

Abstract
Sepsis is a leading cause of death, which is characterized by uncontrolled inflammatory response. In this study, we report that caveolin-1, a major component of caveolae, is a critical survival factor of sepsis. We induced sepsis using a well established sepsis animal model, cecal ligation and puncture (CLP). CLP induced 67% fatality in caveolin-1 null mice, but only 27% fatality in wild type littermates (p = 0.015). Further studies revealed that mice deficient in caveolin-1 exhibited marked increase in tumor necrosis factor-α and interleukin-6 production 20 h following CLP treatment, indicating uncontrolled inflammatory responses in the absence of caveolin-1. Caveolin-1 null mice also had a significant increase in bacteria number recovered from liver and spleen, indicating elevated bacterial burdens. In addition, caveolin-1 null mice had a 2-fold increase in thymocyte apoptosis compared with wild type littermates, indicating caveolin-1 as a critical modulator of thymocyte apoptosis during sepsis. In conclusion, our findings demonstrate that caveolin-1 is a critical protective modulator of sepsis in mice. Caveolin-1 exerts its protective function likely through its roles in modulating inflammatory response, alleviating bacterial burdens, and suppressing thymocyte apoptosis.
Keywords: Apoptosis, Inflammation, Innate Immunity, Lipopolysaccharide (LPS), Lymphocyte, Thymocyte, Caveolin, Cecal Ligation and Puncture, Sepsis
Introduction
Sepsis is one of the major causes of death, which claims over 215,000 lives and costs $16.7 billion per year in America alone (1–3). The death rate from sepsis is high, exceeding 50%, due to poor understanding of the disease (4). Identifying molecules involved in sepsis, especially endogenous protective modulators, is of great importance, not only in understanding the mechanisms but also in providing new insights for efficient therapies.
Caveolae, a subset of lipid rafts, are microdomains of the plasma membrane that are enriched in cholesterol and sphingolipids (5). In addition to its specific lipid compositions, caveolae also contain abundant signaling molecules such as nitric-oxide synthase (NOS)3 and TLR4 and Src family tyrosine kinases, which provide a platform for signal transduction. Caveolin-1, a 24-kDa protein, is a major component of caveolae and has been used as a marker protein of caveolae (6). Disruption of the caveolin-1 gene leads to loss of caveolae (7), indicating an essential role of caveolin-1 in caveolae formation. Given the importance of caveolae in signal transduction, it is not surprising that caveolin-1 has been implicated in a variety of cellular processes such as endocytosis, phagocytosis, and cholesterol trafficking (5–6, 8).
Evidence from the caveolin-1 null mouse model has established an inhibitory role of caveolin-1 in cell proliferation and tissue homeostasis. For example, mice deficient in caveolin-1 display hypercellularity in lungs and heart (7, 9–10). A number of studies also suggest a role of caveolin-1 in apoptosis. However, the role of caveolin-1 in regulating apoptotic cell death is controversial and seems to be cell type-specific and depend on stimuli. For example, knockdown of caveolin-1 by short hairpin RNA sensitized TRAIL-induced apoptosis in HepG2 cells, and overexpression of caveolin-1 partially blocks TRAIL-induced apoptosis (11). However, deletion of caveolin-1 protects hyperoxia-induced apoptosis via modulation of survivin expression (12). Using a γ-irradiation-induced apoptosis model, Li et al. (13) reported an increase in the number of apoptotic cells in the thymus and spleen of caveolin-1 null mice following γ-irradiation. Interestingly, Volonte et al. (14) showed that caveolin-1 is required for doxorubicin-induced apoptosis in the atria. Lymphocyte apoptosis is a major event in sepsis. Whether caveolin-1 plays a role in lymphocyte apoptosis during sepsis is unknown.
Recent studies revealed that caveolin-1 may play a role in immunity. Caveolin-1 expression is markedly up-regulated in response to LPS stimulus in macrophages (15), and the expression of caveolin-1 attenuates LPS-induced cytokine production in macrophages (16). Using an endotoxemia animal model, Garrean et al. (17) reported that mice lacking caveolin-1 are more resistant to LPS-induced inflammatory injury to the lung. Paradoxically, two recent studies revealed that caveolin-1 null mice display a significant decrease in survival when challenged with Salmonella enterica sv. Typhimurium (16) or Pseudomonas aeruginosa (18). These studies suggest that caveolin-1 may respond to LPS and bacterial infections differently, and caveolin-1 may actually play a protective role in sepsis. Although the endotoxemia animal model is widely used for the study of sepsis, and LPS plays an essential role in sepsis, the endotoxemia animal model does not fully mimic the changes observed in sepsis (19). For example, TLR4 mutant mice are resistant to LPS-induced endotoxic animal death but are highly susceptible to bacteria-induced septic death (20–22). Given the distinct differences between endotoxemia and sepsis, and a limited number of studies aimed to clarify the significance of caveolin-1 in sepsis, we have assessed the role of caveolin in sepsis using a well established and more clinically relevant sepsis animal model, cecal ligation and puncture (CLP) (23). We demonstrate that mice deficient in caveolin-1 are more susceptible to polymicrobial septic death than wild type littermates. Further studies reveal that caveolin-1 protects against septic death likely through its roles in modulating inflammatory response, alleviating bacterial burdens and suppressing thymocyte apoptosis.
