

Abstract
The bacterial metabolism of epoxypropane formed from propylene oxidation uses the atypical cofactor coenzyme M (CoM, 2-mercaptoethanesulfonate) as the nucleophile for epoxide ring opening and as a carrier of intermediates that undergo dehydrogenation, reductive cleavage, and carboxylation to form acetoacetate in a three-step metabolic pathway. 2-Ketopropyl-CoM carboxylase/oxidoreductase (2-KPCC), the terminal enzyme of this pathway, is the only known member of the disulfide oxidoreductase family of enzymes that is a carboxylase. In the present work, the CoM analog 2-bromoethanesulfonate (BES) is shown to be a reversible inhibitor of 2-KPCC and hydroxypropyl-CoM dehydrogenase but not of epoxyalkane:CoM transferase. Further investigations revealed that BES is a time-dependent inactivator of dithiothreitol-reduced 2-KPCC, where the redox active cysteines are in the free thiol forms. BES did not inactivate air-oxidized 2-KPCC, where the redox active cysteine pair is in the disulfide form. The inactivation of 2-KPCC exhibited saturation kinetics, and CoM slowed the rate of inactivation. Mass spectral analysis demonstrated that BES inactivation of reduced 2-KPCC occurs with covalent modification of the interchange thiol (Cys82) by a group with a molecular mass identical to that of ethylsulfonate. The flavin thiol Cys87 was not alkylated by BES under reducing conditions, and no amino acid residues were modified by BES in the oxidized enzyme. The UV-visible spectrum of BES-modifed 2-KPCC showed the characteristic charge transfer absorbance expected with alkylation at Cys82. These results identify BES as a reactive CoM analog that specifically alkylates the interchange thiol that facilitates thioether bond cleavage and enolacetone formation during catalysis.
Keywords: Acetoacetate, Carboxylation, Disulfide, Enzyme Kinetics, Enzyme Mechanisms, Epoxygenase Pathway, Flavin, Mass Spectrometry (MS), Protein Chemical Modification, Thiol
Introduction
The bacterial metabolism of epoxides formed from short-chain aliphatic alkene epoxidation occurs by a three-step metabolic pathway that uses the atypical cofactor coenzyme M (CoM, 2-mercaptoethanesulfonate)3 as a nucleophile for epoxide ring opening and as a carrier of intermediates that undergo dehydrogenation, reductive cleavage, and carboxylation (Fig. 1) (1–3). Epoxide catabolism from (R)- and (S)-epoxypropane converges at the intermediate 2-ketopropyl-CoM (2-KPC), which is a substrate for 2-ketopropyl-CoM carboxylase/oxidoreductase (2-KPCC), a unique member of the disulfide oxidoreductase (DSOR) family of enzymes (3–5).
FIGURE 1.

Pathway of bacterial propylene metabolism.
Like other members of the DSOR family, 2-KPCC uses NADPH as the reductant to reduce a bound flavin. The bound flavin in turn reduces a disulfide bond to free thiols designated as the flavin thiol and the interchange thiol (Fig. 2A) (4–6). For the canonical members of the DSOR family such as glutathione reductase and dihydrolipoamide dehydrogenase, the interchange thiol facilitates substrate disulfide bond reduction to the free thiols by forming a mixed disulfide between the interchange thiol and a substrate molecule, followed by reduction and release of the bound substrate with reformation of the cysteine disulfide (7). Unlike these prototype enzymes, 2-KPCC uses the redox active cysteine pair to facilitate thioether rather than disulfide bond cleavage, forming a mixed disulfide of the interchange thiol and CoM along with enolacetone, which undergoes carboxylation to form acetoacetate (Fig. 2C) (4–6). 2-KPCC is the only known DSOR enzyme to catalyze thioether bond cleavage and substrate carboxylation and thus is of general interest as a new class of dual function oxidoreductase/ligase.
FIGURE 2.

Reactions catalyzed by 2-KPCC. A, reduction of the redox active disulfide by NADPH. B, reduction of the redox active disulfide by DTT. C, mechanism of 2-KPC carboxylation. D, mechanism of inactivation of 2-KPCC by BES. The reduced form of the flavin thiol is shown as the thiolate to highlight the charge transfer, with increased absorbance at 540 nm, resulting from interaction with FAD. To experimentally observe maximal charge transfer in BES-modified 2-KPCC it was necessary to increase the pH from a value of 7.4 to 11.0.
Although kinetic, mechanistic, and structural studies of 2-KPCC have revealed important details of the reaction mechanism, much remains to be learned about how this enzyme operates. Central to the mechanism of this enzyme are the unique properties of CoM, the simplest known organic cofactor, consisting of sulfonate and thiol functional groups separated by an ethyl linker (Fig. 1). Aside from epoxide carboxylation, the only known function of CoM is in archaeal methanogenesis, where CoM carries the methyl group that is subsequently reduced to methane by the nickel-containing enzyme methyl-CoM reductase (8–11).
The CoM analog 2-bromoethanesulfonate (BES) was identified as a potent inhibitor of methanogenesis (12–14), and found to specifically inactivate MCR by binding as a CoM analog and rendering the enzyme inactive by oxidizing the nickel tetrapyrrole cofactor from the +1 to +2 oxidation state (15). Based on these studies of BES inhibition of methanogens and MCR, BES was studied as a possible inhibitor of bacterial alkene metabolism and found to specifically inhibit growth of Xanthobacter autotrophicus Py2 and Rhodococcus rhodochrous B276 with propylene, but not with carbon sources that do not use the CoM-dependent pathway and enzymes of Fig. 1 (16). Treatment of propylene-grown X. autotrophicus with BES resulted in loss of epoxide carboxylation activity that could only be restored by new protein synthesis. The addition of purified 2-KPCC restored epoxide carboxylase activity in cell extracts inactivated with BES, suggesting that 2-KPCC is the target of BES inactivation (16).
BES is the first specific irreversible inactivator of epoxide carboxylation to be identified, and as such the molecular basis of inhibition and inactivation is of significant interest. In the present work, BES is shown to be a reversible rapid equilibrium inhibitor of 2-hydroyxpropyl-CoM dehydrogenase and 2-KPCC, and a time-dependent irreversible inactivator of 2-KPCC. Inactivation of 2-KPCC results from the highly specific alkylation of the interchange thiol of 2-KPCC, identifying BES as the first affinity label and active site probe of this unusual member of the DSOR family of enzymes.
EXPERIMENTAL PROCEDURES
Materials
All solid chemicals, solvents, and endoproteinases used for liquid chromatography-mass spectrometry (LC-MS) studies were obtained from Sigma and were LC-MS grade or the next highest purity available. HPLC resins were obtained from Agilent (Santa Clara, CA).
