140.4, 148.0, 171.8, 173.7.
7-{3-[1-(3,5-Difluorophenyl)-1H-[1,2,3]triazol-4-yl]phenylcarbamoyl}heptanoic acid methyl ester (yc-5–204, 3e)
Compound 3e (yield 59%) was prepared from 1 and 1-azido-3, 5-difluorobenzene (2e) according to the methodology described for the preparation of compound 3b. Rf = 0.55 (1:1 Hexane:EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ = 1.32 (br s, 4H), 1.55 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 2.28–2.35 (m, 4H), 3.57 (s, 3H), 7.48–7.40 (m, 2H), 7.53–7.59 (m, 2H), 7.85 (d, J = 8.0 Hz, 2H), 8.29 (s, 1H), 9.36 (s, 1H), 10.03 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.7, 25.3, 28.6, 28.7, 33.6, 36.7, 51.6, 104.1, 104.4, 116.3, 119.5, 120.6, 120.6, 125.8, 129.8, 130.6, 140.4, 148.0, 161.9, 162.1, 171.8, 173.8.
7-{3-[1-(4-Hydroxymethylphenyl)-1H-[1,2,3]triazol-4-yl]phenylcarbamoyl}heptanoic acid methyl ester (yc-5–210, 3f)
Compound 3f (yield 69%) was prepared from 1 and (4-azidophenyl)methanol (2f) according to the methodology described for the preparation of compound 3b. Rf = 0.57 (EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ = 1.32 (br s, 4H), 1.53 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 2.28–2.35 (m, 4H), 3.57 (s, 3H), 4.59 (s, 2H), 5.37 (br s, 1H), 7.40 (t, J = 8.0 Hz, 1H), 7.54–7.61 (m, 4H), 7.93 (d, J = 8.0 Hz, 2H), 8,26 (s, 1H), 9.23 (s, 1H), 10.01 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.7, 25.3, 28.6, 28.7, 33.6, 36.7, 51.6, 62.6, 116.3, 119.3, 119.9, 120.2, 120.6, 125.8, 128.0, 129.7, 131.1, 135.6, 140.3, 143.7, 147.6, 171.8, 173.8.
7-{3-[1-(3,5-Bis-hydroxymethylphenyl)-1H-[1,2,3]triazol-4-yl]phenylcarbamoyl}heptanoic acid methyl ester (yc-5–214, 3g)
Compound 3g (yield 40%) was prepared from 1 and (3-azido-5-hydroxymethylphenyl)methanol (2g) according to the methodology described for the preparation of compound 3b. Rf = 0.26 (EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ = 1.32 (br s, 4H), 1.54 (t, J = 8.0 Hz, 2H), 1.61 (t, J = 8.0 Hz, 2H), 2.28–2.35 (m, 4H), 3.58 (s, 3H), 4.62 (s, 4H), 7.38–7.41 (m, 2H), 7.59 (d, J = 8.0 Hz, 2H), 7.79 (s, 2H), 8.29 (s, 1H), 9.27 (s, 1H), 10.00 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.7, 25.3, 28.6, 28.7, 33.6, 36.7, 51.6, 62.8, 116.3, 116.4, 119.3, 119.9, 120.7, 124.6, 129.7, 131.1, 136.9, 140.3, 145.0, 147.6, 171.8, 173.8.
7-{3-[1-(3-Fluoro-4-methoxyphenyl)-1H-[1,2,3]triazol-4-yl]phenylcarbamoyl}heptanoic acid methyl ester (yc-5–217, 3h)
Compound 3h (yield 70%) was prepared from 1 and 4-azido-2-fluoro-1-methoxybenzene (2h) according to the methodology described for the preparation of compound 3b. Rf = 0.25 (1:1 Hexane:EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ = 1.32 (br s, 4H), 1.54 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 2.28–2.34 (m, 4H), 3.57 (s, 3H), 3.93 (s, 1H), 7.38–7.44 (m, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 4.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.93 (dd, J = 12.0 and 4.0 Hz, 1H), 8.25 (s, 1H), 9.20 (s, 1H), 10.01 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.7, 25.3, 28.6, 28.7, 33.6, 36.7, 51.6, 56.8, 109.1, 109.3, 114.9, 115.0, 116.2, 116.8, 119.3, 120.1, 120.6, 129.7, 130.0, 130.1, 131.0, 140.3, 147.6, 147.7, 150.5, 152.9, 171.8, 173.7
7-{3-[1-(4-tert-Butylphenyl)-1H-[1,2,3]triazol-4-yl]phenylcarbamoyl}heptanoic acid methyl ester (yc-5–207, 3i)
Compound 3i (yield 58%) was prepared from 1 and 1-azido-4-tert-butylbenzene (2i) according to the methodology described for the preparation of compound 3b. Rf = 0.47 (1:1 Hexane:EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ = 1.35–1.40 (m, 13H), 1.51 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 2.28–2.35 (m, 4H), 3.57 (s, 3H), 7.40 (t, J = 8.0 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.60–7.64 (m, 3H), 7.88 (d, J = 8.0 Hz, 2H), 8.25 (s, 1H), 9.20 (s, 1H), 10.01 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.7, 25.3, 28.6, 28.7, 31.4, 33.6, 34.9, 36.7, 51.6, 116.3, 119.3, 119.9, 120.1, 120.7, 125.8, 127.0, 129.7, 131.1, 134.7, 140.3, 147.6, 151.7, 171.8, 173.7.
7-[3-(1-Cyclohexyl-1H-[1,2,3]triazol-4-yl)phenylcarbamoyl]heptanoic acid methyl ester (yc-5–192, 3j)
Compound 3j (yield 45%) was prepared from 1 and azidocyclohexane (2j) according to the methodology described for the preparation of compound 3b. Rf = 0.30 (1:1 Hexane:EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ = 1.20–1.32 (m, 5H), 1.40–1.60 (m, 6H), 1.68 (d, J = 12.0 Hz, 1H), 1.79–1.85 (m, 4H), 2.11 (d, J = 12.0 Hz, 2H), 2.27–2.32 (m, 4H), 3.57 (s, 3H), 4.50 (t, J = 8.0 Hz, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 8.14 (s, 1H), 8.56 (s, 1H), 9.95 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.7, 25.0, 25.1, 25.3, 28.6, 28.7, 33.2, 33.6, 36.7, 51.6, 59.5, 116.1, 118.8, 119.9, 120.4, 129.6, 131.8, 140.2, 146.4, 171.7, 173.7.