EXPERIMENTAL PROCEDURES
Materials
Fluorescent-conjugated antibody against CD3 (17A2), CD4 (GK1.5), CD8 (53–6.7) or CD19 (ID3), and blood agar plate (MacConkey II) were from BD Biosciences. ELISA kits for quantifying TNF-α and IL-6 were from eBioscience. The ELISA kit for quantifying nitrite/nitrate (NOx) kit was from Cayman Chemical, and the ELISA kit for quantifying LPS was from Charles River. Annexin V/PI and TUNEL apoptotic assay kits were from Roche Applied Science.
Caveolin-1 Null Mice
Caveolin-1 (Cav-1) null mice on C57BL/6 × 129 background were obtained from the Jackson ImmunoResearch Laboratories. To eliminate the effect of background, Cav-1+/− mice were used for breeding, and Cav-1−/− and Cav-1+/+ littermates were used for this study. PCR genotyping was performed using a protocol provided by Jackson ImmunoResearch Laboratories. The animals were fed with a standard laboratory chow diet. Animal care and experiments were approved by the Institutional Animal Care and Use Committee of the University of Kentucky.
CLP Septic Animal Model
CLP was performed as described previously (24). Briefly, 10- to 12-week-old mice were anesthetized by inhalation of 2–5% isoflurane in 100% oxygen using anesthesia equipment. A midline incision (1.0 cm) was made below the diaphragm. The cecum was isolated, fully ligated, punctured twice with a 22-gauge needle, and gently compressed to extrude a small amount of cecal material. The cecum was returned to the abdomen, and the muscle and skin incisions were closed with 6–0 Ethilon suture material. The mice were resuscitated subsequently with 1 ml phosphate-buffered saline subcutaneously. Sham animals were similarly treated without ligation/puncture of the cecum.
Determination of Cytokine and Nitric Oxide Levels
10- to 12-week-old mice were euthanized by CO2 inhalation 2, 4, 8, and 20 h following CLP. The blood was obtained by cardiac puncture. The serum nitrite/nitrate (NOx) levels were measured with a nitrite/nitrate kit, which were used to estimate the generation of NO in vivo; the serum TNF-α, IL-6, corticosterone, or LPS levels were quantified with corresponding ELISA kits.
Myeloperoxidase (MPO) Activity Assay
10- to 12-week-old mice were euthanized by CO2 inhalation 2, 4, 8, and 20 h following CLP. The lungs were collected, and 50 mg of lung was homogenized in 1 ml of potassium phosphate-buffered solution (50 mm, pH 6) containing 50 mm hexadecyltrimethylammonium bromide. The homogenates were sonicated, run through two freeze-thaw cycles, and centrifuged at 14,000 rpm for 20 min. The supernatant was then collected and mixed 1/30 (v/v) with assay buffer (0.167 mg/ml o-dianisidine hydrochloride and 0.0005% H2O2). Absorbance change was recorded at 460 nm for 3 min, and MPO activity was calculated as the change in absorbance per min and normalized by wet lung weight.
Determination of Pulmonary Microvascular Permeability to Liquid
Caveolin-1 null and wild type littermates were challenged with CLP for 8 h. Then, the mice were anesthetized with an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (2.5 mg/kg). The lungs and trachea were surgically exposed. A tracheal cannula was inserted for positive-pressure ventilation (rate, 120 breaths/min; peak inspiration pressure, 8–10 cm H2O). A polyethylene perfusion cannula (PE-90) was advanced into the pulmonary artery via a pulmonic valve that was accessed through a small incision in the right ventricle, and the cannula was secured by means of a suture encompassing the pulmonary artery and aorta. The lungs were perfused with RPMI 1640 medium at a constant flow rate (2 ml/min) using a peristaltic pump. The lung and heart were rapidly excised from thoracic cavity and suspended from a steel wire hook attached to a force displacement transducer (FT03, Grass Telefactor, West Warwick, RI). The lung weight was zeroed, and subsequent weight change due to gain or loss of fluid from the lung was recorded.
Analysis of Bacteremia and Bacterial Burden
The assays were performed as described previously (24). Briefly, the blood and tissues were collected from caveolin-1 null and wild type littermates 20 h following CLP challenge. To determine bacteriaemia, the blood was diluted 1:100 and 1:1000 with phosphate-buffered saline, and 20 μl of the diluted blood was plated on a MacConkey II agar plate. After a 24-h incubation at 37 °C, the number of clones was counted. To determine the bacterial burden, total DNA was isolated from 30 mg of liver or spleen using DNeasy kit (Qiagen), and quantitative PCR was performed with bacteria specific primers (forward, 5′-GAGGAAGGIGIGGAIGACGT-3′; reverse, 5′-AGGAGGTGATCCAACCGCA-3′) using a certain number of DH-5α bacteria as standard.
Flow Cytometry Analysis of Lymphocyte Apoptosis and Homeostasis during Sepsis
Massive lymphocytes die during sepsis due to apoptosis. We analyzed lymphocyte apoptosis with two independent methods, annexin V/PI and TUNEL staining. We also determined T and B lymphocyte homeostasis. Briefly, CLP was performed on 10- to 12-week-old mice. After 18 h, the thymi and spleens were harvested, and single cell suspension was prepared using Stomacher 80 (Seward). The splenocytes were incubated in ACK lysing buffer to remove red blood cells. The cells were then labeled with annexin V/PI, TUNEL, and lymphocyte markers following the manufacturer's instructions. Apoptotic cells were quantified with flow cytometer FACSCalibur (BD Biosciences).
