

Abstract
Straight chain fatty acid α-oxidation increases during differentiation of 3T3-L1 adipocytes, leading to a marked accumulation of odd chain length fatty acyl moieties. Potential roles of this pathway in adipocyte differentiation and lipogenesis are unknown. Mammalian fatty acid 2-hydroxylase (FA2H) was recently identified and suggested to catalyze the initial step of straight chain fatty acid α-oxidation. Accordingly, we examined whether FA2H modulates adipocyte differentiation and lipogenesis in mature adipocytes. FA2H level markedly increases during differentiation of 3T3-L1 adipocytes, and small interfering RNAs against FA2H inhibit the differentiation process. In mature adipocytes, depletion of FA2H inhibits basal and insulin-stimulated glucose uptake and lipogenesis, which are partially rescued by the enzymatic product of FA2H, 2-hydroxy palmitic acid. Expression of fatty-acid synthase and SCD1 was decreased in FA2H-depleted cells, and levels of GLUT4 and insulin receptor proteins were reduced. 2-Hydroxy fatty acids are enriched in cellular sphingolipids, which are components of membrane rafts. Accelerated diffusional mobility of raft-associated lipids was shown to enhance degradation of GLUT4 and insulin receptor in adipocytes. Consistent with this, depletion of FA2H appeared to increase raft lipid mobility as it significantly accelerated the rates of fluorescence recovery after photobleaching measurements of lipid rafts labeled with Alexa 488-conjugated cholera toxin subunit B. Moreover, the enhanced recovery rates were partially reversed by treatment with 2-hydroxy palmitic acid. In conclusion, our findings document the novel role of FA2H in adipocyte lipogenesis possibly by modulation of raft fluidity and level of GLUT4.
Keywords: Adipocyte, Fatty Acid Oxidation, Fluorescence, Gene Silencing, Glucose Transport, Lipid Raft, Lipogenesis, Lysosomes, Fatty Acid 2-Hydroxylation
Introduction
In addition to mitochondrial β-oxidation, fatty acids (FA)2 can also be oxidized in peroxisomes, where they can undergo β- as well as α-oxidation. Peroxisomal α-oxidation produces fatty acyl moieties with one less carbon (odd chain length) without generating acetyl-CoA (1). It is well known that peroxisomal α-oxidation is important for catabolism of branched-chain FA and also for very long chain FA (2). Its contribution to catabolism of other long chain FA is not known, so its role in whole cell energy homeostasis remains unclear. There is evidence for up-regulation of FA α-oxidation during the preadipocyte to adipocyte transition as cells begin accumulating lipids. Using electrospray ionization-mass spectrometry and gas chromatography-coupled mass spectrometry, we and others demonstrated during differentiation of 3T3-L1 cells a marked increase in activity of peroxisomal FA α-oxidation, which was reflected in accumulation of odd chain length acyl moieties in major lipid species (3, 4). The underlying mechanisms for this change and the functions of the α-oxidation pathway in adipocyte differentiation and lipid metabolism in mature adipocytes have not been explored.
Although straight chain FA α-oxidation was first reported in 1964 (5), a detailed understanding of the enzymatic pathway is still unclear. It was postulated that α-oxidation is initiated by 2-hydroxylation of free FA, followed by thioesterification, formation of aldehyde, and dehydrogenation to form an FA with one less carbon. The FA can then undergo more α-oxidation cycles or be incorporated into lipid species (1, 6). Another effect of active α-oxidation is to increase the proportion of lipids with shorter acyl chains and acyl chains with modifications (e.g. 2-hydroxy FA (2-OH FA)). The physiological significance of these changes in lipid composition has not been explored.
In mammalian cells, two types of fatty acid 2-hydroxylase have been identified, phytanoyl-CoA hydroxylase (7) and fatty acid 2-hydroxylase (FA2H) (8, 9). Phytanoyl-CoA hydroxylase is an α-ketoglutarate-dependent acyl-CoA 2-hydroxylase, and its primary function is the degradation of 3-methyl FA, which cannot be degraded by β-oxidation pathway (7). Previous reports on the enzymatic activity of phytanoyl-CoA hydroxylase on straight chain fatty acids or their CoA esters are controversial (10–12). Thus, its roles in 2-hydroxylation and α-oxidation of straight chain FA are not clear. Mammalian FA2H is an NAD(P)H-dependent monooxygenase that was cloned independently by two research groups (8, 9). Mammalian sphingolipids are enriched in 2-OH FA generated by FA2H (6). Sphingolipids are important constituents of membrane raft domains that are thought to coordinate intracellular trafficking and signaling (13). FA2H is highly expressed in brain and epidermis. Absence of 2-OH FA sphingolipids has no effect on neural development but causes late-onset axon and myelin sheath degeneration (14). In the epidermis, 2-OH FA ceramides are essential for the permeability barrier function, and FA2H provides the precursor 2-OH FA for their synthesis. Interestingly, FA2H is required for epidermal lamellar membrane formation during keratinocyte differentiation (15). Physiological functions of FA2H in other cell types/organs are largely unknown.