7-{3-[1-(4-Nitrophenyl)-1H-[1,2,3]triazol-4-yl]phenylcarbamoyl}heptanoic acid methyl ester (yc-5–294, 3k)
Compound 3k (yield 44%) was prepared from 1 and 1-azido-4-nitrobenzene (2k) according to the methodology described for the preparation of compound 3b. Rf = 0.60 (1:1 Hexane:EtOAc); 1H NMR (DMSO-d6, 500 MHz): δ = 1.31 (br s, 4H), 1.52–1.55 (m, 2H), 1.59–1.62 (m, 2H), 2.29–2.35 (m, 4H), 3.58 (s, 3H), 7.42 (t, J = 7.5 Hz, 1H), 7.59 (d, J = 6.5 Hz, 2H), 7.93 (t, J = 8.0 Hz, 1H), 8.31 (s, 1H), 8.35 (d, J = 8.5 Hz, 1H), 8.49 (d, J = 8.5 Hz, 1H), 8.82 (s, 1H), 9.53 (s, 1H), 10.03 (s, 1H); 13C NMR (DMSO-d6, 125 MHz): δ = 24.3, 24.9, 28.2, 28.3, 33.2, 36.3, 51.1, 114.6, 115.9, 119.1, 120.0, 120.2, 123.0, 125.9, 129.3, 130.2, 131.5, 137.2, 139.9, 147.6, 148.5, 171.3, 173.3.
7-{3-[1-(4-Aminophenyl)-1H-[1,2,3]triazol-4-yl]phenylcarbamoyl}heptanoic acid methyl ester (yc-5–295, 3l)
A suspension of compound 3l (0.080 g, 0.17 mmol) and Pd/C (10 wt.%, 20 mg) in EtOAc (20 mL) was stirred under hydrogen atmosphere at room temperature for 5 h. The catalyst was removed by filtration through a pad of Celite and washed thoroughly with MeOH. The solvent was evaporated. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 2:1) to give compound 3l (0.054 g, 72%). Rf = 0.30 (1:1 Hexane:EtOAc); 1H NMR (CD3OD, 500 MHz): δ = 1.40 (br s, 4H), 1.61–1.67 (m, 2H), 1.71–1.75 (m, 2H), 2.34 (t, J = 7.5 Hz, 2H), 2.41 (t, J = 8.0 Hz, 2H), 3.65 (s, 3H), 4.61 (br s, 2H), 6.81 (d, J = 8.0 Hz, 1H), 7.10 (d, J = 8.0 Hz, 1H), 7.21 (s, 1H), 7.27 (t, J = 8.0 Hz, 1H), 7.41 (t, J = 8.0 Hz, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.64 (d, J = 7.5 Hz, 1H), 8.11 (s, 1H), 8.74 (s, 1H); 13C NMR (CD3OD, 125 MHz): δ = 25.8, 26.7, 29.9, 30.0, 34.7, 37.9, 52.0, 107.5, 109.9, 116.4, 118.4, 120.4, 121.2, 122.5, 130.5, 131.4, 132.1, 139.2, 140.5, 149.0, 150.9, 174.7, 176.0.
Octanedioic acid {3-[1-(4-fluorophenyl)-1H-[1,2,3]triazol-4-yl]phenyl}amide hydroxyamide (yc-5–199, 4b)
To a solution of hydroxylamine hydrochloride (1.96 g, 28 mmol) in 20 mL of MeOH, KOH (1.58 g, 28 mmol) was added at 40 °C for 10 min. The reaction mixture was cooled to 0 °C and filtered. Compound 3b (60 mg, 0.14 mmol) was added to the filtrate followed by KOH (0.158 g, 2.8 mmol) at room temperature for 30 min. The reaction mixture was extracted with EtOAc. The organic layer was washed with saturated NH4Cl aqueous solution and brine, dried over Na2SO4, filtered and concentrated. The residue was purified by preparative HPLC to give compound 4b (31 mg, 51%). HPLC purity: 14.1 min, 95.6% (method 2). 1H NMR (DMSO-d6, 400 MHz): δ = 1.29 (br s, 4H), 1.51 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 1.94 (t, J = 8.0 Hz, 2H), 2.32 (t, J = 8.0 Hz, 2H), 7.40 (t, J = 8.0 Hz, 1H), 7.49 (t, J = 8.0 Hz, 2H), 7.55 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 8.01–8.04 (m, 2H), 8.26 (s, 1H), 8.65 (s, 1H), 9.24 (s, 1H), 10.02 (s, 1H), 10.33 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.4, 28.8, 32.6, 36.8, 116.3, 117.0, 117.3, 171.8, 119.4, 120.3, 120.6, 122.7, 122.8, 129.7, 131.0, 133.6, 140.3, 147.7, 169.5. ESI-HRMS calculated for [C22H24FN5O3 − H]−: 424.1790; found: 424.1790.
Octanedioic acid hydroxyamide {3-[1-(4-iodophenyl)-1H-[1,2,3]triazol-4-yl]phenyl}amide (yc-5–177, 4c)
Compound 4c (yield 27%) was prepared according to the methodology described for the preparation of compound 4b. HPLC purity: 15.6 min, 95.2% (method 2). 1H NMR (DMSO-d6, 400 MHz): δ = 1.29 (br s, 4H), 1.50 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 1.94 (t, J = 8.0 Hz, 2H), 2.33 (t, J = 8.0 Hz, 2H), 7.40 (t, J = 8.0 Hz, 1H), 7.54–7.60 (m, 2H), 7.81 (d, J = 8.0 Hz, 2H), 7.99 (d, J = 8.0 Hz, 2H), 8.26 (s, 1H), 8.65 (s, 1H), 9.29 (s, 1H), 10.01 (s, 1H), 10.33 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 172.2, 169.8, 148.2, 140.7, 139.4, 137.0, 131.2, 130.1, 122.6, 121.0, 120.3, 119.8, 116.6, 37.1, 33.0, 29.2, 25.8. ESI-HRMS calculated for [C22H24IN5O3 − H]−: 532.0851; found: 532.0849.