Statistical Analysis
The survival assay was analyzed by a Log-Rank x2 test using SAS software. Significance in experiments comparing two groups was determined by a two-tailed Student's t test. Values are reported as the mean ± S.D. A value of p < 0.05 was considered significant.
RESULTS AND DISCUSSION
Caveolin-1 Protects against Septic Death in Mice
We employed a CLP sepsis model to determine the role of caveolin-1 in polymicrobial sepsis. In contrast to resistant to LPS-induced endotoxic animal death in caveolin-1 null mice (17), caveolin-1 null mice were more susceptible to CLP-induced septic death (p = 0.015, Fig. 1). CLP treatment induced 67% fatality in caveolin-1 null mice but only 27% fatality in wild type littermates. This finding indicates that caveolin-1 is a critical protective factor of sepsis, and caveolin-1 plays distinct roles in endotoxemia and sepsis.
FIGURE 1.
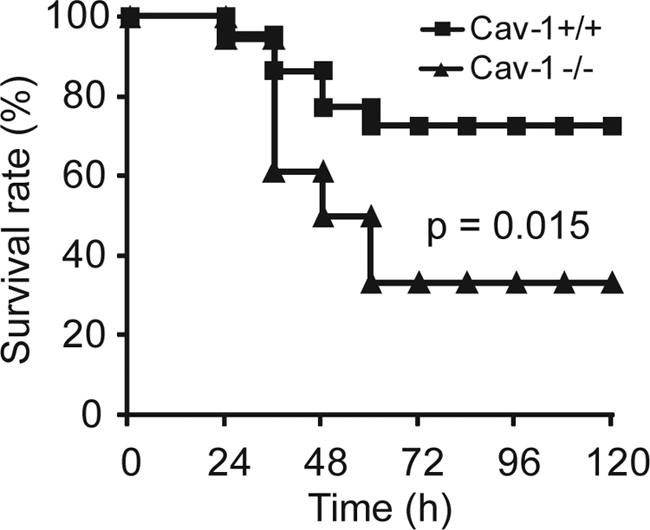
Caveolin-1 protects against septic death in mice. Caveolin-1 null mice (solid triangle, n = 18) and wild type littermates (solid square, n = 22) were treated with CLP, and survival was observed for 5 days. The data are expressed as the percentage of mice surviving at indicated times. p = 0.015 versus wild type.
Effect of Caveolin-1 on CLP-induced Lung Injury
Caveolin-1 is abundantly expressed in lungs. Using an LPS-induced endotoxemia animal model, Garrean et al. (17) showed that the deficiency of caveolin-1 protects against LPS-induced lung injury as assessed by lower MPO activity, less edema formation, and lower micro vascular permeability. We assessed the effect of caveolin-1 on CLP-induced lung injury by quantifying MPO activity, edema formation, and microvascular permeability. As shown in Fig. 2a, MPO activity was markedly increased 2 h following CLP treatment. However, there was no difference in MPO activity between caveolin-1 null and wild type control mice. Edema formation was determined by measuring the lung wet to dry ratio 8 h following CLP treatment. No increase in the wet/dry ratio was observed in CLP-treated wild type mice compared with sham-treated wild type mice (Fig. 2b). This finding is consistent with early studies showing that CLP does not induce edema formation in wild type mice (25). Interestingly, there was no increase in wet/dry ratio in CLP-treated caveolin-1 null compared with sham-treated caveolin-1 null mice (Fig. 2b), indicating no edema formation in caveolin-1 null mice during sepsis. We then used the isolated-perfused lung model to determine lung microvessel infiltration coefficient (Kf,c), a quantitative measurement of vascular endothelial permeability to liquid. As shown in Fig. 2c, no changes in Kf,c value were observed between sham- and CLP-treated cavolin-1 null mice. We observed a small increase in the Kf,c value in lungs from CLP-treated wild type mice compared with sham-treated wild type mice, but the difference was not statistically significant. Of note, compared with the marked increase in Kf,c value in LPS-challenged wild type mice (17), the Kf,c value observed in CLP-challenged wild type mice was 10-fold less than the Kf,c value observed in LPS-treated mice, which indicates that the lung injury in CLP-treated wild type mice is milder compared with LPS-treated wild type mice. Taken together, the above data indicate that CLP treatment does not cause significant lung injury in caveolin-1 null mice. Therefore, it is unlikely that lung injury accounts for the increased fatality observed in caveolin-1 null mice.
FIGURE 2.