In this study, we focused on FA2H, which catalyzes the initial step of the FA α-oxidation pathway, resulting in production of 2-OH FA. We studied the regulation of FA2H during adipocyte differentiation and asked whether it influences lipogenesis in mature adipocytes. Lateral mobility of raft-associated lipids might be important in adipogenesis because enhanced raft lipid mobility accelerates the trafficking of GLUT4 to lysosomes and its degradation in 3T3-L1 adipocytes (16). Accordingly, we also evaluated the importance of FA2H in regulating lateral mobility of raft-associated lipids, co-localization of GLUT4 with lysosomes and GLUT4 protein levels.
EXPERIMENTAL PROCEDURES
Materials
Fetal bovine serum, calf serum, Dulbecco's modified Eagle's medium (DMEM), SilencerTM siRNA construction kit, LipofectamineTM RNAiMAX, and Alexa 568 phalloidin were ordered from Invitrogen. 1-Oleoyl-2-{6-[(7-nitro-2–1,3-benzoxadiazol-4-yl)amino]hexanoyl}-sn-glycero-3-phosphocholine was obtained from Avanti Polar Lipids, Inc. (Alabaster, AL). Rabbit anti-GLUT1 and -GLUT4 antibodies were kindly provided by Dr. Michael Mueckler (Washington University, St. Louis, MO). Rabbit anti-insulin receptor β-subunit was obtained from Upstate (Charlottesville, VA). Rabbit anti-FABP4/AP2 antibody was ordered from Abcam (Cambridge, MA). Rat anti-LAMP1 antibody was obtained from Developmental Studies Hybridoma Bank (Iowa City, IA). Other reagents were obtained from Sigma.
Cell Culture of 3T3-L1 Cells and Differentiation into the Adipocyte Phenotype
3T3-L1 cells (from American Type Culture Collection) were cultured to confluence in DMEM containing 20% calf serum with medium change every 2 days as described previously (17). Two days after cell confluence, differentiation was initiated by adding differentiation medium (0.5 mm isobutylmethylxanthine, 0.25 μm dexamethasone, 1 μg/ml insulin in DMEM containing 10% fetal bovine serum). Two days later, isobutylmethylxanthine and dexamethasone were removed, and insulin (1 μg/ml) was maintained for 2 more days. Thereafter, cells were grown in DMEM containing 10% fetal bovine serum with media replacement every 2 days.
siRNA Construction and Transfection
siRNAs directed against mouse FA2H were constructed and purified employing the SilencerTM siRNA construction kit. The sequences specific for mouse FA2H 5′-CCGCAGGATCCCACAGAGA-3′ (sequence 1) and 5′-GCACTAAACTGTGGGATTA-3′ (sequence 2) were selected based upon their potency to inhibit the target gene expression. Transfection of siRNA in 3T3-L1 fibroblasts was performed using LipofectamineTM RNAiMAX according to the manufacturer's instructions. Transfection of siRNA (10 nm final concentration) in adipocytes was performed exactly as described previously (18). A scrambled siRNA (Invitrogen) was used as a negative control. The transfection media were replaced with growing media on the 2nd day. Most of the experiments were performed two days later.
Quantitative Gas Chromatography Analysis of TAG
Lipids were extracted by the method of Bligh-Dyer in the presence of internal standard (T21:0 TAG, 100 nmol/mg protein) and separated on silica gel 60-Å plates. Spots corresponding to TAG were visualized with 0.01% rhodamine 6G and identified with TAG standard. The bands were scraped and extracted with chloroform/methanol, 2:1. FA methyl esters of the TAG fractions were prepared and analyzed by quantitative gas chromatography as described previously (18).
Lipogenesis Assay
Lipogenesis was assayed as described previously (19). Briefly, cells after the indicated treatment were incubated with 5 mm d-[U-14C]glucose (1 μCi per well) for 60 min at 37 °C. Cells were then washed on ice three times with cold phosphate-buffered saline, scraped into 1 ml of phosphate-buffered saline, and shaken vigorously with 5 ml of Betafluor scintillant (National Diagnostics, Manville, NJ). The samples were settled overnight, and radioactivity in the organic phase was determined by liquid scintillation counting. Activities of lipogenesis were normalized to protein concentrations.
Glucose Uptake Assay
Glucose uptake assay was measured as described previously (20). Briefly, cells were serum-starved with DMEM containing 0.2% bovine serum albumin for 4 h at 37 °C. Cells were then treated with 100 nm insulin for 15 min. Glucose uptake was initiated by adding 2-[3H]deoxyglucose and measured as described previously (20). Nonspecific background uptake in the presence of the inhibitor cytochalasin B (20 μm) was subtracted from all values. Glucose uptake activities were normalized to protein concentrations.
Protein Extraction and Western Blot
Cell monolayers were washed twice with ice-cold phosphate-buffered saline and lysed at 4 °C for 30 min with a buffer containing 50 mm Tris·HCl, pH 7.6, 1% Triton X-100, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 10 mm NaF, 1 mm sodium pyrophosphate, 1 mm sodium orthovanadate, and 1% protease inhibitor mixture solution (Sigma). The cell lysates were clarified by centrifugation at 10,000 × g for 10 min at 4 °C prior to separation by SDS-PAGE. The resolved proteins were transferred to nitrocellulose membranes, and the membranes were blocked with powdered milk solution (5% (w/v)). Individual proteins were detected with specific antibodies and visualized by blotting with horseradish peroxidase-conjugated secondary antibodies as described previously (17).