Octanedioic acid hydroxyamide {3-[1-(4-trifluoromethylphenyl)-1H-[1,2,3]triazol-4-yl]phenyl}amide (yc-5–202, 4d)
Compound 4d (yield 39%) was prepared according to the methodology described for the preparation of compound 4b. HPLC purity: 15.6 min, 98.6% (method 2). 1H NMR (DMSO-d6, 400 MHz): δ = 1.29 (br s, 4H), 1.50 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 1.95 (t, J = 8.0 Hz, 2H), 2.33 (t, J = 8.0 Hz, 2H), 7.42 (t, J = 8.0 Hz, 1H), 7.59 (t, J = 8.0 Hz, 2H), 8.03 (d, J = 8.0 Hz, 2H), 8.25 (d, J = 8.0 Hz, 2H), 8.66 (s, 1H), 8.30 (s, 1H), 9.43 (s, 1H), 10.03 (s, 1H), 10.33 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.4, 28.8, 32.6, 36.8, 116.3, 119.5, 120.2, 120.7, 120.8, 125.8, 127.6, 127.7, 128.9, 129.8, 130.7, 139.8, 140.4, 148.0, 169.5, 171.8. ESI-HRMS calculated for [C23H24F3N5O3 − H]−: 474.1759; found: 474.1757.
Octanedioic acid {3-[1-(3,5-difluorophenyl)-1H-[1,2,3]triazol-4-yl]phenyl}amide hydroxyamide (yc-5–205, 4e)
Compound 4e (yield 41%) was prepared according to the methodology described for the preparation of compound 4b. HPLC purity: 15.0 min, 96% (method 2). 1H NMR (DMSO-d6, 400 MHz): δ = 1.29 (br s, 4H), 1.49 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 1.94 (t, J = 8.0 Hz, 2H), 2.33 (t, J = 8.0 Hz, 2H), 7.40–7.48 (m, 2H), 7.53–7.59 (m, 2H), 7.86 (d, J = 8.0 Hz, 2H), 8.29 (s, 1H), 8.65 (s, 1H), 9.36 (s, 1H), 10.03 (s, 1H), 10.33 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.4, 28.8, 32.6, 36.8, 104.1, 104.3, 104.4, 104.6, 116.3, 119.5, 120.3, 120.6, 129.8, 130.6, 138.8, 138.9, 140.4, 148.0, 161.9, 162.1, 164.4, 164.5, 169.5, 171.8. ESI-HRMS calculated for [C22H23F2N5O3 − H]−: 442.1696; found: 442.1696.
Octanedioic acid hydroxyamide {3-[1-(4-hydroxymethylphenyl)-1H-[1,2,3]triazol-4-yl]phenyl}amide (yc-5–211, 4f)
Compound 4f (yield 45%) was prepared according to the methodology described for the preparation of compound 4b. HPLC purity: 12.2 min, 95.1% (method 2). 1H NMR (DMSO-d6, 400 MHz): δ = 1.29 (br s, 4H), 1.50 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 1.94 (t, J = 8.0 Hz, 2H), 2.33 (t, J = 8.0 Hz, 2H), 4.59 (d, J = 4.0 Hz, 2H), 5.36 (t, J = 8.0 Hz, 1H), 7.40 (t, J = 8.0 Hz, 1H), 7.54–7.61 (m, 4H), 7.93 (d, J = 8.0 Hz, 2H), 8.26 (s, 1H), 8.65 (s, 1H), 9.24 (s, 1H), 10.02 (s, 1H), 10.33 (s,1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.4, 28.8, 32.6, 36.8, 62.6, 116.3, 119.3, 119.9, 120.2, 120.6, 125.8, 128.0, 129.7, 131.1, 135.6, 140.3, 143.7, 147.6, 169.5, 171.8. ESI-HRMS calculated for [C23H27N5O4− H]−: 436.1990; found: 436.1989.
Octanedioic acid {3-[1-(3,5-bis(hydroxymethyl)phenyl)-1H-[1,2,3]-triazol-4-yl]phenyl}amide hydroxyamide (yc-5–215, 4g)
Compound 4g (yield 43%) was prepared according to the methodology described for the preparation of compound 4b. HPLC purity: 11.4 min, 95.2% (method 2). 1H NMR (DMSO-d6, 400 MHz): δ = 1.29 (br s, 4H), 1.50 (t, J = 6.8 Hz, 2H), 1.60 (t, J = 6.4 Hz, 2H), 1.94 (t, J = 7.2 Hz, 2H), 2.32 (t, J = 7.6 Hz, 2H), 4.62 (d, J = 5.6 Hz, 4H), 5.41 (t, J = 6.0 Hz, 2H), 7.38–7.42 (m, 2H), 7.58 (dd, J = 8.0 and 4.0 Hz, 2H), 7.79 (s, 2H), 8.29 (s, 1H), 8.65 (d, J = 4.0 Hz, 1H), 9.27 (s, 1H), 10.01 (s, 1H), 10.33 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.4, 28.8, 32.6, 36.8, 62.8, 116.3, 116.4, 119.3, 119.9, 120.7, 124.6, 125.8, 129.7, 131.1, 136.9, 140.3, 145.0, 147.6, 169.5, 171.8. ESI-HRMS calculated for [C24H29N5O5 − H]−: 466.2096; found: 466.2095.
Octanedioic acid {3-[1-(3-fluoro-4-methoxyphenyl)-1H-[1,2,3]-triazol-4-yl]phenyl}amide hydroxyamide (yc-5–218, 4h)
Compound 4h (yield 41%) was prepared according to the methodology described for the preparation of compound 4b. HPLC purity: 14.1 min, 96% (method 2). 1H NMR (DMSO-d6, 400 MHz): δ = 1.29 (br s, 4H), 1.50 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 1.94 (t, J = 8.0 Hz, 2H), 2.33 (t, J = 8.0 Hz, 2H), 3.93 (s, 3H), 7.38–7.44 (m, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 12.0 Hz, 1H), 8.26 (s, 1H), 8.65 (s, 1H), 9.20 (s, 1H), 10.02 (s, 1H), 10.33 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.0, 25.1, 28.4, 32.3, 36.4, 56.4, 108.7, 108.9, 114.6, 115.9, 116.4, 118.9, 119.7, 120.2, 129.4, 129.6, 130.6, 140.0, 147.3, 150.1, 152.5, 169.1, 171.4. ESI-HRMS calculated for [C23H26FN5O4 + H]+: 456.2042; found: 456.2034.