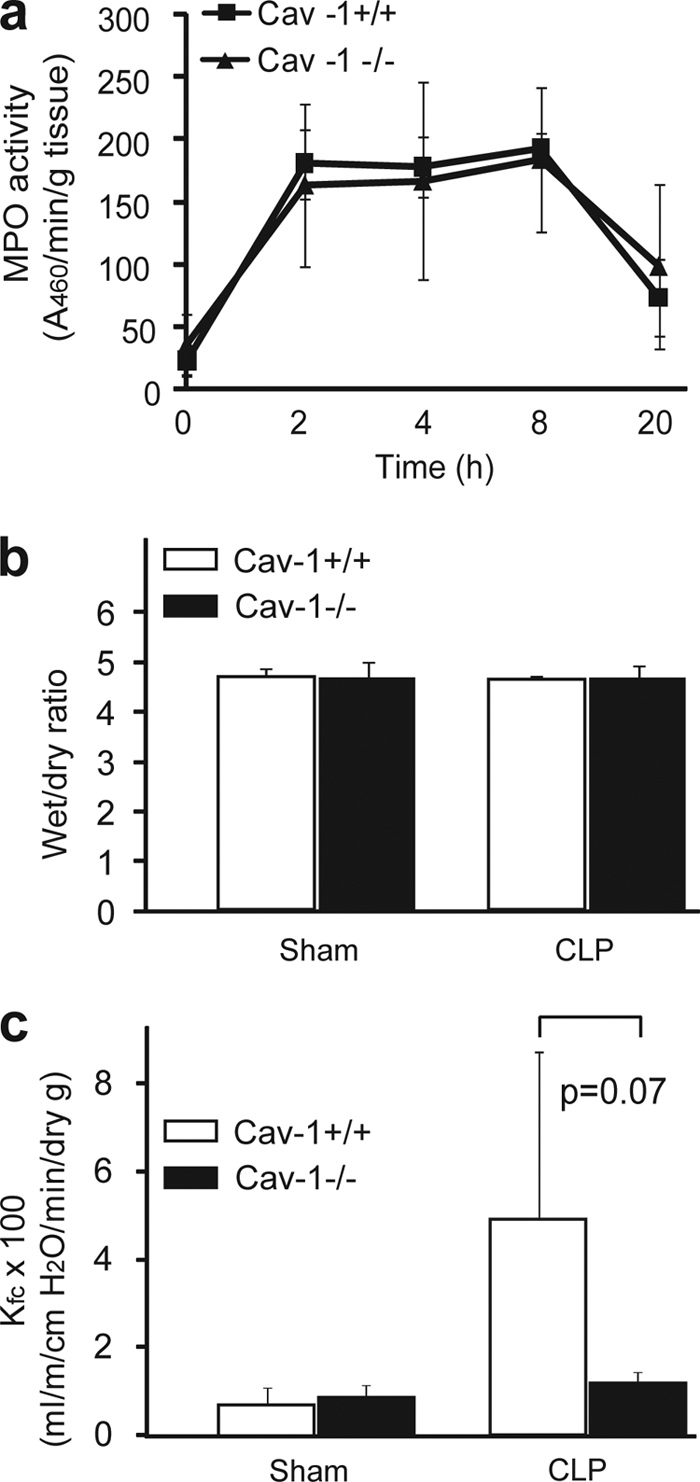
Effect of caveolin-1 on CLP-induced lung injury. a, effect of caveolin-1 on MPO activity. Caveolin-1 null mice (solid triangle) and wild type littermates (solid square) were treated with CLP for 2, 4, 8, and 20 h, and MPO activity in lung was quantified (n = 6 per group, mean ± S.D.). b, effect of caveolin-1 on pulmonary edema formation. Caveolin-1 null mice (solid) and wild type littermates (open) were treated with CLP or sham. After 8 h, the lung was collected, and wet/dry ratio was determined (n = 5 per group, mean ± S.D.). c, effect of caveolin-1 on pulmonary microvascular permeability. Caveolin-1 null mice (solid) and wild type littermates (open) were treated with CLP or sham for 8 h. Then, the lungs were isolated, and lung microvascular permeability was assessed by measuring microvessel infiltration coefficient (Kf,c) (n = 5 per group, mean ± S.D.).
Uncontrolled Inflammatory Response in Caveolin-1 Null Mice during Sepsis
Rapid innate immune response that generates high levels of inflammatory cytokines constitutes the first line of a self defense system against bacterial infections. However, uncontrolled production of cytokines is a major cause of septic death (26). To understand why caveolin-1 null mice are susceptible to septic death, we assessed inflammatory cytokine production in CLP-treated caveolin-1 null and wild type littermates. We treated the mice with CLP for 2, 4, 8, and 20 h and determined the serum cytokine levels. As shown in Fig. 3, caveolin-1 null and wild type mice exhibited distinct inflammatory responses during sepsis. Wild type mice exhibited a typical acute phase response with a rapid induction of TNF-α and IL-6 in the early stage of sepsis (2 to 4 h) and significant decrease in TNF-α and IL-6 levels thereafter. In contrast, caveolin-1 null mice exhibited exaggerated inflammatory cytokine generation during sepsis. The serum TNF-α and IL-6 levels continued to increase 4 h after CLP treatment in caveolin-1 null mice and 3–4-fold higher than that of wild type littermates by 20 h, which indicates a prolonged and uncontrolled cytokine generation in the absence of caveolin-1 (Fig. 3, a and b).
FIGURE 3.