Fluorescence Recovery after Photobleaching (FRAP)
3T3-L1 adipocytes on 8-well chambered coverglass (Fisher) were transfected with siRNAs as described above. Prior to imaging, the cells were incubated with 1 μg/ml Alexa 488-labeled CTxB (Invitrogen) at 4 °C for 30 min and then washed three times in chilled phenol red-free medium supplemented with 25 mm HEPES buffer, pH 7.4. Images were acquired at room temperature on a Zeiss LSM 510 confocal microscope (Carl Zeiss MicroImaging, Thornwood, NY) with a C-Apochromat 40×, 1.2 numerical aperture, water-immersion objective. The region of interest (ROI) for bleaching was placed in the rim of the plasma membrane (PM) of the labeled adipocytes. Photobleaching of fluorescent CTxB in the ROIs was performed for 100 iterations using the 488-nm laser line of an argon laser at full power. In the post-bleach period, the recovery of fluorescence was recorded at 15-s intervals for 200 s with 5% laser intensity. For data analysis, using FRAP profiler plugin included with MBF-ImageJ software, fluorescence intensity in the ROIs at each time point was automatically quantified, background subtracted, corrected for loss of fluorescence during imaging, and normalized to the pre-bleach intensity. The recovery curves were fitted by a nonlinear least squares technique with origin 7.5 software, assuming a one-phase exponential equation: F(t) = F0 + (FE − F0) × (1 − exp(−τ t)), where F0 is the intensity of the ROI at t = 0 s after photobleaching; FE is the end value of the recovered fluorescence intensity; and τ is the fitted parameter (16). The mobile fraction (Mf) was calculated with the equation Mf = (FE − F0)/(FI − F0), where FI is the initial fluorescence intensity before photobleaching. Diffusion coefficients (D) were calculated as described previously (21).
Immunofluorescent Microscopy
Cells grown on coverslips were fixed with 3% paraformaldehyde, quenched with 50 mm ammonium chloride, permeabilized, and then blocked with goat serum. Following incubation with primary antibodies and then Alexa 594- or -488-conjugated secondary antibodies, the coverslips were mounted as described previously (22). Confocal microscopy was performed on a Zeiss LSM 510 confocal microscope (Carl Zeiss MicroImaging, Thornwood, NY). Images in two channels were acquired by sequential scanning under the same parameters. Quantitative co-localization analyses were performed using ImageJ software with JACoP plugin as described previously (23). Co-localization was analyzed within the entire images. The same threshold values were used for all images, and Manders' M1 and M2 co-localization coefficients were calculated (24). To obtain the volume of tubulin and actin, 3T3-L1 adipocytes were cultured on the coverslips and then fixed, permeabilized, and stained as described in TIRF microscopy. The Z-series confocal images of tubulin and actin were captured with 0.5-μm increments on an Olympus BX52 microscope (Olympus America, Center Valley, PA) equipped with a 100×/1.35 NA UPlanApo oil-immersion objective lens by using QED imaging software (Media Cybernetics). Volume of an entire cell and volumes of tubulin and actin in one cell were calculated by using voxel counter plugin included with the ImageJ software.
Fixed Cell Total Internal Reflection Fluorescence (TIRF) Microscopy
Cells were fixed in 3% paraformaldehyde, quenched, permeabilized, blocked, and incubated with Alexa 568-conjugated phalloidin for actin in blocking solution for 1 h. Cells were washed four times with phosphate-buffered saline and imaged on an Olympus IX-81 inverted fluorescence microscope (Olympus America, Center Valley, PA) with a 60×/1.45 NA PlanApo oil immersion objective lens. To avoid user bias, cells were randomly selected by bright field illumination before TIRF imaging as described previously (25). Image analysis was performed by using ImageJ software.
RNA Extraction and Real Time Quantitative PCR
Real time quantitative PCR (RT-PCR) analyses of RNA message from cultured cells were performed as described previously (26). In brief, RNA was isolated utilizing TRIzol (Invitrogen), washed in 75% ethanol, dried, and redissolved in water. Reverse transcription was performed using the SuperScript III first strand synthesis system (Invitrogen). RT-PCR assays were performed on an ABI 7500 fast real time PCR system (Applied Biosystems) using SYBR Green PCR master mix. Acidic ribosomal phosphoprotein (36B4) RNA was used for normalization of expression. Primers for quantitative PCR analyses are listed in supplemental Table S1.
Statistical Analyses
The data are presented as means ± S.E. The results were analyzed by one-way analysis of variance followed by post hoc t test. A p value < 0.05 was considered statistically significant.