Octanedioic acid {3-[1-(4-tert-butylphenyl)-1H-[1,2,3]triazol-4-yl]phenyl}amide hydroxyamide (yc-5–208, 4i)
Compound 4i (yield 33%) was prepared according to the methodology described for the preparation of compound 4b. HPLC purity: 16.3 min, 95.5% (method 2). 1H NMR (DMSO-d6, 400 MHz): δ = 1.30 (br s, 4H), 1.33 (s, 9H), 1.50 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 1.95 (t, J = 8.0 Hz, 2H), 2.33 (t, J = 8.0 Hz, 2H), 7.40 (t, J = 8.0 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.57–7.64 (m, 3H), 7.88 (d, J = 8.0 Hz, 2H), 8.25 (s, 1H), 8.65 (s, 1H), 9.20 (s, 1H), 10.02 (s, 1H), 10.34 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.4, 28.8, 31.4, 32.6, 34.9, 36.8, 116.3, 119.3, 119.9, 120.1, 120.7, 127.0, 129.7, 131.1, 134.7, 140.3, 147.6, 151.7, 169.5, 171.8. ESI-HRMS calculated for [C26H33N5O3 + H]+: 464.2656; found: 464.2653.
Octanedioic acid [3-(1-cyclohexyl-1H-[1,2,3]triazol-4-yl)phenyl]amide hydroxyamide (yc-5–193, 4j)
Compound 4j (yield 27%) was prepared according to the methodology described for the preparation of compound 4b. HPLC purity: 7.9 min, 97.4% (method 1). 1H NMR (DMSO-d6, 400 MHz): δ = 1.20–1.35 (m, 5H), 1.38–1.53 (m, 4H), 1.59 (t, J = 8.0 Hz, 2H), 1.67 (d, J = 12.0 Hz, 1H), 1.78–1.84 (m, 4H), 1.94 (d, J = 8.0 Hz, 2H), 2.09 (d, J = 12.0 Hz, 2H), 2.31 (t, J = 8.0 Hz, 2H), 4.46–4.52 (m, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 8.16 (s, 1H), 8.56 (s, 1H), 9.96 (s, 1H), 10.35 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.0, 25.1, 25.4, 28.8, 32.6, 33.2, 36.8, 59.6, 116.1, 118.8, 119.8, 120.4, 129.6, 131.8, 140.2, 146.4, 169.5, 171.7. ESI-HRMS calculated for [C22H31N5O3 − H]−: 412.2354; found: 412.2352.
Octanedioic acid {3-[1-(3-aminophenyl)-1H-[1,2,3]triazol-4-yl]phenyl}amide hydroxyamide (yc-5–292, 4l)
Compound 4l (yield 26%) was prepared according to the methodology described for the preparation of compound 4b. HPLC purity: 10.6 min, 97.1% (method 3). 1H NMR (DMSO-d6, 400 MHz): δ = 1.28 (br s, 4H), 1.49–1.70 (m, 4H), 1.94 (t, J = 8.0 Hz, 2H), 2.32 (t, J = 8.0 Hz, 2H), 5.58 (br s, 2H), 6.66 (d, J = 8.0 Hz, 1H), 7.00 (d, J = 8.0 Hz, 1H), 7.15 (s, 1H), 7.21 (dd, J = 8.0, 8.0 Hz, 1H), 7.38 (dd, J = 8.0, 8.0 Hz, 1H), 7.54–7.72 (m, 2H), 8.25 (s, 1H), 8.66 (br s, 1H), 9.09 (s, 1H), 10.00 (s, 1H), 10.33 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.7, 28.1, 31.9, 36.0, 104.5, 106.5, 113.6, 115.5, 118.4, 119.0, 119.9, 128.9, 129.8, 130.4, 137.1, 139.5, 146.7, 149.7, 168.7, 171.0. ESI-HRMS
7-(3-tert-Butoxycarbonylaminophenylcarbamoyl)heptanoic acid methyl ester (yc-5–179, 6)
To a solution of 5 (1.00 g, 4.8 mmol) and suberic acid monomethyl ester (0.903 g, 4.8 mmol) in 15 mL of dry pyridine at 0 °C was added 0.883 g (5.7 mmol) of POCl3 dropwise. This solution was stirred at 0 °C for 1h, then diluted with EtOAc and washed thoroughly with saturated aqueous KHSO4 and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:3) to give compound 6 (0.847 g, 46%). Rf = 0.55 (1:1 Hexane:EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ = 1.28 (br s, 4H), 1.46–1.55 (m, 13H), 2.24–2.30 (m, 4H), 3.57 (s, 3H), 7.00 (d, J = 8.0 Hz, 1H), 7.11 (t, J = 8.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.76 (s, 1H), 9.30 (s, 1H), 9.79 (S, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.7, 25.4, 28.5, 28.6, 28.7, 33.6, 36.7, 79.3, 109.5, 113.5, 129.0, 140.0, 140.1, 153.1, 171.5, 173.7.
7-(3-Aminophenylcarbamoyl)heptanoic acid methyl ester (yc-5–181, 7)
To a solution of compound 6 (0.800 g, 2.1 mmol) in CH2Cl2 (15 mL) at 0 °C, TFA (5 mL) was added. After 1 h, the reaction mixture was concentrated in vacuo. The crude product 7 (0.49 g, 83%) was used for next reaction directly. The analytical sample was purified by preparative thin-layer chromatography (hexanes/EtOAc 1:1). 1H NMR (DMSO-d6, 400 MHz): δ = 1.37–1.38 (m, 4H), 1.63–1.67 (m, 2H), 1.68–1.73 (m, 2H), 2.31 (t, J = 7.2 Hz, 4H), 3.67 (s, 3H), 6.43 (d, J = 7.2 Hz, 1H), 6.67 (d, J = 7.2 Hz, 1H), 7.07 (t, J = 7.6 Hz, 1H), 7.20 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.6, 25.3, 28.7, 33.9, 37.6, 51.5, 106.6, 109.6, 111.0, 129.6, 171.2, 174.2.