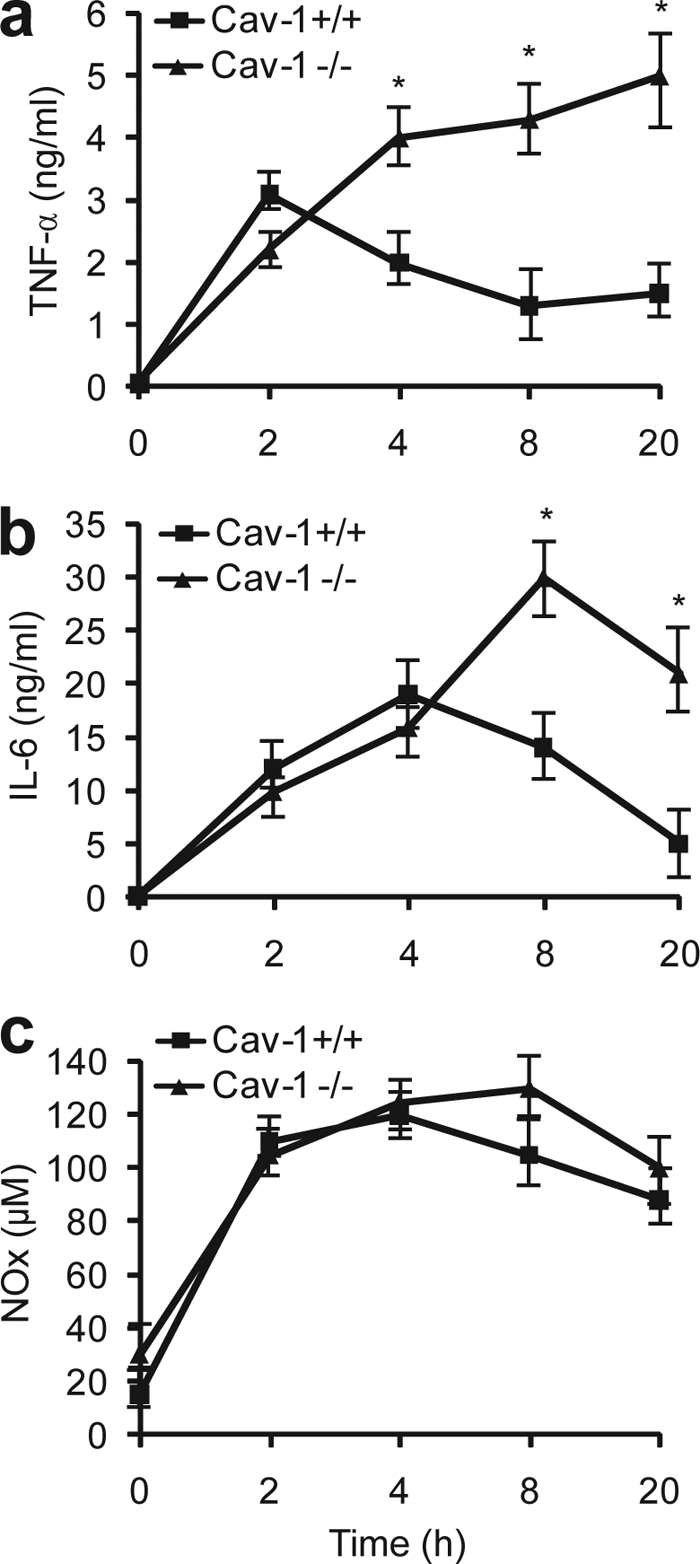
Uncontrolled inflammatory response in caveolin-1 null mice during sepsis. Caveolin-1 null mice and wild type littermates were treated with CLP for 2, 4, 8, and 20 h, and the serum concentrations of TNF-α (a), IL-6 (b), and NOx (c) were quantified. n = 6 per group with triplicate measurements, mean ± S.D. *, p < 0.05 versus wild type.
Like cytokines, NO is another key molecule in innate immune response during sepsis (27). Upon bacterial infections, iNOS expression in macrophages is dramatically induced to generate high level of NO, which is used to kill bacteria and modulate immune reactions (27). However, too much NO is deleterious and can cause oxidative cell damage (28). Given the fact that caveolin-1 inhibits eNOS activity through its binding to eNOS (29), and there is a temporal reduction in iNOS expression in LPS-treated eNOS null mice (30), it is plausible to predict that mice deficient in caveolin-1 may have augmented NO production during sepsis. We measured serum nitrite and nitrate (NOx) levels to determine whether caveolin-1 affects NO production in sepsis. Unexpectedly, caveolin-1 null mice exhibited similar NO production compared with wild type littermates during sepsis. Both caveolin-1 null mice and wild type littermates showed a rapid production of NO in the early stage of sepsis (2 to 8 h) and gradual decrease in NO levels by 20 h (Fig. 3c). Our data indicate that the NO production is unlikely responsible for the increased septic death observed in caveolin-1 null mice.
Available evidence suggests that caveolin-1 may regulate inflammatory response through its role in macrophages. The expression of caveolin-1 in macrophages is up-regulated in response to LPS stimulus (15). Furthermore, numerous studies demonstrated that caveolin-1 attenuates LPS-induced cytokine production in macrophages (16, 31–32). Utilizing RAW264 and peritoneal macrophages, Wang et al. (31–32) demonstrated that caveolin-1 attenuates inflammatory cytokine generation. The authors further showed that caveolin-1 inhibits LPS-TLR4 signaling by directly binding to TLR4, which prevents TLR4 association with MyD88 and TIR-domain-containing adapter-inducing interferon-β, and by suppressing the activation of extracellular signal-regulated kinase (ERK)1/2 MAPK, c-Jun N-terminal kinase (JNK) MAPK, and phosphatidylinositol 3-kinase. To test whether caveolin-1 plays a role in macrophages in vivo, we elucidated the effect of caveolin-1 on inflammatory cytokine expression in liver, the largest pool of macrophages in vivo. Quantitative real time-PCR analysis revealed that caveolin-1 null mice have a >2-fold increase in TNF-α and IL-6 expression than that of wild type controls 4 h post-CLP challenge. Given that macrophages are major cells producing inflammatory cytokines during sepsis (33–34), these in vitro and in vivo data provide a mechanistic explanation, at least partly, for our findings that caveolin-1 attenuates systemic inflammatory response during sepsis and protects against septic death.