Bacterial Cell Growth and Enzyme Purification
X. autotrophicus strain Py2 and Escherichia coli strains containing expression vectors were cultured as described previously (17–20). Recombinant epoxyalkane:CoM transferase (EaCoMT), recombinant R-hydroxypropyl-CoM dehydrogenase (R-HPCDH), and native 2-KPCC were purified to homogeneity (>95% pure by SDS-PAGE analysis) as described previously (17, 19, 20). Recombinant β-hydroxybutyrate dehydrogenase (β-HBDH) from Rhodobacter sp. DSMZ 12077 (21) was expressed in E. coli and purified as described previously (18).
EaCoMT Activity Assay
The activity of EaCoMT was determined by monitoring the depletion of epoxypropane from the head space of sealed assay vials by FID gas chromatography as described previously (20). Assays were conducted in a 1-ml total assay volume in 9-ml serum vials containing 50 mm Tris buffer, pH 7.4, 5 mm CoM, and 0.03 mg of EaCoMT. BES (5 mm) was included as indicated. The assays were initiated by the addition of epoxypropane (4 mm) from a 500 mm stock solution prepared in H2O.
R-HPCDH Assay
The activity of R-HPCDH was measured with R-hydroxypropyl-CoM (R-HPC) as substrate by monitoring the reduction of NAD+ spectrophotometrically (19). Assays were conducted in 2-ml anaerobic quartz cuvettes that contained 1 ml of assay components. Buffer and stock solutions were made anoxic by sparging or by repeated evacuation and flushing with N2 on a vacuum manifold. Assay components were added from stock solutions to sealed cuvettes (2-ml) that had been sparged with nitrogen using gas-tight syringes. Assays contained 50 mm Tris buffer, pH 7.4, 10 mm NAD+, 1 mm EDTA, and varying concentrations of R-HPC (23.3–1000 μm) and BES (0–7.5 mm). Assays were initiated by the addition of enzyme (1.4 μg of R-HPCDH). NADH formation (ϵ340 = 6.22 mm−1 cm−1) was monitored using a Shimadzu UV-2401 spectrophotometer containing a water-jacketed cell holder maintained at 30 °C. Rates were determined from the linear portions of progress curves (typically within the first 60 s of assays).
Coupled Spectrophotometric Assay of 2-KPCC-catalyzed 2-KPC Carboxylation
A continuous spectrophotometric assay was developed that couples acetoacetate production by 2-KPCC to acetoacetate reduction and NADH oxidation by β-HBDH. Assays were conducted in 2-ml anaerobic quartz cuvettes that contained 1 ml of assay components. Buffer and stock solutions were made anoxic by sparging or by repeated evacuation and flushing with N2 on a vacuum manifold, except for potassium bicarbonate solutions, which were prepared by adding anoxic water to a degassed vial containing KHCO3 solid. Assay components were added from stock solutions to sealed cuvettes as described above. The concentrations of assay components in the cuvettes were 50 mm Tris buffer, pH 7.4, 10 mm DTT, 0.2 mm NADH, 40 mm KHCO3, 10 mm CO2, 0–10 mm BES, 0.345 mg of β-HBDH (90 units of enzyme activity), varying concentrations of 2-KPC (0 to 0.70 mm), and 2-KPCC (0.05–0.1 mg). The amount of β-HBDH included in assays was severalfold higher than that required for saturation of rates. The cuvettes were equilibrated at 30 °C prior to initiating assays by the addition of 2-KPCC. Rates were determined from the linear portions of progress curves (typically 20–120 s). The change in absorbance at 340 nm over time was used to calculate rates of β-hydroxybutyrate production using the extinction coefficient for NADH (6.22 mm−1 cm−1).
Assay of 2-KPCC-catalyzed 2-KPC Protonation
The 2-KPCC-catalyzed formation of acetone from 2-KPC was determined by gas chromatography using the assay procedure described previously (4). Buffer and stock solutions were made anoxic and depleted of residual CO2 by sparging and/or repeated evacuation and flushing with N2 that had been passed through a column of Ascarite II (Thomas Scientific, Swedesboro, NJ). Assays were conducted in a 1-ml total assay volume in 9-ml serum vials containing 50 mm Tris buffer, pH 7.4, 5 mm NADPH, 3 mm 2-KPC, and 0.2 mg of 2-KPCC. The assays were initiated by the addition of 2-KPC.
Analysis of Initial Velocity Data
Initial velocities were plotted versus substrate concentration and fit to the standard form of the Michaelis-Menten equation as described by Cleland (22) using the software SigmaPlot 11. For analysis of enzyme inhibition, data were fit to the following form of the Michaelis-Menten equation, which describes the effect of a noncompetitive (mixed) inhibitor (22).
Removal of BES from Aqueous Solutions with AG1-X8 Ion-exchange Resin
AG1-X8 (Bio-Rad, chloride form, mesh size 100–200) resin was rehydrated in 10 volumes of 50 mm Tris buffer, pH 7.4, and filtered under vacuum through 7-cm diameter Whatman No. 1 filter paper. This process was repeated 3 times before the resin was finally vacuum dried for 10 min. To assess the ability of AG1-X8 resin to quantitatively remove BES from solutions, BES (150 μl of a 125 mm stock solution) was added to 15 mg of dried AG1-X8 resin in a microcentrifuge tube followed by incubation for 3 min at room temperature. The sample was briefly spun in a microcentrifuge (5 s pulse) to pellet the resin. Portions of the AG-1X8-treated BES solution (0–25 μl) were then transferred to assay cuvettes to assess their effects on R-HPCDH and 2-KPC assays, conducted as described above. No inhibition of 2-KPCC or R-HPCDH activity was observed, demonstrating that the AG1-X8 resin removed a sufficient amount of BES to prevent rapid equilibrium inhibition effects in 2-KPCC and R-HPCDH assays.
Assay of Time-dependent Inactivation of 2-KPCC by BES
Samples of 2-KPCC (2.28 mg) were incubated anoxically at 30 °C in 3-ml serum vials containing a total volume of 1.25 ml of buffer (50 mm Tris, pH 7.4) either containing or lacking DTT (10 mm) and BES (0–10 mm). Incubations were initiated by the addition of BES from a stock solution. At the desired time points, 0.15-ml samples were removed with gas-tight syringes, transferred to 1.5-ml microcentrifuge tubes containing 15 mg of AG1-X8 ion-exchange resin to remove residual BES, and then incubated for 3 min and pulse-centrifuged as described above. Fifty-μl samples were then removed from the supernatant and used as the source of 2-KPCC for coupled spectrophotometric assays of 2-KPCC activity, conducted as described above. 2-KPCC activity assays were conducted in duplicate for each incubation time point. The apparent first-order rate constants (kobs) for each individual BES concentration were determined by fitting the data to Equation 2,
where υ is the observed velocity at any given time, υ0 is the velocity at incubation t = 0 (υ/υ0 is the fractional velocity remaining after a given pre-incubation), and t is time in min. The maximal rate of enzyme inactivation (kinact) and the concentration of inhibitor required to reach the half-maximal rate of inactivation (Ki) were determined by fitting the kobs values for individual BES concentrations ([I]) to Equation 3 (23) using the software SigmaPlot.