7-(3-Azidophenylcarbamoyl)heptanoic acid methyl ester (yc-5–182, 8)
To a solution of 7 (0.450 g, 1.6 mmol) in a 1:3 mixture of acetic acid and water (15 mL) was added NaNO2 (0.446 g, 6.4 mmol) at 0 °C and the mixture was stirred for 30 min. To this was added NaN3 (0.420 g, 6.4 mmol) at the same temperature and stirring was continued for 1h. To this was added saturated aqueous NaHCO3 solution followed by NaHCO3 powder until the mixture reached ca. pH 7. The mixture was extracted with EtOAc and the combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:4) to give compound 8 (0.28 g, 56%). Rf = 0.69 (1:1 Hexane:EtOAc); 1H NMR (CD3OD, 400 MHz): δ = 1.38–1.40 (m, 4H), 1.65 (t, J = 6.8 Hz, 2H), 1.70 (t, J = 6.4 Hz, 2H), 2.32–2.39 (m, 4H), 3.65 (s, 3H), 6.79 (d, J = 8.0 Hz, 1H), 7.25–7.33 (m, 2H), 7.50 (d, J = 1.6 Hz, 1H), 9.88 (br s, 1H); 13C NMR (CD3OD, 100 MHz): δ = 24.1, 25.1, 28.4, 33.2, 36.4, 50.5, 109.9, 113.9, 115.9, 125.8, 129.7, 140.1, 140.5, 173.3, 174.5.
7-[3-(4-Phenyl-[1,2,3]triazol-1-yl)-phenylcarbamoyl]-heptanoic acid methyl ester (yc-5–183, 10a)
In a mixture of 8 (0.12 g, 0.39 mmol) and phenylacetylene (0.06 g, 0.59 mmol) in water and ethyl alcohol (v/v = 1:1, 10 mL), sodium ascorbate (31 mg, 0.15 mmol, dissolved in 1 mL of water) was added, followed by the addition of copper 33 sulfate pentahydrate (19 mg, 0.08 mmol, dissolved in 1 mL of water). The heterogeneous mixture was stirred vigorously overnight at room temperature. The reaction mixture was diluted with EtOAc and washed thoroughly with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (hexane/EtOAc, 1:1) to afford 0.078 g (48%) of 10a. Rf = 0.31 (1:1 Hexane:EtOAc); 1H NMR (400 MHz, CD3OD): δ = 1.31 (br s, 4H), 1.53 (t, J = 8.0 Hz, 2H), 1.60 (t, J = 8.0 Hz, 2H), 2.30 (t, J = 8.0 Hz, 2H), 2.35 (t, J = 8.0 Hz, 2H), 3.57 (s, 3H), 7.39 (t, J = 8.0 Hz, 1H), 7.50 (t, J = 8.0 Hz, 2H), 7.54–7.56 (m, 2H), 7.64 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 2H), 8.36 (s, 1H), 9.26 (s, 1H), 10.22 (s, 1H); 13C NMR (DMSO-d6, 90 MHz): δ = 172.5, 141.4, 137.6, 131.0, 129.8, 129.0, 126.1, 120.4, 119.7, 115.1, 111.2, 52.0, 37.2, 34.0, 29.1, 29.0, 25.6, 25.1.
7-[3-(4-Cyclohexyl-[1,2,3]triazol-1-yl)phenylcarbamoyl]heptanoic acid methyl ester (yc-5–184, 10b)
Compound 10b (yield 40%) was prepared from 8 and Cyclohexylacetylene according to the methodology described for the preparation of compound 10a. Rf = 0.41 (1:1 Hexane:EtOAc); 1H NMR (400 MHz, CD3OD): δ = 1.40–1.59 (m, 8H), 1.61–1.69 (m, 2H), 1.71–1.80 (m, 3H), 1.86–1.89 (m, 2H), 2.10 (d, J = 12.0 Hz, 2H), 2.34 (t, J = 8.0 Hz, 2H), 2.42 (t, J = 8.0 Hz, 2H), 2.80–2.90 (m, 1H), 3.65 (s, 3H), 7.50–7.60 (m, 3H), 8.22 (s, 1H), 8,26 (s, 1H), 10.07 (br s, 1H); 13C NMR (100 MHz, CD3OD): δ = 24.7, 25.2, 26.0, 26.1, 28.6, 28.7, 32.8, 33.6, 35.0, 36.7, 51.6, 110.8, 114.6, 118.9, 119.3, 173.7, 130.5, 137.5, 140.9, 153.7, 172.0.
Octanedioic acid hydroxyamide [3-(4-phenyl-[1,2,3]triazol-1-yl)phenyl] amide (yc-5–185, 11a)
Compound 11a (yield 35%) was prepared according to the methodology described for the preparation of compound 4a. HPLC purity: 13.7 min, 99% (method 2). 1H NMR (DMSO-d6, 400 MHz): δ = 1.29 (br s, 4H), 1.50 (t, J = 8.0 Hz, 2H), 1.61 (t, J = 8.0 Hz, 2H), 1.94 (t, J = 8.0 Hz, 2H), 2.35 (t, J = 8.0 Hz, 2H), 7.39 (t, J = 4.0 Hz, 1H), 7.48–7.54 (m, 3H), 7.56 (s, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 2H), 8.36 (s, 1H), 8.65 (s, 1H), 9.26 (s, 1H), 10.22 (s, 1H), 10.33 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.3, 25.4, 28.8, 32.6, 36.8, 110.9, 114.8, 119.3, 120.0, 125.8, 128.6, 129.4, 130.6, 137.3, 147.0, 147.7, 169.5, 172.1. ESI-HRMS calculated for [C22H25N5O3 + H]+: 408.2030; found: 408.2030.