RESULTS
Regulation of FA2H during Hormone-induced Differentiation of 3T3-L1 Adipocytes
During hormone-induced differentiation of 3T3-L1 adipocytes, FA α-oxidation was activated, and the amounts and percentages of odd chain length acyl moieties were dramatically increased in major lipid species (3, 4). Recent studies suggest that FA2H catalyzes FA 2-hydroxylation, an initial step of the FA α-oxidation pathway to generate 2-OH FA (Fig. 1A). Accordingly, we measured levels of FA2H during adipocyte differentiation. Fig. 1B shows that FA2H mRNA levels dramatically increase during differentiation of 3T3-L1 adipocytes. We next explored whether increased FA2H is required for adipocyte differentiation. Accordingly, we synthesized two siRNAs against FA2H, which significantly decrease FA2H levels in 3T3-L1 cells (Fig. 1C). Sequence 2 is more potent than sequence 1 in depleting FA2H. Both siRNAs decreased the expression of markers for adipocyte differentiation, including peroxisome proliferator-activated receptor γ, CD36, and SCD1 (Fig. 1E). In contrast, the levels of pref-1, an inhibitory factor for adipocyte differentiation (27), are much higher in cells pretreated with FA2H siRNAs (Fig. 1D). Consistent with its potency in depleting FA2H, sequence 2 blocks the expression of adipocyte markers more efficiently (Fig. 1E). We further measured the accumulation of TAG using quantitative gas chromatography. Both FA2H siRNAs block the accumulation of TAG during adipocyte differentiation, and sequence 2 has a more potent effect (Fig. 2A). The fatty acid composition analysis also shows the decrease of the percentage of monounsaturated fatty acids in TAG (Fig. 2B), which agrees with the decrease of SCD1 levels by FA2H siRNAs during differentiation (Fig. 1E). Interestingly, percentages of polyunsaturated FA in TAG increase (Fig. 2D), whereas their total amounts decrease by FA2H siRNAs (Fig. 2C). Collectively, these data suggest that FA2H is important for full differentiation of 3T3-L1 cells into adipocytes.
FIGURE 1.
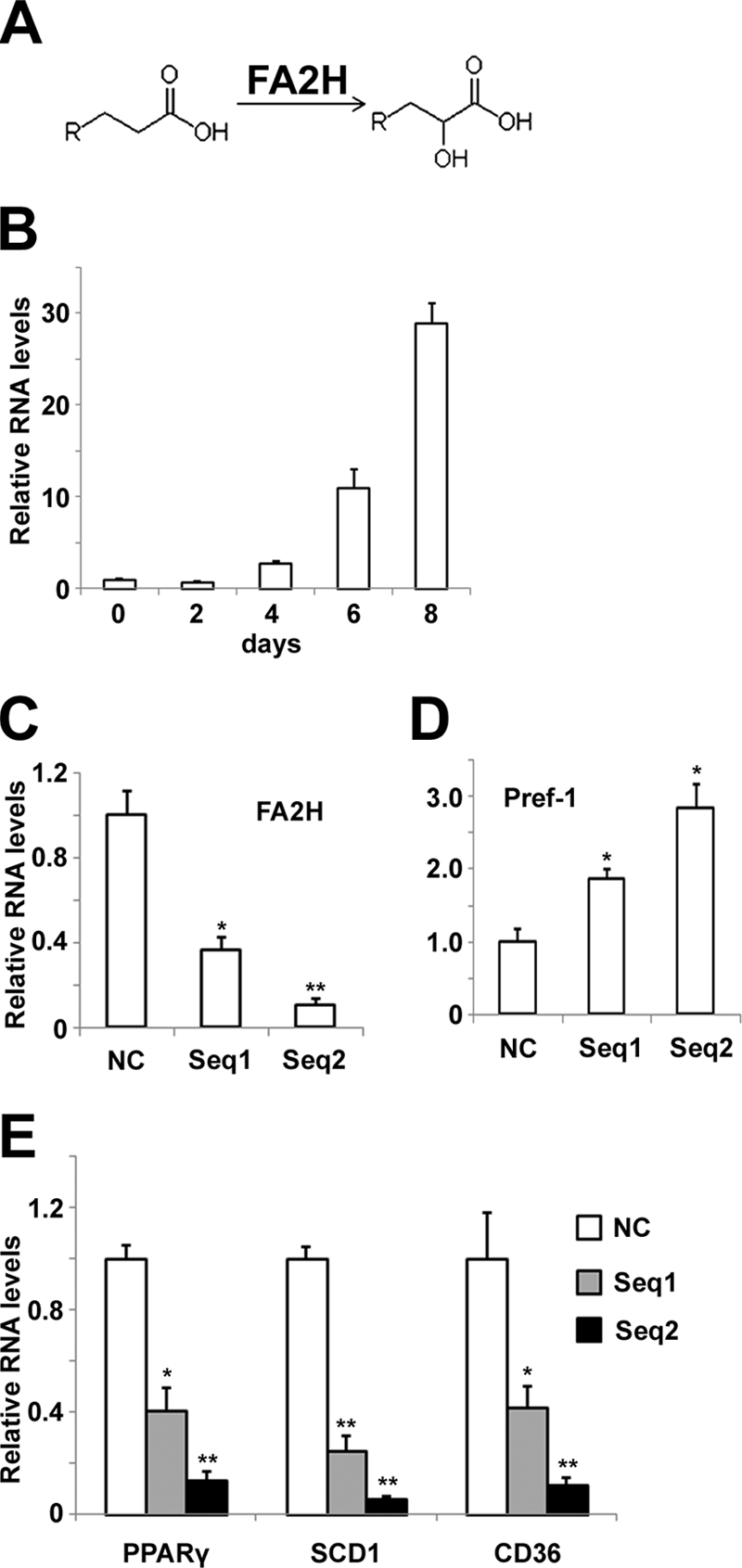
Treatment of siRNAs against FA2H blocks hormone-induced differentiation of 3T3-L1 adipocytes. A, FA 2-hydroxylation catalyzed by FA2H (8). B, mRNA samples from differentiating 3T3-L1 cells at the indicated stages were prepared, and the FA2H levels were analyzed by RT-PCR as described under “Experimental Procedures.” C, 3T3-L1 cells were treated with a negative control (NC) siRNA or siRNAs recognizing FA2H (sequence (seq) 1 and sequence 2), and mRNA samples were prepared 2 days after transfection. FA2H levels were analyzed by RT-PCR. D and E, 3T3-L1 cells pretreated with a negative control siRNA or siRNAs recognizing FA2H (sequence 1 and sequence 2) were induced to differentiation. On day 8, mRNA samples were prepared, and the Pref1, peroxisome proliferator-activated receptor γ, SCD1, and CD36 levels were analyzed by RT-PCR. The results represent means ± S.E. of three independent experiments. *, p < 0.05; **, p < 0.01 (compared with negative control).