Assay of Time-dependent Inactivation of R-HPCDH by BES
Samples of R-HPCDH (0.273 mg) were tested for possible time-dependent inactivation by BES, essentially as described above for 2-KPCC, with the following modifications. The total volume of the assay mixture was reduced to 0.15 ml, and the entire reaction mixture was removed and subjected to AG1-X8 treatment after a 200-min incubation time. Fifty μl of the AG-1X8-treated sample was then diluted 8-fold by the addition of buffer. Five μl of the diluted enzyme sample was used for assaying R-HPCDH activity using 1 mm R-HPC as substrate as described above.
UV-Visible Spectral Analysis of 2-KPCC
Samples of 2-KPCC (1.8 mg/ml) were incubated anoxically as described above in the presence of 10 mm DTT and with or without 10 mm BES. After a 4-h incubation, the sample with BES had lost 98% activity based on activity assays conducted as described above. At that time, the samples were desalted using prepacked columns of Sephadex G-25 (Pharmacia, PD-10) equilibrated in 100 mm lysine buffer, pH 11, containing 100 mm NaCl and 0.1 mm EDTA. Desalted samples were air oxidized for 30 min before recording UV-visible spectra on a Shimadzu UV-2401PC spectrophotometer. The concentrations of samples were normalized by extracting and quantifying FAD using sodium dodecyl sulfate as described by Aliverti and co-workers (24).
Preparation of Acrylamide Protein Plugs for LC-MS Analysis
2-KPCC (30.3 mg/ml stock) was diluted with 100 mm Tris, pH 7.4, to a concentration of 50 ng/μl. Three 1-ml samples of the diluted 2-KPC were prepared in sealed 4-ml serum vials as follows: the first sample was prepared aerobically and contained no additional components, the second sample was prepared aerobically and contained 5 mm BES, and the third sample was prepared anaerobically and contained 10 mm DTT and 5 mm BES. This sample was reduced with DTT before the addition of BES. All samples were allowed to incubate at room temperature for 6.5 h. After incubation, the samples were transferred to 1.5-ml microcentrifuge tubes containing 15 mg of AG1-X8 resin. The resin was pelleted and the enzyme solution was then decanted away from the resin and prepared for SDS-PAGE.
Four μl of the 50 ng/μl solution was diluted to 14 μl with 2× sample loading mixture. These samples were boiled and loaded onto a 12% acrylamide gel that had been poured 24 h in advance and allowed to sit at room temperature. Individual bands, containing 200 ng of protein, were cut into 4 equal slices and placed into 1.5-ml microcentrifuge tubes. Two hundred μl of neat HPLC grade acetonitrile was added to the tubes and the protein samples were incubated at room temperature for 5 min. Acetonitrile was decanted from the dehydrated SDS-PAGE gel slices, and the samples were dried in a SpeedVac concentrator at room temperature.
Preparation of Liquid 2-KPCC Samples for LC-MS Analysis
2-KPCC (30.3 mg/ml) was diluted with 100 mm Tris buffer, pH 7.4, to a concentration of 2 mg/ml. Three samples (0.15 ml each) were prepared aerobically with 5 mm BES, and anaerobically with 10 mm DTT and 5 mm BES as described above. The samples were incubated for 4 h at room temperature before the addition of 15 mg of AG1-X8 resin. The resin was pelleted, and the samples were decanted and desalted by passing them over a 1 × 5-cm column of Sephadex G-25 (PD-10 Amersham Biosciences), which had previously been equilibrated with 10 mm NH4HCO3. The samples were eluted with 10 mm NH4HCO3 (volume ∼3 ml) and placed in a spin column concentrator (Centricon YM 30, Amicon), which had been previously washed with 10 mm NH4HCO3. The samples were concentrated to 0.15 ml and flash frozen in liquid N2.
Instrumentation Used for Mass Spectral Sample Analysis
All samples were analyzed with an Agilent 1100 HPLC connected to a ThermoFinnigan LCQ Advantage ion-trap mass spectrometer equipped with a nanospray ion source. Samples were loaded onto nanospray columns either offline via a pressure bomb (ProXeon), or online via the autosampler with the aid of an inline C8 sample trap (Michrom Bioresources microcap trap) fitted to the LCQ divert valve. All columns were sprayed at 250 nl/min with the spray voltage set at 2.2 kV and the heated capillary set at 200 °C.
Determination of BES Modification Stoichiometry by Mass Spectrometry
2-KPCC was diluted 1:100 into 2% (v/v) acetonitrile containing 0.1% formic acid (Buffer A). Approximately 50 ng was loaded offline onto a 75-μm inner diameter × 10-cm PicoFrit column (New Objective) packed with Zorbax 300SB-C8 (Agilent). The column was developed with a 10-min hold of buffer A, followed by a 90-min linear gradient from 0–90% buffer B (neat acetonitrile, 0.1% formic acid), and a 20-min hold at 90% buffer B. Native molecular mass was determined by averaging all MS spectra (across a width of one-half of the peak height) for a 2-KPCC polypeptide. The charge envelope was deconvoluted using both ProMass Deconvolution 2.0 and Bioworks 3.1 algorithms to give two separate average mass values for 2-KPCC.
In-Gel Digestion of 2-KPCC
Two hundred nanograms of 2-KPCC, for each condition, was run in triplicate on 12% SDS-PAGE gels. Gels were stained with Coomassie Blue and destained using standard protocols. Each protein band was excised, cut into four pieces, and placed into a microcentrifuge tube. To each tube, 200 μl of neat acetonitrile was added. After 5 min acetonitrile was discarded and samples were dried in a SpeedVac for storage at 4 °C until processing. Gel samples were processed using a standard in-gel digestion procedure incorporating tris(2-carboxyethyl)phosphine reduction and cysteine alkylation with iodoacetamide. To achieve sequence coverage of all six 2-KPCC cysteine residues, three parallel digests were performed for each condition. To the processed and dried gel cores, proteases were added from stocks in digestion buffer (50 mm NH4HCO3, 5% (v/v) acetonitrile) as follows: trypsin (600 ng), trypsin + Pro-C (300 ng each), and Glu-C + Lys-C (300 ng each). Gel cores were allowed to swell with the added protease(s) for 15 min, followed by addition of 25 μl of digestion buffer, and incubation at 37 °C for ∼18 h. Peptides were extracted by shaking with 100 μl of 5% (v/v) formic acid for 30 min at 30 °C. Samples were then microcentrifuged at 14,000 × g for 2 min. Twenty-five microliters of the supernatant from the trypsin digest was loaded on-line, with 5 μl of the other digests loaded off-line; in an attempt to capture less hydrophobic peptides that could have been lost by use of the C8 sample trap.