Octanedioic acid [3-(4-cyclohexyl-[1,2,3]triazol-1-yl)-phenyl]-amide hydroxyamide (yc-5–186, 11b)
Compound 11b (yield 30%) was prepared according to the methodology described for the preparation of compound 4a. HPLC purity: 9.1 min, 96.1% (method 1). 1H NMR (DMSO-d6, 400 MHz): δ = 1.29 (br s, 4H), 1.40–1.49 (m, 5H), 1.59 (t, J = 8.0 Hz, 2H), 1.68 (d, J = 12.0 Hz, 1H), 1.76 (d, J = 12.0 Hz, 2H), 1.94 (t, J = 8.0 Hz, 2H), 2.01 (d, J = 12.0 Hz, 2H), 2.33 (t, J = 8.0 Hz, 2H), 2.74 (t, J = 8.0 Hz, 1H), 7.47 (d, J = 4.0 Hz, 2H), 7.58 (d, J = 4.0 Hz, 1H), 8.25 (s, 1H), 8.47 (s, 1H), 10.16 (s, 1H), 10.32 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.3, 25.4, 26.0, 28.8, 32.6, 32.8, 35.0, 36.8, 110.8, 114.6, 118.9, 119.3, 130.5, 137.5, 140.9, 153.7, 169.5, 172.1. ESI-HRMS calculated for [C22H31N5O3 − H]−: 412.2354; found: 412.2357.
7-[3-(1-Phenyl-1H-[1,2,3]triazol-4-yl)phenylcarbamoyl]heptanoic acid (yc-5–168, 13)
To a solution of compound 12 (0.200 g, 0.49 mmol) in a mixture of MeOH (10 mL) and water (10 mL) was added LiOH·H2O (0.412 g, 9.84 mmol), and the mixture was stirred at room temperature for 1 h. The reaction mixture was acidfied with 1N HCl dropwise to pH 5 and extracted with EtOAc. The organic layer was washed with water and brine, dried over Na2SO4, and then filtered. The solvent was evaporated to give compound 13 (0.172 g, 89%). 1H NMR (DMSO-d6, 400 MHz): δ = 1.32 (br s, 4H), 1.51 (t, J = 8.0 Hz, 2H), 1.61 (t, J = 8.0 Hz, 2H), 2.20 (d, J = 8.0 Hz, 2H), 2.31 (t, J = 8.0 Hz, 2H), 7.40 (t, J = 8.0 Hz, 1H), 7.52–7.65 (m, 5H), 7.98 (d, J = 8.0 Hz, 2H), 8.26 (s, 1H), 9.26 (s, 1H), 10.01 (s, 1H), 11.92 (br s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 24.8, 25.4, 28.7, 28.8, 34.0, 36.8, 116.3, 119.3, 120.4, 120.7, 129.7, 130.3, 131.0, 137.0, 140.3, 171.7, 174.9. ESI-HRMS calculated for [C22H24N4O3 − H]−: 391.1776; found: 391.1774.
Octanedioic acid (2-aminophenyl)amide [3-(1-phenyl-1H-[1,2,3] triazol-4-yl)phenyl]amide (yc-5–169, 14)
To a stirred solution of compound 13 (0.1 g, 0.25 mmol) and 1, 2-phenyldiamine (0.275 g, 2.54 mmol) in dry DMF (10 mL) at room temperature, HOAt (0.113 g, 0.76 mmol), triethylamine (0.35 mL, 2.54 mmol) and DMAP (0.031 g, 0.25 mmol) were added sequentially and stirring was continued overnight. The reaction mixture was diluted with ethyl acetate, washed with water, saturated NaHCO3 solution, saturated NH4Cl solution, and brine, and dried over Na2SO4, filtered, and concentrated. The crude material was purified by preparative HPLC to give compound 14 (0.042 g, 34%). 1H NMR (DMSO-d6, 400 MHz): δ = 1.36 (br s, 4H), 1.61–1.63 (m, 4H), 2.29–2.36 (m, 4H), 4.81 (br s, 1H), 6.53 (t, J = 8.0 Hz, 1H), 6.71 (d, J = 8.0 Hz, 1H), 6.88 (t, J = 8.0 Hz, 1H), 7.14 (d, J = 4.0 Hz, 1H), 7.41 (t, J = 8.0 Hz, 1H), 7.50–7.65 (m, 5H), 7.98 (d, J = 8.0 Hz, 2H), 8.27 (s, 1H), 9.09 (s, 1H), 9.26 (s, 1H), 10.03 (S, 1H); 13C NMR (DMSO-d6, 100 MHz): δ = 25.5, 25.6, 28.9, 36.1, 36.8, 116.3, 116.6, 119.3, 120.4, 120.7, 124.0, 125.7, 125.8, 126.1, 129.1, 129.7, 130.3, 131.0, 137.0, 140.3, 142.3, 147.7, 171.5, 171.8. ESI-HRMS calculated for [C28H30N6O2 + H]+: 483.2503; found: 483.2502.
Biological Methods
HDACs Inhibition Assay
Purified HDACs were incubated with 1 µM carboxyfluorescein (FAM)-labeled acetylated peptide substrate and test compound for 17 hours at 25 °C in HDAC assay buffer containing 100 mM HEPES (pH 7.5), 25 mM KCl, 0.1% BSA and 0.01% Triton X-100. Reactions were terminated by the addition of buffer containing 0.078% SDS for a final SDS concentration of 0.05%. Substrate and product were separated electrophoretically using a Caliper LabChip 3000 system with blue laser excitation and green fluorescence detection (CCD2). The fluorescence intensity in the substrate and product peaks was determined using the Well Analyzer software on the Caliper system. The reactions were performed in duplicate for each sample. IC50 values were automatically calculated using the IDBS XLFit version 4.2.1 plug-in for Microsoft Excel and the XLFit 4 Parameter Logistic Model (Sigmoidal Dose-response Model): ((A + ((B−A)/1 + ((C/x)^D)))), where x is compound concentration, A is the estimated minimum and B is the estimated maximum of % inhibition, C is the inflection point and D is the Hill slope of the sigmoidal curve. The standard errors of the IC50s were automatically calculated using the IDBS XLFit version 4.2.1 plug-in for Microsoft Excel and the formula xf4_FitResultStdError().