FIGURE 2.

Treatment of siRNAs against FA2H blocks TAG accumulation during differentiation of 3T3-L1 adipocytes. A, 3T3-L1 cells pretreated with a negative control (NC) siRNA or siRNAs recognizing FA2H (sequence (seq) 1 and sequence 2) were induced to differentiation. On day 8, total lipids were extracted, and TAG were measured by quantitative gas chromatography and normalized to total protein as described under “Experimental Procedures.” Results are the means ± S.E. of three independent experiments. *, p < 0.01 (compared with negative control). B, total saturated and monosaturated (Mono) FA were presented. C, total polyunsaturated FA (PUFA) were presented. D, percentages of polyunsaturated FA were presented.
Depletion of FA2H Impairs Lipogenesis and Glucose Uptake in Adipocytes
We next examined whether FA2H is critical for lipogenesis, which is important to adipocyte differentiation. To avoid effects due to alterations in differentiation, we transfected mature adipocytes with FA2H siRNA and accomplished about 80% knockdown of FA2H levels using sequence 2 (Fig. 3A). Cells were then starved for 4 h, stimulated with 100 nm insulin, and incubated with d-[U-14C]glucose for 1 h to access rates of lipogenesis. Interestingly, depletion of FA2H decreases lipogenesis under both basal and insulin-stimulated conditions (Fig. 3B). To further confirm the specificity of FA2H knockdown, we examined whether 2-OH FA, the enzymatic product of the FA2H, could reverse its inhibitory effects on lipogenesis. Accordingly, cells depleted with FA2H were pretreated with 100 μm 2-OH palmitic acid for 2 days prior to the assay for lipogenesis. As shown (Fig. 3B), 2-OH palmitic acid partially reversed the inhibitory effects of FA2H depletion on both basal and insulin-stimulated lipogenesis. Glucose uptake is the major driving force for lipogenesis in adipocytes, so we examined whether FA2H knockdown inhibits glucose uptake and whether this effect could be reversed by 2-OH palmitic acid. Cells pretreated with control siRNA or siRNA against FA2H were starved and stimulated with insulin. Glucose uptake was initiated by incubation with 2-[3H]deoxyglucose for 5 min. Similar to its effect on lipogenesis, FA2H depletion significantly reduced glucose uptake in cells depleted with FA2H under both basal and stimulated conditions (Fig. 4). The inhibitory effects were partially reversed by preincubation with 100 μm 2-OH palmitic acid (Fig. 4).
FIGURE 3.

FA2H depletion impairs basal and insulin-stimulated lipogenesis in adipocytes. 3T3-L1 adipocytes were treated with a negative control (NC) siRNA or an siRNA recognizing FA2H. A, 2-day post-transfection, mRNA samples were prepared and FA2H levels were analyzed by RT-PCR as described under “Experimental Procedures.” B, 100 μm 2-OH palmitic acid (2-OH) was added 2 days before assay as indicated. Cells were starved for 4 h and then stimulated with 100 nm insulin for 15 min. Lipogenesis was assayed using 5 mm d-[U-14C]glucose (1 μCi per well) and normalized to total protein. *a, p < 0.05 (compared with negative control). *b, p < 0.05 (compared with FA2H). Results are the means ± S.E. of three independent experiments. AU, arbitrary unit.
FIGURE 4.
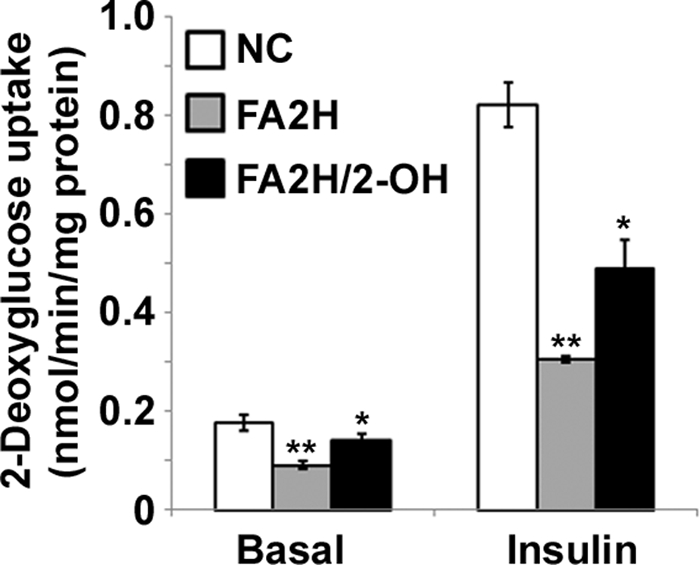
FA2H depletion impairs basal and insulin-stimulated glucose uptake in adipocytes. 3T3-L1 adipocytes were treated with a negative control (NC) siRNA or an siRNA recognizing FA2H. 100 μm 2-OH palmitic acid (2-OH) was added as indicated. Cells were starved for 4 h and then stimulated with 100 nm insulin for 15 min. Glucose uptake was assayed as described under “Experimental Procedures.” **, p < 0.01 (compared with negative control); *, p < 0.05 (compared with FA2H). Results are the means ± S.E. of three independent experiments.