Mapping Location of BES Modification
All protein and peptide identifications were made using SEQUEST as part of Bioworks 3.1. The protein data base used for searching MS/MS spectra contained five protein sequences: 2-KPCC (Q56839), Lys-C (Q7M135), Glu-C (1814271A), porcine trypsin (P00761), and endoproteinase Pro-C (P27195) nested within 100 random decoy sequences. The generation of random decoys was accomplished using ExPASy RandSeq using the amino acid sequence of 2-KPCC. Using SEQUEST, the criteria for DTA file generation was set as follows: group scan = 20, group count = 2, minimum ion count = 20. All other parameters were default. Peptide hits to the data base were filtered by charge state and Xcorr score (+1, ≥3.0; +2 ≥ 4.0; +3, ≥ 5.0). Additionally, peptide identifications were required to have a dCN score of ≥0.1. Six differential modifications were used in the data base searches and included oxidized methionine, iodoacetamide-modified cysteine, acrylamide-modified cysteine, bromoethanesulfonate-modified cysteine, and oxidized bromoethanesulfonate-modified cysteine.
RESULTS
Development of a Continuous Spectrophotometric Assay for Measurement of 2-KPCC Carboxylation
The physiologically important reaction for 2-KPCC is the carboxylation of 2-KPC using NADPH as reductant as shown in Fig. 1 and summarized in Equation 4.
 |
In the absence of CO2, 2-KPCC catalyzes the protonation of 2-KPC to form acetone in a fortuitous reaction that occurs at a rate that is about 25% of the rate of 2-KPC carboxylation,
Both of these reactions have been exploited in previous studies of 2-KPCC to measure enzyme activity. For the carboxylation assay, activity was determined by measuring the incorporation of radiolabeled 14CO2 into acetoacetate, whereas for the protonation assay, activity was determined by measuring acetone production by gas chromatography. Both of these assays are discontinuous, making them laborious for the kinetic analyses necessary for the studies described below. 2-KPCC activity can be measured by monitoring the loss of absorbance from oxidation of NADPH, but this requires observable changes in NADPH concentration, resulting in changes of rate during the assay time courses. It was thus of interest to develop a continuous spectrophotometric assay for 2-KPCC where reductant could be present at a saturating level throughout the course of the assay. To achieve this, we relied on the fact that DTT can serve as an alternative reductant for 2-KPCC by directly reducing the redox active disulfide required for catalysis, bypassing the need for NADPH and the intermediacy of FADH2 (see Fig. 2, A and B). In the presence of DTT the carboxylation of 2-KPC thus occurs as shown in Equation 6.
 |
For the development of the coupled assay, excess β-HBDH and NADH were included in the assay to remove the acetoacetate, in the process forming NAD+ as shown in Equation 7.
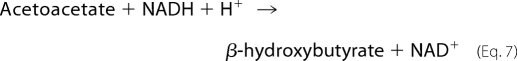 |
The success of this coupled assay relies on the fact that 2-KPCC does not use NADH as a reductant (i.e. the enzyme is specific for NADPH, or the alternate reductant DTT). β-HBDH, when present in large excess, efficiently removes acetoacetate at an NADH concentration of 0.2 mm, allowing NAD+ formation to be measured spectrophotometrically according to Equation 7. β-HBDH has also been successfully used as a coupling enzyme for measuring the activity of acetone carboxylase, which catalyzes the ATP-dependent carboxylation of acetone to form acetoacetate (18).
Using the newly developed coupled enzyme assay, the observed rates of NADH oxidation were linear over the course of the assays and directly proportional to the amount of 2-KPCC present in the assays. The rates at near saturating concentrations of 2-KPC were the same as in the discontinuous assay with DTT as reductant, which are about 60% of the discontinuous rates when NADPH is used as reductant.
BES Inhibition of the Enzymes of (R)-Epoxypropane Metabolism
BES was tested as a possible inhibitor of the three individual enzymes that together carboxylate the (R)-isomer of epoxypropane to acetoacetate (Fig. 1). As shown in Table 1, when present at a concentration comparable with that of the substrate, BES was an inhibitor of both R-HPCDH (70% inhibition) and 2-KPCC (40% inhibition) but not of EaCoMT. For both R-HPCDH and 2-KPCC, which were assayed using continuous spectrophotometric assays over short time frames (20–120 s), the progress curves for product formation were largely linear in the absence and presence of BES, demonstrating that the inhibition is largely rapid equilibrium over these short time courses. For 2-KPCC there was a small amount of time-dependent loss of activity (loss of progress curve linearity) that was more pronounced when assays were allowed to proceed for longer time periods (more than 2 min), suggesting that 2-KPCC might be subject to time-dependent inactivation as well as rapid equilibrium inhibition. It should be noted that BES did not inhibit the activity of the coupling enzyme β-HBDH when assayed directly with acetoacetate as the substrate, demonstrating that the inhibition is directed toward 2-KPCC.
TABLE 1.
BES inhibition of enzymes involved in (R)-epoxypropane metabolism
Enzyme assays were performed as described under “Experimental Procedures.” Activities are percentages relative to assays conducted in the absence of BES ± S.E. The concentrations of substrate in each assay were as follows: (R)-epoxypropane, 4 mm; R-HPC, 4 mm; and 2-KPC, 3 mm. Assays are the averages of triplicate measurements.
| Enzyme | Activity in presence of 5 mm BES |
|---|---|
| % | |
| EaCoMT | 100 |
| R-HPCDH | 32.7 ± 3.6 |
| 2-KPCC | 61.2 ± 4.3 |
Kinetic Characterization of BES Inhibition of R-HPCDH
A kinetic analysis of the inhibition of R-HPCDH by BES was conducted by varying the concentrations of R-HPC and BES at a fixed, saturating concentration of NAD+ (10 mm). As shown in Fig. 3, BES behaved as a mixed (noncompetitive) inhibitor of R-HPC oxidation with a fit of the experimental data providing the following kinetic parameters: kcat = 29.0 ± 0.6 s−1, Km = 124 ± 10 μm, Kis = 1,710 ± 280 μm, and Kii = 2,300 ± 200 μm. By comparison, CoM and ethanethiol (a CoM analog lacking the sulfonate group) were shown in a previous study to be mixed inhibitors of R-HPC oxidation by R-HPCDH as well (19). The inhibition constants for CoM were very similar to those determined here for BES (Kis = 2,250 μm; Kii = 3,620 μm), whereas the inhibition constants for ethanethiol were ∼10-fold higher (Kis = 26,700 μm; Kii = 38,900 μm) (19).