Elevated Bacterial Burdens in Caveolin-1 Null Mice in Sepsis
Caveolae have been implicated in endocytosis and phagocytosis. A number of in vitro studies showed that caveolin-1 is involved in the uptake of bacteria (13, 18, 35). A more recent report showed that mice deficient in caveolin-1 have elevated bacterial burden when infected with P. aeruginosa (18). These findings suggest that caveolin-1 might play a role in bacterial clearance in sepsis. To further explore the mechanisms underlying caveolin-1 protection against polymicrobial sepsis, we investigated the effects of caveolin-1 on bacterial burden. We collected blood from CLP-challenged mice and quantified bacteria using blood agar culture. As shown in Fig. 4a, caveolin-1 null mice tended to have a higher bacteria number compared with wild type littermates 20 h after CLP challenge, though the difference is not statistically significant due to variation among mice (p = 0.28). We further determined bacterial burdens in the liver and spleen in CLP-challenged mice. Caveolin-1 null mice had a significant increase in bacteria number in the liver and spleen compared with wild type littermates (Fig. 4, b and c).
FIGURE 4.

Elevated bacterial burdens in caveolin-1 null mice in sepsis. Caveolin-1 null mice (solid) and wild type littermates (open) were treated with CLP for 20 h. The number of bacteria in blood (a), liver (b), and spleen (c) were determined as described under “Experimental Procedures.” n = 10 per group with triplicate measurements, mean ± S.D. *, p < 0.05 versus wild type.
We notice that there is a disconnection between enhanced inflammation and compromised bacterial killing in CLP-challenged caveolin-1 null mice. Two important functions of caveolin-1 may contribute to this paradoxical phenomenon. Utilizing RAW264 and peritoneal macrophages, Wang et al. (31–32) demonstrated that caveolin-1 attenuates inflammatory cytokine generation. Macrophages are major cells responsible for cytokine generation. These in vitro data provide an explanation for enhanced systemic inflammatory response observed in mice deficient in caveolin-1 during sepsis; utilizing in vitro phagocytosis assay, Li et al. (13) demonstrated that peritoneal macrophages from caveolin-1 null mice exhibit impaired phagocytosis of Escherichia coli. Macrophages also play a key role in bacterial clearance. This decrease in phagocytic ability observed in caveolin-1 null macrophages provides an explanation for increased bacterial burdens during sepsis. Our findings uncover a role of caveolin-1 in alleviating bacterial burdens in sepsis and warrant further effort to determine how caveolin-1 regulates bacteria uptake and removal during sepsis.
Elevated Thymocyte Apoptosis and Altered Lymphocyte Homeostasis in Caveolin-1 Null Mice during Sepsis
During sepsis, massive lymphocytes die from apoptosis, which is a major problem in sepsis (36–37). Available evidence shows that alleviating apoptosis provides protection against septic animal death (38). We investigated lymphocyte apoptosis in caveolin-1 null and wild type littermates 18 h following CLP treatment with both annexin V/PI and TUNEL assays. As shown in Fig. 5, cavaeolin-1 null mice exhibited a significant increase in apoptosis in thymus as assessed by a 2-fold increase in annexin V+/PI− thymocytes (Fig. 5, a and c). TUNEL staining also clearly revealed a 2-fold increase in thymocyte apoptosis in the thymus of caveolin-1 null mice (Fig. 5, b and c). Furthermore, compared with wild type littermates, caveolin-1 null mice had a significant decrease in CD4+CD8+ thymocytes ratio, and increases in CD4−CD8− and CD4−CD8+ thymocytes ratio in thymus (Fig. 5d), indicating altered T lymphocyte homeostasis in the absence of caveolin-1 during sepsis. Although there was no difference in splenocyte apoptosis in spleen (Fig. 5e), caveolin-1 null mice exhibited altered lymphocyte homeostasis illustrated by a significant increase in CD19+ B cell ratio and a decrease in CD3+ T cell ratio in spleen (Fig. 5f). To examine whether caveolin-1 affects lymphocyte homeostasis in normal circumstances, we analyzed the number and ratio of lymphocyte in the thymus and spleen from caveolin-1 null and wild type littermates without CLP challenge. No significant difference was observed (data not shown). These findings indicate caveolin-1 specifically modulates lymphocyte apoptosis and homeostasis in sepsis. To the best of our knowledge, this is the first report showing caveolin-1 as a negative modulator of lymphocyte apoptosis during sepsis. Adaptive immune cells temper initial innate responses (39–40). It is possible that the marked increase in lymphocyte apoptosis observed in caveolin-1 null mice may have rendered this negative regulatory mechanism ineffective, which contributes to enhanced inflammatory response during sepsis.
FIGURE 5.

Elevated thymocyte apoptosis and altered lymphocyte homeostasis in caveolin-1 null mice during sepsis. Caveolin-1 null mice and wild type littermates were treated with CLP for 18 h. Apoptosis in thymus (a–c) and in spleen (e) was analyzed with annexin V/PI (AV) and TUNEL methods. Annexin V+PI− (AV+PI−) and TUNEL+ staining indicate apoptotic cells. The lymphocyte homeostasis in thymus (d) and spleen (f) was analyzed with a flow cytometer. n = 6 per group, mean ± S.D. *, p < 0.05 and **, p < 0.01 versus wild type.