MTS cell proliferation assay
All the pancreatic cancer cell lines were obtained from ATCC (Rockville, MD) and were maintained in a humidified environment at 37 °C with 5% CO2. BxPc-3 and HupT3 cells were grown in RPMI 1640 (Mediatech, Hercules, CA) containing 10% fetal calf serum (FBS). Panc 04.03 was grown in RPMI 1640 medium with 15% FBS. MiaPaca-2 was grown in DMEM (Mediatech, Hercules, CA) with 10% FBS. For cytotoxicity assay, cultured cells were detached with trypsin, washed and counted. An aliquot (3–5 × 103 cells) was placed in triplicate into 6-wells of a 96-well microtiter plate in a total volume of 150 µL. Four-hours post plating, 50 µL of the culture medium containing either diluent (DMSO) or varying concentrations of the indicated HDACIs from a concentration of 1 nm to 50 µM was added to each well. Cytotoxicity was measured at time 0 and 72 h post treatment using the CellTiter 96® Aqueous Non-radioactive Cell Proliferation Assay kit (Promega, Madison, WI) according to the manufacturer's instructions. The absorbance of the product formazan, which is considered to be directly proportional to the number of living cells in the culture, was measured at 490 nm using a SpectraMax M2 Microplate Reader (Molecular Devices, Sunnyvale, CA). The IC50s were calculated using XLfit (IDBS Limited, Guildford, UK).
Xenograft tumor model
Female athymic nude mice (8–10 weeks old) were inoculated subcutaneously with 3 × 106 Su86.86 (left flank) and Panc04.03 (right flank) pancreatic cancer cells mixed with Matrigel (BD Biosciences). Two weeks after injection, tumors were size matched and mice were randomized into three treatment groups: (a) control DMSO, 4 mice; (b) compound 4a (10 mg/kg), 4 mice; (c) compound 4a (100 mg/kg), 4 mice. Established SU86.86 and Panc04.03 xenografts (tumor volume 150–200 mm3) were treated by i.p. injections with Diluent (50 µL DMSO) or compound 4a (10 and 100 mg/kg; every 12 h for 48 h). After cessation of therapy, tumors were dissected from sacrificed animals. Tumor proteins were extracted from fresh tumor tissues taken from mouse, separated by SDS-PAGE (50 µg/well), transferred to polyvinylidene difluoride membrane, and probed with antibodies.
Figure 4.

HDACI 4a re-activates expression of the CDK inhibitor p21 in BXPC3 pancreatic cancer cells. The pancreatic cancer cell line BXPC3 was treated with vehicle (DMSO) or compound 4a (5 µM) for 18 h. Cell lysates were prepared, separated by SDS-PAGE, transferred to PVDF membrane and immunoblotted as indicated.
ACKNOWLEDGMENT
This work was supported in part by gift funds from an anonymous donor, by grant (no. 271210) from the ADDF/Elan, and by the Mayo Foundation and the Pancreatic Cancer SPORE P50 CA102701.
Abbreviations
- HDAC
histone deacetylase
- HDACI
histone deacetylase inhibitor
- HAT
histone acetyltransferase
- ZBG
zinc binding group
- SAHA
suberoylanilide hydroxamic acid
- TSA
trichostatin A
- CTCL
cutaneous T-cell lymphoma
- NAD
nicotinamide adenine dinucleotide
- CDK
cyclin-dependent kinase
- DNA
Deoxyribonucleic Acid
- TLC
thin-layer chromatography
- MTT
3-[4,5-dimethylthiazol-2-yl]2,5-diphenyltetrazolium bromide
REFERENCES
- 1.Butler KV, Kozikowski AP. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr. Pharm. Des. 2008;14:505–528. doi: 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]
- 2.Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat. Rev. Neurosci. 2006;7:784–796. doi: 10.1038/nrn1989. [DOI] [PubMed] [Google Scholar]
- 3.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 4.(a) Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]; (b) Kurdistani SK, Grunstein M. Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol. 2003;4:276–284. doi: 10.1038/nrm1075. [DOI] [PubMed] [Google Scholar]; (c) Struhl K, Moqtaderi Z. The TAFs in the HAT. Cell. 1998;94:1–4. doi: 10.1016/s0092-8674(00)81213-1. [DOI] [PubMed] [Google Scholar]; (d) Wolffe AP, Guschin D. Review: chromatin structural features and targets that regulate transcription. J. Struct. Biol. 2000;129:102–122. doi: 10.1006/jsbi.2000.4217. [DOI] [PubMed] [Google Scholar]
- 5.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 6.Kummar S, Gutierrez M, Gardner ER, Donovan E, Hwang K, Chung EJ, Lee MJ, Maynard K, Kalnitskiy M, Chen A, Melillo G, Ryan QC, Conley B, Figg WD, Trepel JB, Zwiebel J, Doroshow JH, Murgo AJ. Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clin. Cancer Res. 2007;13:5411–5417. doi: 10.1158/1078-0432.CCR-07-0791. [DOI] [PubMed] [Google Scholar]
- 7.Kozikowski AP, Chen Y, Gaysin A, Chen B, D'Annibale MA, Suto CM, Langley BC. Functional Differences in Epigenetic Modulators-Superiority of Mercaptoacetamide-Based Histone Deacetylase Inhibitors Relative to Hydroxamates in Cortical Neuron Neuroprotection Studies. J Med. Chem. 2007;50:3054–3061. doi: 10.1021/jm070178x. [DOI] [PubMed] [Google Scholar]
- 8.Kinzel O, Llauger-Bufi L, Pescatore G, Rowley M, Schultz-Fademrecht C, Monteagudo E, Fonsi M, Gonzalez PO, Fiore F, Steinkühler C, Jones P. Discovery of a Potent Class I Selective Ketone Histone Deacetylase Inhibitor with Antitumor Activity in Vivo and Optimized Pharmacokinetic Properties. J. Med. Chem. 2009;52:3453–3456. doi: 10.1021/jm9004303. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kelly WK, Richon VM, O'Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin. Cancer Res. 2003;9:3578–3588. [PubMed] [Google Scholar]; (b) Carducci MA, Gilbert J, Bowling MK, Noe D, Eisenberger MA, Sinibaldi V, Zabelina Y, Chen TL, Grochow LB, Donehower RC. A Phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin. Cancer Res. 2001;7:3047–3055. [PubMed] [Google Scholar]; (c) Sasakawa Y, Naoe Y, Inoue T, Sasakawa T, Matsuo M, Manda T, Mutoh S. Effects of FK228, a novel histone deacetylase inhibitor, on human lymphoma U-937 cells in vitro and in vivo. Biochem. Pharmacol. 2002;64:1079–1090. doi: 10.1016/s0006-2952(02)01261-3. [DOI] [PubMed] [Google Scholar]