FIGURE 3.

Noncompetitive rapid-equilibrium inhibition of R-HPCDH-catalyzed R-HPC oxidation by BES. Assays of R-HPC oxidation were performed as described under “Experimental Procedures.” The double-reciprocal plots for assays performed in the presence of different concentrations of BES are shown. Data points represent the average of duplicate experiments. The solid lines were generated by nonlinear least-square fits of the v versus S data to the equation for a rectangular hyperbola using Sigmaplot. Symbols: ●, 0 mm BES; ■, 2 mm BES; ▴, 3 mm BES; ♦, 5 mm BES; ▾, 7 mm BES.
To determine whether R-HPCDH was subjected to time-dependent inactivation by BES under non-turnover conditions, R-HPCDH samples were incubated with BES for 200 min and then assayed for activity after removal of BES. To remove BES prior to conducting enzyme assays, AG1-X8, an anion exchange resin with a high affinity for small molecules containing a sulfonate functional group, was added to protein samples and then removed by microcentrifugation. This procedure was found to rapidly and quantitatively remove all non-protein-bound BES from samples. For R-HPCDH, preincubation of the enzyme with BES for 200 min resulted in no loss of enzymatic activity once BES was removed, demonstrating that BES is not an irreversible inactivator.
Kinetic Characterization of Rapid Equilibrium Inhibition of 2-KPCC by BES
As noted above, the progress curves for 2-KPCC were largely linear over short time courses in the absence or presence of 5 mm BES, demonstrating that there is a rapid equilibrium component to the inhibition. As shown in Fig. 4, for these short time courses BES behaved as a mixed inhibitor of 2-KPC carboxylation at a fixed saturating concentration of DTT and CO2, providing the following kinetic parameters: kcat = 12.7 ± 0.3 min−1, Km = 35.0 ± 3.7 μm, Kis = 2980 ± 710 μm, and Kii = 4460 ± 330 μm.
FIGURE 4.

Noncompetitive rapid equilibrium inhibition of 2-KPCC-catalyzed 2-KPC oxidation by BES. Assays of 2-KPC carboxylation were performed as described under “Experimental Procedures.” The double-reciprocal plots for assays performed in the presence of different concentrations of BES are shown. Data points represent the average of duplicate experiments. The solid lines were generated by nonlinear least-square fits of the v versus S data to the equation for a rectangular hyperbola using Sigmaplot. Symbols: ●, 0 mm BES; ■, 2.5 mm BES; ▴, 5 mm BES; ▾, 7.5 mm BES.
Time- and Concentration-dependent Inactivation of 2-KPCC by BES
A previous study showed that the addition of BES to cell extracts of X. autotrophicus resulted in an irreversible loss of epoxide carboxylation activity that could only be restored by adding purified active 2-KPCC back to the cell extracts, suggesting that 2-KPCC is irreversibly inactivated by BES (16). To investigate this in more detail, 2-KPCC was incubated anoxically in the presence of DTT with various concentrations of BES, and samples were removed at various time points and assayed for activity after removal of BES as described above. To ensure that no significant abiotic reaction of BES and DTT occurred (i.e. alkylation of one or both of the thiols of DTT), the following experiment was performed. DTT (10 mm) was incubated anoxically with and without BES (20 mm) at room temperature for 30 h in 50 mm Tris, pH 7.4. Samples were then removed and added directly to 2-KPCC activity assays. The BES from the samples preincubated with and without DTT inhibited 2-KPCC identically, and to an extent consistent with the concentrations of BES present at the outset of the incubations, showing that DTT and BES do not react abiotically to any significant degree under these conditions.
As shown in Fig. 5A, in the presence of DTT 2-KPCC exhibited a time-dependent loss of activity that followed apparent first-order kinetics and was proportional to the amount of BES present in the samples. Fig. 5B shows the relationship between the apparent first-order rate constants (kobs) for inactivation derived from the data in Fig. 5A and the BES concentration. The inactivation exhibited saturation kinetics, as would be expected for an affinity label that rapidly forms a reversible EI complex that undergoes slow, irreversible inactivation according to Scheme 1.
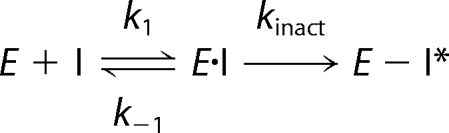 |
A fit of the data to Equation 3 provided the following kinetic parameters: kinact (the theoretical maximal rate of inactivation) = 0.012 ± 0.001 min−1 and Ki (the concentration of BES giving the half-maximal rate of inactivation) = 1.4 ± 0.3 mm.
FIGURE 5.

2-Bromoethanesulfonate is a time- and concentration-dependent inactivator of 2-KPCC. Panel A, time- and concentration-dependent inactivation of 2-KPCC by BES. 2-KPCC was incubated with various concentrations of BES, followed by removal of residual BES and assay of 2-KPCC activity as described under “Experimental Procedures.” Symbols: ●, no BES; ■, 0.125 mm BES; □, 0.50 mm BES; Δ, 1.0 mm BES; 224, 2.0 mm BES; ▴, 3.0 mm BES; ▾, 5.0 mm BES; ○, 9.43 mm BES. Pseudo first-order rate constants (kobs) were determined by fitting the data in panel A to Equation 2. Panel B, plot of pseudo first-order rate constants for inactivation (kobs) versus BES concentration. The line through the data points was generated by nonlinear least-squares fit to the rectangular hyperbola in Equation 3. Panel B, inset, double-reciprocal plot of the data. The line through the data points was derived from the non-linear fit of kobs versus [BES].
The fact that inactivation of 2-KPCC exhibits saturation kinetics indicates that BES is an affinity label mimicking the CoM portion of the substrate 2-KPC, forming a Michaelis complex with the enzyme prior to irreversible inactivation. A hallmark of affinity labels is partial protection from inactivation by substrates and substrate analogs that bind reversibly. Under the reducing conditions used for these incubations 2-KPC undergoes turnover to produce acetone as the product (see Equation 5; DTT can be used in place of NADPH for this reaction as well). Therefore, CoM, the product of the reaction, was tested for its ability to protect 2-KPCC from BES inactivation. In the presence of 5 mm BES and 5 mm CoM, 2-KPCC retained 55% activity after a 190-min incubation, whereas 2-KPCC incubated identically but in the absence of CoM retained only 21% activity. A variety of other brominated organic compounds, including bromoethane, 1-bromopropane, and 3-bromopropionate, were not inhibitors or inactivators of 2-KPCC, highlighting the importance of the sulfonate in the specificity of BES.