Depletion of FA2H Decreases Protein Levels of GLUT4 and Insulin Receptor (IR) in Adipocytes
Glucose uptake in adipocytes is dependent on the activity of GLUT4 and its membrane recruitment by insulin signaling following insulin binding to the IR. We examined the effect of FA2H depletion on protein levels of GLUT4 and IR. Protein levels of both decreased significantly in FA2H-depleted cells (Fig. 5, A–C). Interestingly, their decreased protein levels were partially reversed by preincubation with 100 μm 2-OH palmitic acid (Fig. 5, A–C). This is not due to dedifferentiation of adipocytes because the level of FABP4/AP2, a marker for adipocyte differentiation, is not changed (Fig. 5A). Moreover, levels of GLUT1 are not changed, suggesting FA2H depletion regulates degradation of specific proteins, not global degradative pathways (Fig. 5A). A dose response of 2-OH palmitic acid for reversing GLUT4 expression was performed. Significant effects were observed at 20 μm reaching a maximum at 100 μm (supplemental Fig. S1). The lower effects at 200 μm may be due to toxicity of high level of 2-OH palmitic acid. The decreased GLUT4 and IR levels likely contribute to the decreased insulin-stimulated glucose uptake and lipogenesis in cells depleted with FA2H.
FIGURE 5.

FA2H depletion decreases GLUT4 and IR levels. A, 3T3-L1 adipocytes were treated with a negative control (NC) siRNA or an siRNA recognizing FA2H. 100 μm 2-OH palmitic acid (2-OH) was added as indicated. Whole cell lysates were prepared and analyzed by immunoblotting (IB) using antibodies recognizing GLUT4, IR, tubulin, FABP4/AP2, or GLUT1 as described under “Experimental Procedures.” Band intensities of GLUT4 (B) and IR (C) were quantified with ImageJ software. The graph was acquired from four independent experiments. **, p < 0.01 (compared with negative control); *, p < 0.05 (compared with FA2H). The data represent the means ± S.E.
Regulation of FAS and SCD1 Gene Expression by FA2H Depletion in Adipocytes
Because high carbohydrate and insulin stimulation increase lipogenic gene expression in adipocytes (28, 29), we examined whether chronic decrease of glucose uptake by FA2H depletion may also modulate lipogenic gene expression as a secondary effect leading to impaired lipogenesis. Accordingly, mRNA samples from control cells and cells depleted of FA2H were prepared. To confirm whether 2-OH palmitic acid could reverse the effect of FA2H depletion on lipogenic gene expression, mRNA from FA2H-depleted cells pretreated with 2-OH palmitic acid was also purified. As expected, FA2H depletion inhibited FAS and SCD1 expression, and this inhibitory effect could be partially reversed by 2-OH palmitic acid (Fig. 6).
FIGURE 6.
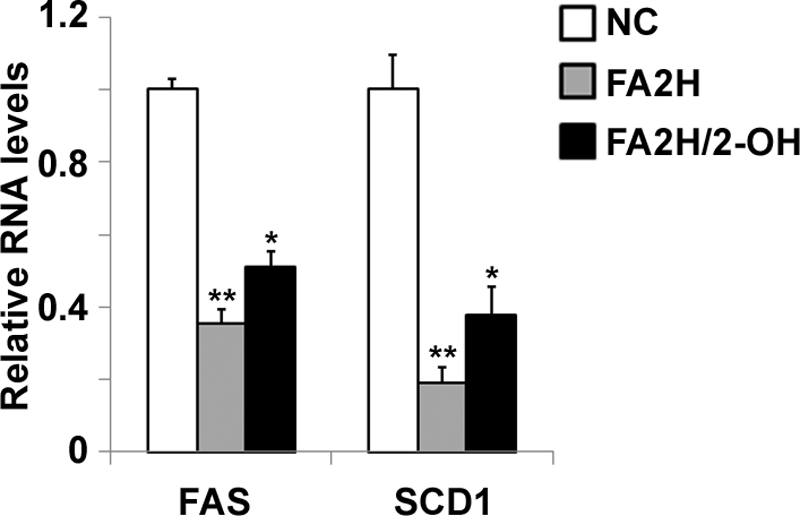
FA2H depletion decreases expression of FAS and SCD1. 3T3-L1 adipocytes were treated with a negative control (NC) siRNA or an siRNA recognizing FA2H. 100 μm 2-OH palmitic acid (2-OH) was added as indicated. mRNA samples were prepared, and the FAS and SCD1 levels were analyzed by RT-PCR as described under “Experimental Procedures.” **, p < 0.01 (compared with negative control); *, p < 0.05 (compared with FA2H). The data represent the means ± S.E. of three independent experiments.