- 10.(a) Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]; (b) Lin HS, Hu CY, Chan HY, Liew YY, Huang HP, Lepescheux L, Bastianelli E, Baron R, Rawadi G, Clâement-Lacroix P. Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br. J. Pharmacol. 2007;150:862–872. doi: 10.1038/sj.bjp.0707165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]; (c) Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 12.Sasakawa Y, Naoe Y, Noto T, Inoue T, Sasakawa T, Matsuo M, Manda T, Mutoh S. Antitumor efficacy of FK228, a novel histone deacetylase inhibitor, depends on the effect on expression of angiogenesis factors. Biochem. Pharmacol. 2003;66:897–906. doi: 10.1016/s0006-2952(03)00411-8. [DOI] [PubMed] [Google Scholar]
- 13.(a) Dow GS, Chen YF, Andrews KT, Caridha D, Gerena L, Gettayacamin M, Johnson J, Li Q, Melendez V, Obaldia N, Tran TN, Kozikowski AP. Antimalarial Activity of Phenylthiazolyl-Bearing Hydroxamate-Based Histone Deacetylase Inhibitors. Antimicrob. Agents Chemother. 2008;52:3467–3477. doi: 10.1128/AAC.00439-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ouaissi M, Ouaissi A. Histone deacetylase enzymes as potential drug targets in cancer and parasitic diseases. J. Biomed. Biotechnol. 2006;2006:13474. doi: 10.1155/JBB/2006/13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]; (b) Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv. Cancer Res. 2004;91:137–168. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]
- 16.Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, de los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 2003;307:720–728. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- 17.Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, Nguyen D, Wright KL, Atadja PW, Bhalla K, Pinilla-Ibarz J, Seto E, Sotomayor EM. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009;10:92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acharya MR, Sparreboom A, Venitz J, Figg WD. Rational Development of Histone Deacetylase Inhibitors as Anti-cancer Agents: A Review. Mol. Pharmacol. 2005;68:917–932. doi: 10.1124/mol.105.014167. [DOI] [PubMed] [Google Scholar]
- 20.Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002;277:25748–25755. doi: 10.1074/jbc.M111871200. [DOI] [PubMed] [Google Scholar]
- 21.(a) Guo L, Han A, Bates DL, Cao J, Chen L. Crystal structure of a conserved N-terminal domain of histone deacetylase 4 reveals functional insights into glutamine-rich domains. Proc. Natl. Acad. Sci. U.S.A. 2007;104:4297–4302. doi: 10.1073/pnas.0608041104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Neddermann P, De Francesco R, Steinkühler C, Gallinari P, Carfí A. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J. Biol. Chem. 2008;283:26694–26704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivieccio MA, Brochier C, Willis DE, Walker BA, D'Annibale MA, McLaughlin K, Siddiq A, Kozikowski AP, Jaffrey SR, Twiss JL, Ratan RR, Langley B. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. U S A. 2009;106:19599–19604. doi: 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang B, West EJ, Van KC, Gurkoff GG, Zhou J, Zhang XM, Kozikowski AP, Lyeth BG. HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 2008;1226c:181–191. doi: 10.1016/j.brainres.2008.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD. Use of the Nitrile Oxide Cycloaddition (NOC) Reaction for Molecular Probe Generation: A New Class of Enzyme Selective Histone Deacetylase Inhibitors (HDACIs) Showing Picomolar Activity at HDAC6. J. Med. Chem. 2008;51:4370–4373. doi: 10.1021/jm8002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Lopez-Sanchez M, Savoy DN, Billadeau DD, Dow GS, Kozikowski AP. A series of potent and selective, triazolylphenyl-based histone deacetylases inhibitors with activity against pancreatic cancer cells and Plasmodium falciparum. J. Med. Chem. 2008;51:3437–3448. doi: 10.1021/jm701606b. [DOI] [PubMed] [Google Scholar]
- 26.(a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (b) Torn²e CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y, He R, Chen Y, D'Annibale MA, Langley B, Kozikowski AP. Studies of benzamide- and thiol-based histone deacetylase inhibitors in models of oxidative-stress-induced neuronal death: identification of some HDAC3-selective inhibitors. ChemMedChem. 2009;4:842–852. doi: 10.1002/cmdc.200800461. [DOI] [PubMed] [Google Scholar]
- 28.Arnold NB, Arkus N, Gunn J, Korc M. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances gemcitabine-induced cell death in pancreatic cancer. Clin. Cancer Res. 2007;13:18–26. doi: 10.1158/1078-0432.CCR-06-0914. [DOI] [PubMed] [Google Scholar]
- 29.Kumagai T, Wakimoto N, Yin D, Gery S, Kawamata N, Takai N, Komatsu N, Chumakov A, Imai Y, Koeffler HP. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int. J. Cancer. 2007;121:656–665. doi: 10.1002/ijc.22558. [DOI] [PubMed] [Google Scholar]
- 30.Karagianni P, Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene. 2007;26:5439–5449. doi: 10.1038/sj.onc.1210612. [DOI] [PubMed] [Google Scholar]
- 31.Lâopez-Soto A, Folgueras AR, Seto E, Gonzalez S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: potential implications for the immunosurveillance of cancer. Oncogene. 2009;28:2370–2382. doi: 10.1038/onc.2009.117. [DOI] [PubMed] [Google Scholar]
- 32.Schneider G, Reichert M, Saur D, Hamacher R, Fritsch R, Schmid RM. HDAC3 is linked to cell cycle machinery in MiaPaCa2 cells by regulating transcription of skp2. Cell Prolif. 2007;40:522–531. doi: 10.1111/j.1365-2184.2007.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanda T, Okamoto T, Uchida Y, Nakagawa H, Iida S, Kayukawa S, Suzuki T, Oshizawa T, Suzuki T, Miyata N, Ueda R. Proteome analyses of the growth inhibitory effects of NCH-51, a novel histone deacetylase inhibitor, on lymphoid malignant cells. Leukemia. 2007;21:2344–2353. doi: 10.1038/sj.leu.2404902. [DOI] [PubMed] [Google Scholar]