Reduced but Not Oxidized 2-KPCC Is Inactivated by BES
The target of inactivation by BES is potentially one or more of the reduced cysteine thiols of 2-KPCC, which could undergo alkylation by transfer of the ethylsulfonate moiety (ES) as shown in Equation 8.
To provide evidence for this, air-oxidized 2-KPCC was incubated with 5 mm BES under conditions identical to those described above, but aerobically and in the absence of DTT. Under these conditions, no inactivation of 2-KPCC activity was observed over a 4-h time course, suggesting that one or more thiols that can be oxidized to the disulfide form is the target of BES.
The target of BES inactivation was further refined by examining BES inactivation of 2-KPCC using NADPH rather than DTT as the reductant. The addition of 5 mm NADPH resulted in inactivation of 2-KPCC at rates identical to those observed in the presence of DTT. Because NADPH specifically reduces the redox active disulfide at the active site of 2-KPCC through the intermediacy of FAD, this suggests that either the interchange thiol, the flavin thiol, or both are targets of BES inactivation.
Determination of BES Modification Stoichiometry
Table 2 summarizes the results for the 2-KPCC average molecular mass determinations under oxidized and DTT-reduced conditions, and in the absence and presence of BES, using two different deconvolution algorithms. All trends appeared consistent between the results of the two algorithms, with ProMass 2.0 typically generating lower mass values. Analytical reproducibility was demonstrated for un-treated 2-KPCC by running the analysis two separate times (runs 1 and 2, Table 2). When 2-KPCC was pre-treated with BES in the absence of DTT, no significant mass change was observed over that of untreated 2-KPCC. However, when 2-KPCC was pre-treated with BES in the presence of DTT, a corresponding average mass increase of 109.0 Da, compared with BES-treated oxidized 2-KPCC, was observed (Table 2). As the average molecular mass for a BES modification was expected to be 107.1 Da, the observed increase was highly suggestive of a single BES modification.
TABLE 2.
Average molecular mass determinations of 2-KPCC using LC-MS and two spectral deconvolution algorithms
Under the conditions utilized for analysis, 2-KPCC was observed as the FAD-depleted monomer.
| Sample | Theoretical average molecular mass | Measured average molecular mass by deconvolution algorithm | Mass accuracy (Meas-Theor) (for BIOMASS 3.1) | Mass change with BES modification (2-KPCC + BES + DTT) − (2-KPCC + BES) (for BIOMASS 3.1) | ||
|---|---|---|---|---|---|---|
| BIOMASS 3.1 | ProMass 2.0 | |||||
| M | M ± S.D. | ΔDa | Dppm | ΔDa | ||
| Oxidized 2-KPCC (run 1) | 57,345.6a | 57,365.0 | 57,362.1 ± 12.9 | +19.4 | 338 | |
| Oxidized 2-KPCC (run 2) | 57,345.6a | 57,358.0 | 57,355.7 ± 6.9 | +12.4 | 216 | |
| Oxidized 2-KPCC + BES | 57,345.6b | 57,361.0 | 57,358.4 ± 11.0 | +15.4 | 268 | |
| DTT-reduced 2-KPCC + BES | 57,454.7c | 57,470.0 | 57,463.3 ± 10.2 | +15.3 | 266 | 109.0 |
a Calculated using the ExPASy Compute pI/MW tool using the 2-KPCC protein sequence (Q56839) minus the average atomic mass of two hydrogens.
b Calculated awith no BES modification assumed.
c The average mass with BES modification of a single cysteine (+C2H4SO3 = 108.1; calculated using the average atomic masses of the elements, with the sum being rounded to the nearest 0.1 Da). The calculation assumed the SO3 moiety of BES and the Cys87 thiol of 2-KPCC to be deprotonated and protonated, respectively, under the conditions used for the analysis (in a solvent containing 0.1% (v/v) formic acid).
Mapping the Location of the BES Modification
With evidence for a single BES modification, experiments were performed to map the position of this modification. Because the modification only appeared to take place under reducing conditions, the modification was presumed to be occurring on a reactive cysteine residue, as noted above. Thus, all six of the cysteine residues present in 2-KPCC must be represented among proteolytic peptides for subsequent MS/MS analysis.
Using the proteolytic enzyme combinations described under “Experimental Procedures,” 73.2% total sequence coverage (% by # amino acids) was achieved for 2-KPCC pre-treated with BES and DTT. This coverage resulted in representation of all six cysteines. Interestingly, no proline-specific cleavages of 2-KPCC were observed, despite the abundance of proline residues in 2-KPCC and identification of several Pro-C autolytic peptides. As such, the trypsin + Pro-C digest yielded no new information beyond that of the digestion with trypsin alone.
Table 3, part A, lists the cysteine-containing peptides identified from at least two separate MS/MS spectra. As expected, most of the cysteines were modified by iodoacetamide as part of the in-gel digestion procedure. In one case, an acrylamide-modified cysteine was observed, which was likely the result of using an insufficiently polymerized acrylamide gel.
TABLE 3.
Part A, cysteine containing peptides identified from MS/MS spectra with selected SEQUEST parameters shown for each peptide. Part B, Bioworks ShowOut view for peptide #1 in panel A, depicting the next two closest matches to the database. Subscripts denote the position of the cysteine within the 2-KPCC sequence. Symbols represent modifications to cysteine and are as follows: #, iodoacetamide; ∼, acrylamide; and @, BES

a Calculated charge state based on the observed m/z value.
b The Xcorr value is the cross-correlation value from the search, or a measure of how closely an experimental MS/MS spectrum matches to a theoretical MS/MS spectrum for a given peptide in the database.
c Delta Correlation value, dCn indicates how different the first hit is from the second hit in a database search. It is calculated as follows: (Xcorr#1 − Xcorr#2)/Xcorr#1. A general rule of thumb is that a dCn of 0.1 or greater is good.
d Relative Sp score. Sp is the preliminary score. After finding all peptides that match within the selected mass tolerance, SEQUEST dose a preliminary scoring of these candidates to reduce the list down to 500 for the final XScorr analysis. Ideally the RSp value should be 1 on the best matches.