Depletion of FA2H Increases Recovery Rate of FRAP Measurement of Alexa 488-labeled CTxB in Adipocytes
2-OH FA containing lipid species participate in intramolecular and intermolecular hydrogen bonding and enhance the formation of sterol- and sphingolipid-enriched lipid rafts (30, 31). Lipid composition plays an important role in regulating membrane mobility, and enhanced lateral mobility of raft-associated lipids decreases protein levels of GLUT4 and insulin receptor in 3T3-L1 adipocytes (16). Accordingly, we examined whether FA2H may modulate levels of GLUT4 and IR in adipocytes by influencing lipid-lipid interaction in lipid rafts. To confirm whether FA2H has direct effects on lipid rafts, we measured diffusional mobility of raft-associated lipids using FRAP experiments in control and FA2H knockdown adipocytes pre-labeled with Alexa 488-labeled CTxB. CTxB internalizes rapidly at 37 °C (32), and mobility of CTxB bound to PM are much slower as compared with membrane proteins and lipids (16, 33). Accordingly, the FRAP experiments were performed at room temperature to minimize internalization during fluorescence recovery as suggested previously (16). We photo-bleached an area of the rim of the plasma membrane using high intensity laser. The diffusive exchange of photobleached CTxB with nearby unbleached labeled molecules was then followed for 200 s. Recovery into the bleached region was higher in FA2H-depleted adipocytes than in adipocytes treated with control siRNA, and this effect was reversed by preincubation with 2-OH palmitic acid (Fig. 7, A and B). The fluorescence recovery of individual experiments was shown in supplemental Fig. S2. The mean Mf values ± S.E. calculated from 12 independent experiments were increased from 6.7 ± 1.1 to 27 ± 2.5% in control and FA2H knockdown adipocytes (Fig. 7C). Pretreatment with 2-OH palmitic acid in FA2H-depleted adipocytes reversed Mf from 27 ± 2.5 to 13 ± 1.8% (Fig. 7C). Mf represents the percentage of fluorescent molecules that were able to recover into the bleached area over the time course of the experiment. Consistent with this, diffusion coefficients (D, μm2 s−1) were also increased from 0.07 ± 0.01 to 0.23 ± 0.02 in control and FA2H knockdown adipocytes (Fig. 7D). Pretreatment with 2-OH palmitic acid in FA2H-depleted adipocytes reversed D from 0.23 ± 0.02 to 0.13 ± 0.01 (Fig. 7D). In contrast to sphingolipids, phosphatidylcholine (PC) is not enriched in lipid rafts. We performed similar FRAP experiments using 1-oleoyl-2-{6-[(7-nitro-2–1,3-benzoxadiazol-4-yl)amino]hexanoyl}-sn-glycero-3-phosphocholine. Interestingly, FA2H depletion does not affect the diffusion of labeled PC molecules (supplemental Fig. S3). These experiments suggest that FA2H depletion specifically modulates lateral mobility of lipids that are enriched in rafts.
FIGURE 7.

FA2H depletion enhances diffusional mobility of raft-associated lipids. 3T3-L1 adipocytes were treated with a negative control (NC) siRNA or an siRNA recognizing FA2H. 100 μm 2-OH palmitic acid (2-OH) was added as indicated. Labeling with Alexa 488-CTxB and FRAP measurements were performed as described under “Experimental Procedures.” A, selected images from a confocal FRAP experiment in adipocytes labeled with Alexa 488-CTxB at room temperature. The scale bar represents 10 μm. B, kinetics of recovery for Alexa 488-CTxB. Each curve represents means ± S.E. from 12 experiments. C, calculated Mf. D, calculated diffusion coefficients (D). *a, p < 0.01 (compared with negative control). *b, p < 0.01 (compared with FA2H).
FA2H Depletion Does Not Induce Actin Remodeling in Adipocytes
Altered organization of cell actin has been shown to regulate membrane lateral mobility (34). A recent study also identified that actin polymerization controls a distal GLUT4 trafficking event (25). Accordingly, we examined whether FA2H depletion modulates cortical actin network and/or microtubular network. Actin was stained with Alexa 568-phalloidin and tubulin labeled with antibody. Images of three-dimensional projection of confocal Z-sections were generated, and the volumes of actin and tubulin normalized to total cell volumes were calculated. No differences were observed in control cells, cells depleted with FA2H, and cells depleted with FA2H and incubated with 2-OH palmitic acid (Fig. 8, A and B, and supplemental Fig. S4). We further used TIRF microscopy to image actin associated with the PM in response to FA2H depletion, and no differences were observed (Fig. 8C and supplemental Fig. S5). Collectively, our data demonstrated that regulation of mobility of CTxB by FA2H depletion is not due to modulation of actin cytoskeleton organization.
FIGURE 8.

FA2H depletion does not induce actin or tubulin remodeling in adipocytes. 3T3-L1 adipocytes were treated with a negative control (NC) siRNA or an siRNA recognizing FA2H. 100 μm 2-OH palmitic acid (2-OH) was added as indicated. The cells were fixed and labeled, and the entire cell volume was imaged by confocal microscope as described under “Experimental Procedures.” Volumes of actin (A) and tubulin (B) expressed were normalized to total cell volume (n = 5 cells). C, cells were fixed, labeled with Alexa 568-phalloidin, and then imaged by TIRFM as described under “Experimental Procedures.” The fluorescence of cells from 20 experiments is shown. The data represent the means ± S.E. NS, not significant; A.U., arbitrary unit.