Apoptosis is a complex process that involves multiple regulatory mechanisms. A number of key apoptotic signaling molecules have been found in caveolae, such as Fas, tumor necrosis factor receptor 1, Fas-associated death domain protein (FADD), and ceramide (41–43). Caveolae provide a platform for the assembly of these signaling molecules. Given the critical roles of caveolin-1 in caveolae formation and in modulating signaling in caveolae, it is plausible that caveolin-1 may affect apoptosis through modulating signaling in caveolae. In addition to a direct regulation of apoptotic signaling, caveolin-1 may be involved in the clearance of apoptotic cells. Utilizing in vitro phagocytosis assay of apoptotic thymocytes, a recent study by Li et al. (13) showed that the loss of caveolin-1 decreases the phagocytic ability of peritoneal macrophages. It is possible that the increase in thymocyte apoptosis observed in caveolin-1 null mice is caused by a decrease in phagocytic clearance of apoptotic cells by macrophages. Further study is warranted to determine the detailed mechanisms underlying caveolin-1 modulating lymphocyte apoptosis and its contribution to modulating inflammatory response during sepsis.
In summary, we employed a clinically relevant CLP sepsis animal model to determine the role of caveolin-1 in sepsis. We demonstrates that caveolin-1 is a critical survival factor of polymicrobial sepsis in mice. Further studies reveal that caveolin-1 protects against septic death through its roles in modulating inflammatory response, alleviating bacterial burdens, and suppressing lymphocyte apoptosis. Our findings uncover caveolin-1 as a protective modulator of sepsis, which may provide a novel target for the intervention of sepsis.
Acknowledgments
We thank the members of the Kentucky Pediatric Research Institute for invaluable advice and assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM085231 and 3R01GM085231-02S1. This work was also supported by the American Heart Association (0530241N) and the Children's Miracle Network (to X.-A. L.).
- NOS
- nitric-oxide synthase
- Cav-1
- caveolin-1
- CLP
- cecal ligation and puncture
- MPO
- myeloperoxidase
- NOx
- nitrite and nitrate
- PI
- propidium iodide
- TLR4
- Toll-like receptor 4
- TNF
- tumor necrosis factor
- IL
- interleukin
- TUNEL
- terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling
- ELISA
- enzyme-linked immunosorbent assay
- MAPK
- mitogen-activated protein kinase
- iNOS
- inducible nitric-oxide synthase
- eNOS
- endothelial nitric-oxide synthase.
REFERENCES
- 1.Dombrovskiy V. Y., Martin A. A., Sunderram J., Paz H. L. (2007) Crit. Care Med. 35, 1244–1250 [DOI] [PubMed] [Google Scholar]
- 2.Martin G. S., Mannino D. M., Eaton S., Moss M. (2003) N. Engl. J. Med. 348, 1546–1554 [DOI] [PubMed] [Google Scholar]
- 3.Riedemann N. C., Guo R. F., Ward P. A. (2003) J. Clin. Invest. 112, 460–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sessler C. N., Perry J. C., Varney K. L. (2004) Curr. Opin. Crit. Care 10, 354–363 [DOI] [PubMed] [Google Scholar]
- 5.Li X. A., Everson W. V., Smart E. J. (2005) Trends Cardiovasc. Med. 15, 92–96 [DOI] [PubMed] [Google Scholar]
- 6.Williams T. M., Lisanti M. P. (2004) Genome Biol. 5, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drab M., Verkade P., Elger M., Kasper M., Lohn M., Lauterbach B., Menne J., Lindschau C., Mende F., Luft F. C., Schedl A., Haller H., Kurzchalia T. V. (2001) Science 293, 2449–2452 [DOI] [PubMed] [Google Scholar]
- 8.Lajoie P., Goetz J. G., Dennis J. W., Nabi I. R. (2009) J. Cell Biol. 185, 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen A. W., Park D. S., Woodman S. E., Williams T. M., Chandra M., Shirani J., Pereira de Souza A., Kitsis R. N., Russell R. G., Weiss L. M., Tang B., Jelicks L. A., Factor S. M., Shtutin V., Tanowitz H. B., Lisanti M. P. (2003) Am. J. Physiol. Cell Physiol. 284, C457–474 [DOI] [PubMed] [Google Scholar]
- 10.Razani B., Engelman J. A., Wang X. B., Schubert W., Zhang X. L., Marks C. B., Macaluso F., Russell R. G., Li M., Pestell R. G., Di Vizio D., Hou H., Jr., Kneitz B., Lagaud G., Christ G. J., Edelmann W., Lisanti M. P. (2001) J. Biol. Chem. 276, 38121–38138 [DOI] [PubMed] [Google Scholar]
- 11.Zhao X., Liu Y., Ma Q., Wang X., Jin H., Mehrpour M., Chen Q. (2009) Biochem. Biophys. Res. Commun. 378, 21–26 [DOI] [PubMed] [Google Scholar]