The single BES modification was localized to cysteine 82, which is the interchange thiol of 2-KPCC (Table 3, part A). Additional scrutiny of the modification at Cys82 was carried out by examining the ShowOut view for the peptide (Table 3, part B). BES modification at Cys82 gives a significantly higher Xcorr value than for the modification being placed at Cys87 (the flavin thiol). Furthermore, the ion identifications and MS/MS spectra of the BES-modified peptide, shown in Fig. 6, A and B, respectively, indicated that several of the most prominent ions observed are those that favor BES modification at position Cys82 and not Cys87. Of these ions, the most notable is y2–22, observed with an m/z value of 1274.1 (Fig. 6B). Additionally, when oxidized 2-KPCC that had been pre-treated with BES was digested with trypsin and analyzed by MS/MS, iodoacetamide and not BES was found to be the modification at position 82.
FIGURE 6.

A, amino acid sequence with +1 and +2 ion identifications for the peptide containing a BES modification. Symbols, @, BES modification; #, iodoacetamide modification. All underlined ions were experimentally observed, whereas ions also in bold are those that allowed discrimination between assignment of the BES modification at Cys82 or Cys87; note that one of these discriminating ions (y2–22 at m/z 1274.1) is the most prominent ion in the spectrum. B, MS/MS spectra of the BES modified peptide. Inset, full MS spectrum at 95.47 min, with the arrow showing the [M + 3H]3+ BES-modified peptide at m/z 1168.4 that was automatically selected for MS/MS.
UV-Visible Spectroscopic Evidence for Alkylation of Cys82 by BES
A distinguishing feature of DSOR enzymes is the formation of a charge-transfer complex resulting from the interaction of the deprotonated flavin thiolate (Cys87 in the case of 2-KPCC) and the oxidized flavin in the two-electron reduced enzyme (Fig. 2A, product of step 4). Covalent modification of the interchange thiol of 2-KPCC by BES should irreversibly lock oxidized 2-KPCC into the form in which the charge transfer can be observed, as shown in Fig. 2D. To test this, 2-KPCC was incubated with DTT, with or without BES, followed by gel filtration chromatography, air oxidation, and UV-visible spectral analysis. As shown in Fig. 7, 2-KPCC that had been preincubated in the absence of BES showed the normal oxidized flavin spectrum, whereas 2-KPCC preincubated in the presence of BES showed the characteristic spectral changes associated with the charge-transfer complex. These results confirm the mass spectral results showing that the interchange thiol and not the flavin thiol of 2-KPCC is the specific target of BES inactivation and alkylation.
FIGURE 7.

UV-visible spectra of untreated 2-KPCC and BES-modified 2-KPCC. 2-KPCC samples were prepared as described under “Experimental Procedures.” The reduced spectra were recorded after removal of oxygen and the addition of 1 mm dithionite. Trace 1, air-oxidized 2-KPCC; trace 2, air-oxidized 2-KPCC modified by BES; trace 3, dithionite-reduced 2-KPCC; trace 4, dithionite-reduced 2-KPCC modified by BES.
DISCUSSION
The present work expands on our previous work (16) showing BES to be an inactivator of bacterial epoxide metabolism by determining the targets and mechanisms of BES inhibition and inactivation. R-HPCDH and 2-KPCC were reversibly inhibited by BES, whereas 2-KPCC was additionally inactivated by the highly specific and irreversible alkylation of the interchange thiol that interacts with the substrate 2-KPC within the enzyme active site. The latter result is particularly interesting in that it identifies a fundamental new mechanism for inactivation of a CoM-dependent enzyme by BES. By comparison, in the case of methanogenic methyl-CoM reductase, BES inactivation occurs via oxidation of Ni(I) in the nickel tetrapyrrole cofactor F430 to Ni(II), in the process producing a debrominated alkylsulfonate product (15). Bromopropanesulfonate, which is an even more potent inhibitor than BES, oxidizes Ni(I) in F430 as well, but by a different mechanism (15, 25–27).
Although half-maximal inhibition of methyl-CoM reductase was observed at low micromolar concentrations of BES (15), ∼1000-fold higher concentrations were required for the inhibition of R-HPCDH and 2-KPCC. Interestingly, BES was a mixed (noncompetitive) rapid equilibrium inhibitor of both R-HPCDH and 2-KPCC. The inhibition constants reflecting competitive inhibition (Kis) were 14- and 85-fold higher than the Km values for substrates R-HPC and 2-KPC for R-HPCDH and 2-KPCC, respectively. The inhibition constants reflecting binding to the ES complex (Kii) were slightly higher, although similar in magnitude to, the Kis values. These results support the results of x-ray crystallographic studies of R-HPCDH and 2-KPCC showing that the hydroxypropyl and ketopropyl moieties, in addition to the sulfonate of CoM, are important in high affinity substrate binding (5, 28). For methyl-CoM reductase, where the group bound to CoM is the nonpolar methyl group, the ethanesulfonate could be expected to be the major binding determinant, perhaps helping to explain why BES is a much higher affinity inhibitor for this enzyme.
Interestingly, EaCoMT, the only enzyme of the pathway of epoxide carboxylation for which CoM is the substrate (Fig. 1), was not inhibited by BES, even at BES concentrations more than 2 orders of magnitude higher than the Km for CoM (34 μm). EaCoMT is a zinc metalloenzyme, in which the role of zinc is to coordinate the thiol of CoM, thereby lowering its pKa for nucleophilic attack on epoxypropane (20). A calorimetric study of the binding of CoM, ethanesulfonate, and ethanethiol to EaCoMT revealed that the zinc-thiol interaction contributes much more binding energy to the formation of the enzyme-CoM complex than the interaction of the sulfonate with the enzyme (20). It is therefore not surprising that BES is not a CoM analog (and inhibitor) for EaCoMT because the substitution of the thiol group by bromine will result in the loss of this key interaction with zinc.
The kinetics for the time-dependent inactivation of 2-KPCC by BES show that inactivation occurs by a two-step process, the first step of which is rapid, reversible and saturable (Scheme 1). As noted under “Results,” other brominated compounds lacking the sulfonate group did not inhibit 2-KPCC. Thus, as illustrated in Fig. 2D, the interaction of the sulfonate of BES with the two arginine residues that coordinate the sulfonate of the substrate 2-KPC is believed to be key to the selectivity of BES as an alkylating agent.
The highly selective alkylation of the interchange thiol of 2-KPCC makes BES an excellent probe for investigating novel features of this unique member of the DSOR family of enzymes, the only one known to function as a carboxylase. Structural studies of 2-KPCC have revealed a smaller and more hydrophobic active site architecture relative to all other DSOR enzymes due to amino acid insertions (5). Binding of 2-KPC induces a unique conformational change that encapsulates the substrate 2-KPC, at the same time opening a hydrophobic channel believed to allow CO2 entry (5). It will be interesting to see if the ethylsulfonate group bound to the enzyme induces a similar conformational change.