- 12.Zhang M., Lin L., Lee S. J., Mo L., Cao J., Ifedigbo E., Jin Y. (2009) Am. J. Physiol. Lung Cell Mol. Physiol. 297, L945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J., Scherl A., Medina F., Frank P. G., Kitsis R. N., Tanowitz H. B., Sotgia F., Lisanti M. P. (2005) Cell Cycle 4, 1599–1607 [DOI] [PubMed] [Google Scholar]
- 14.Volonte D., McTiernan C. F., Drab M., Kasper M., Galbiati F. (2008) Am. J. Physiol. Heart Circ. Physiol. 294, H392–401 [DOI] [PubMed] [Google Scholar]
- 15.Lei M. G., Tan X., Qureshi N., Morrison D. C. (2005) Infect. Immun. 73, 8136–8143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medina F. A., de Almeida C. J., Dew E., Li J., Bonuccelli G., Williams T. M., Cohen A. W., Pestell R. G., Frank P. G., Tanowitz H. B., Lisanti M. P. (2006) Infect. Immun. 74, 6665–6674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garrean S., Gao X. P., Brovkovych V., Shimizu J., Zhao Y. Y., Vogel S. M., Malik A. B. (2006) J. Immunol. 177, 4853–4860 [DOI] [PubMed] [Google Scholar]
- 18.Gadjeva M., Paradis-Bleau C., Priebe G. P., Fichorova R., Pier G. B. (2010) J. Immunol. 184, 296–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eskandari M. K., Bolgos G., Miller C., Nguyen D. T., DeForge L. E., Remick D. G. (1992) J. Immunol. 148, 2724–2730 [PubMed] [Google Scholar]
- 20.Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 21.Echtenacher B., Freudenberg M. A., Jack R. S., Männel D. N. (2001) Infect. Immun. 69, 7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vazquez-Torres A., Vallance B. A., Bergman M. A., Finlay B. B., Cookson B. T., Jones-Carson J., Fang F. C. (2004) J. Immunol. 172, 6202–6208 [DOI] [PubMed] [Google Scholar]
- 23.Rittirsch D., Huber-Lang M. S., Flierl M. A., Ward P. A. (2009) Nat. Protocols 4, 31–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo L., Song Z., Li M., Wu Q., Wang D., Feng H., Bernard P., Daugherty A., Huang B., Li X. A. (2009) J. Biol. Chem. 284, 19826–19834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayala A., Perrin M. M., Kisala J. M., Ertel W., Chaudry I. H. (1992) Circ. Shock 36, 191–199 [PubMed] [Google Scholar]
- 26.Cohen J. (2002) Nature 420, 885–891 [DOI] [PubMed] [Google Scholar]
- 27.Titheradge M. A. (1999) Biochim. Biophys. Acta 1411, 437–455 [DOI] [PubMed] [Google Scholar]
- 28.Beckman J. S., Koppenol W. H. (1996) Am. J. Physiol. 271, C1424–1437 [DOI] [PubMed] [Google Scholar]
- 29.Feron O., Belhassen L., Kobzik L., Smith T. W., Kelly R. A., Michel T. (1996) J. Biol. Chem. 271, 22810–22814 [DOI] [PubMed] [Google Scholar]
- 30.Connelly L., Madhani M., Hobbs A. J. (2005) J. Biol. Chem. 280, 10040–10046 [DOI] [PubMed] [Google Scholar]
- 31.Wang X. M., Kim H. P., Song R., Choi A. M. K. (2006) Am. J. Respir. Cell Mol. Biol. 34, 434–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X. M., Kim H. P., Nakahira K., Ryter S. W., Choi A. M. (2009) J. Immunol. 182, 3809–3818 [DOI] [PubMed] [Google Scholar]
- 33.Su G. L. (2002) Am J Physiol Gastrointest Liver Physiol 283, G256–265 [DOI] [PubMed] [Google Scholar]
- 34.Uesugi T., Froh M., Arteel G. E., Bradford B. U., Thurman R. G. (2001) Hepatology 34, 101–108 [DOI] [PubMed] [Google Scholar]
- 35.Sukumaran S. K., Quon M. J., Prasadarao N. V. (2002) J. Biol. Chem. 277, 50716–50724 [DOI] [PubMed] [Google Scholar]
- 36.Hotchkiss R. S., Nicholson D. W. (2006) Nat. Rev. Immunol 6, 813–822 [DOI] [PubMed] [Google Scholar]
- 37.Pinheiro da Silva F., Nizet V. (2009) Apoptosis 14, 509–521 [DOI] [PubMed] [Google Scholar]
- 38.Ayala A., Perl M., Venet F., Lomas-Neira J., Swan R., Chung C. S. (2008) Curr Pharm Des 14, 1853–1859 [DOI] [PubMed] [Google Scholar]
- 39.Dong Kim K. D., Zhao J., Auh S., Yang X., Du P., Tang H., Fu Y. X. (2007) Nat Med 13, 1248–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao J., Yang X., Auh S. L., Kim K. D., Tang H., Fu Y. X. (2009) Trends in Immunology 30, 8–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Obeid L. M., Linardic C. M., Karolak L. A., Hannun Y. A. (1993) Science 259, 1769–1771 [DOI] [PubMed] [Google Scholar]
- 42.Lalor D., Liu P., Hayashi J. (2004) Cell. Immunol. 230, 10–16 [DOI] [PubMed] [Google Scholar]
- 43.Ko Y. G., Lee J. S., Kang Y. S., Ahn J. H., Seo J. S. (1999) J. Immunol. 162, 7217–7223 [PubMed] [Google Scholar]