In addition to catalyzing the carboxylation and protonation of 2-KPC, 2-KPCC also catalyzes the reverse of the physiological carboxylation reaction at a slow rate as well as the decarboxylation of acetoacetate and exchange of 14CO2 into acetoacetate, reactions that provided evidence for enolacetone as an intermediate in the reaction mechanism (Fig. 2C) (4). The latter two reactions are interesting in that they are stimulated 72- and 38-fold, respectively, by the presence of a saturating concentration of CoM (4). It will be interesting to see if bound BES mimics the stimulatory effect of CoM on the decarboxylation and exchange reactions.
Finally, the utility of BES alkylation as a mechanistic probe is seen in the charge-transfer absorbance of alkylated 2-KPCC presented in Fig. 7. No charge transfer was observed for the alkylated enzyme at pH 7.4, indicating that the flavin thiol remains protonated at this normal pH value. To observe the maximal charge transfer, it was necessary to increase the pH to a value of 11. In contrast, the charge transfer for “classic” DSOR enzymes such as mercuric reductase alkylated at the interchange thiol by iodoacetamide can be seen at pH 7.4 (29). This observation is compatible with the unique, hydrophobic active site architecture of 2-KPCC that presumably results in a higher pKa value for the flavin thiol, at least in the conformation in which the interchange thiol has been alkylated by the ethylsulfonate group.
This work was supported, in whole or in part, by National Institutes of Health Grant GM51805.
- CoM
- coenzyme M (2-mercaptoethanesulfonate)
- 2-KPC
- 2-ketopropyl-CoM
- 2-KPCC
- 2-ketopropyl-CoM carboxylase/oxidoreductase
- DSOR
- disulfide oxidoreductase
- BES
- 2-bromoethanesulfonate
- EaCoMT
- epoxyalkane:CoM transferase
- R-HPCDH
- R-hydroxypropyl-CoM dehydrogenase
- β-HBDH
- β-hydroxybutyrate dehydrogenase
- R-HPC
- R-hydroxypropyl-CoM
- ES
- ethylsulfonate.
REFERENCES
- 1.Allen J. R., Clark D. D., Krum J. G., Ensign S. A. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 8432–8437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ensign S. A. (2001) Biochemistry 40, 5845–5853 [DOI] [PubMed] [Google Scholar]
- 3.Ensign S. A., Allen J. R. (2003) Annu. Rev. Biochem. 72, 55–76 [DOI] [PubMed] [Google Scholar]
- 4.Clark D. D., Allen J. R., Ensign S. A. (2000) Biochemistry 39, 1294–1304 [DOI] [PubMed] [Google Scholar]
- 5.Nocek B., Jang S. B., Jeong M. S., Clark D. D., Ensign S. A., Peters J. W. (2002) Biochemistry 41, 12907–12913 [DOI] [PubMed] [Google Scholar]
- 6.Pandey A. S., Nocek B., Clark D. D., Ensign S. A., Peters J. W. (2006) Biochemistry 45, 113–120 [DOI] [PubMed] [Google Scholar]
- 7.Pai E. F. (1991) Curr. Opin. Struct. Biol. 1, 796–803 [Google Scholar]
- 8.Taylor C. D., McBride B. C., Wolfe R. S., Bryant M. P. (1974) J. Bacteriol. 120, 974–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiMarco A. A., Bobik T. A., Wolfe R. S. (1990) Annu. Rev. Biochem. 59, 355–394 [DOI] [PubMed] [Google Scholar]
- 10.Wolfe R. S. (1991) Annu. Rev. Microbiol. 45, 1–35 [DOI] [PubMed] [Google Scholar]
- 11.Thauer R. K. (1998) Microbiology 144, 2377–2406 [DOI] [PubMed] [Google Scholar]
- 12.Gunsalus R. P., Romesser J. A., Wolfe R. S. (1978) J. Bacteriol. 17, 2374–2377 [DOI] [PubMed] [Google Scholar]
- 13.Zinder S. H., Anguish T., Cardwell S. C. (1984) Appl. Environ. Microbiol. 47, 1343–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sparling R., Daniels L. (1987) Can. J. Microbiol. 33, 1132–1136 [Google Scholar]
- 15.Goenrich M., Mahlert F., Duin E. C., Bauer C., Jaun B., Thauer R. K. (2004) J. Biol. Inorg. Chem. 9, 691–705 [DOI] [PubMed] [Google Scholar]
- 16.Boyd J. M., Ellsworth A., Ensign S. A. (2006) J. Bacteriol. 188, 8062–8069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allen J. R., Ensign S. A. (1997) J. Bacteriol. 179, 3110–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyd J. M., Ensign S. A. (2005) Biochemistry 44, 8543–8553 [DOI] [PubMed] [Google Scholar]
- 19.Clark D. D., Ensign S. A. (2002) Biochemistry 41, 2727–2740 [DOI] [PubMed] [Google Scholar]
- 20.Krum J. G., Ellsworth H., Sargeant R. R., Rich G., Ensign S. A. (2002) Biochemistry 41, 5005–5014 [DOI] [PubMed] [Google Scholar]
- 21.Krüger K., Lang G., Weidner T., Engel A. M. (1999) Appl. Microbiol. Biotechnol. 52, 666–669 [DOI] [PubMed] [Google Scholar]
- 22.Cleland W. W. (1979) Methods Enzymol. 63, 103–138 [DOI] [PubMed] [Google Scholar]
- 23.Copeland R. A. (2000) Enzymes, 2nd Ed., Wiley-VCH, New York [Google Scholar]
- 24.Aliverti A., Curti B., Vanoni M. A. (1999) in Flavoprotein Protocols (Chapman S. K., Reid G. A. eds) pp. 9–23, Humana Press, Totowa, NJ [Google Scholar]
- 25.Hinderberger D., Piskorski R. R., Goenrich M., Thauer R. K., Schweiger A., Harmer J., Jaun B. (2006) Angew. Chem. Int. Ed. Eng. 45, 3602–3607 [DOI] [PubMed] [Google Scholar]
- 26.Kunz R. C., Horng Y. C., Ragsdale S. W. (2006) J. Biol. Chem. 281, 34663–34676 [DOI] [PubMed] [Google Scholar]
- 27.Kunz R. C., Dey M., Ragsdale S. W. (2008) Biochemistry 47, 2661–2667 [DOI] [PubMed] [Google Scholar]
- 28.Krishnakumar A. M., Nocek B. P., Clark D. D., Ensign S. A., Peters J. W. (2006) Biochemistry 45, 8831–8840 [DOI] [PubMed] [Google Scholar]
- 29.Fox B. S., Walsh C. T. (1983) Biochemistry 22, 4082–4088 [DOI] [PubMed] [Google Scholar]