Co-localization of GLUT4 and Lysosome Marker Is Increased in FA2H-depleted Adipocytes
We further investigated co-localization of GLUT4 with lysosomes, where it is degraded (35). Adipocytes grown on coverslips were pretreated with siRNAs, serum-starved, and fixed. Subcellular distribution of GLUT4 and LAMP1, a lysosome marker, was examined by immunostaining. Consistent with the decrease on GLUT4 protein levels, depletion of FA2H increased GLUT4 co-localization with LAMP1, and this effect was partially reversed by preincubation with 2-OH palmitic acid (Fig. 9, A and B). GLUT4 co-localization with LAMP1 increased from 0.246 ± 0.011 to 0.430 ± 0.010 in control and FA2H knockdown adipocytes (Fig. 9B). Pretreatment with 2-OH palmitic acid in FA2H-depleted adipocytes reversed it from 0.430 ± 0.010 to 0.306 ± 0.011 (Fig. 9B). These data suggest that FA2H-mediated hydroxylation influences intracellular trafficking of membrane proteins to lysosomes for degradation possibly via effects on membrane lipids. Collectively, our results show a new model of how lipogenesis could be modulated by modification of lipid structure by hydroxylation (Fig. 9C).
FIGURE 9.

FA2H depletion increases co-localization of GLUT4 with LAMP1. A, 3T3-L1 adipocytes were treated with a negative control (NC) siRNA or an siRNA recognizing FA2H. 100 μm 2-OH palmitic acid (2-OH) was added as indicated. The cells were starved, fixed, and immunostained for GLUT4 (2° antibody, Alexa 594-IgG) and LAMP1 (2° antibody, Alexa 488-IgG). The scale bar represents 15 μm. B, co-localization between GLUT4 and LAMP1 was quantified with ImageJ software as described under “Experimental Procedures,” and the means ± S.E. of the Manders' co-localization coefficient from 10 images are illustrated in the graph. *a, p < 0.01 (compared with negative control). *b, p < 0.01 (compared with FA2H). C, Model of FA2H-mediated lipogenesis in adipocytes.
DISCUSSION
Lipid rafts are plasma membrane microdomains that are highly enriched with cholesterol and sphingolipids. A variety of receptors, transporters, and other proteins assemble in these nanoscale compartments to provide platforms that function in membrane signaling and trafficking (36–38). Lateral mobility of membrane lipids and proteins plays important roles in regulating their internalization and intracellular trafficking (34, 39). The assembly of the rafts is determined largely by lipid structures and their interactions. In this study, we identified FA2H as a likely regulator of the mobility of raft-associated lipids and showed that this could play an important role in lipogenesis in adipocytes. First, levels of FA2H markedly increase during hormone-induced differentiation of 3T3-L1 adipocytes, and siRNAs against FA2H block this process. Second, depletion of FA2H decreases the protein levels of GLUT4 and IR, leading to impaired glucose uptake and lipogenesis under basal and insulin-stimulated conditions. Third, decreased glucose uptake impairs the lipogenic pathway by reducing FAS and SCD1 gene expression. Fourth, the decreased levels of GLUT4 and IR in FA2H-depleted adipocytes are likely due to increased diffusional mobility of raft-associated lipids, which promotes degradation of GLUT4 and IR. Finally, localization of GLUT4 in lysosomes increases in FA2H-depleted adipocytes. Collectively, these data document a novel role of FA2H and 2-OH FA in modulating adipocyte lipogenesis.
Previous studies (3, 4) demonstrated the presence of a straight chain fatty acid α-oxidation pathway in adipocytes and the activation of this pathway during adipocyte differentiation. 2-OH FA generated in the initial step of FA α-oxidation could be further oxidized or incorporated into lipid species (e.g. sphingolipids). This study demonstrates an important role of straight chain FA 2-hydroxylation by FA2H in adipocyte metabolism. FA2H generation of 2-OH FA modulates protein levels of GLUT4, IR, and thus lipogenesis in adipocytes possibly via altering composition and fluidity of lipid rafts (Fig. 9C). It is not clear how mobility of CTxB molecules is modulated by FA2H as we observed. Altered organization of cell actin has been shown to regulate membrane lateral mobility (34). However, our data demonstrated that regulation of mobility of CTxB by FA2H depletion is not due to modulation of actin cytoskeleton organization. There are many other potential mechanisms that could account for FA2H-mediated mobility of CTxB molecules. For example, FA2H has been shown to regulate cell migration (40), so modulation of extracellular matrix could be one potential mechanism. Other possibilities include the changes of membrane lipid compositions, expression, and recruitment of specific membrane proteins. Moreover, because 2-OH palmitic acid cannot totally reverse the effects of FA2H depletion, FA2H may regulate some hydroxylation-independent pathways. The observation we reported is a novel pathway regulating lipogenesis, and many more experiments are required to understand its detailed biochemical mechanisms.
The relation between membrane mobility and GLUT4 trafficking/degradation was also supported by several previous studies on membrane cholesterol. Lateral diffusion of membrane proteins is reduced by a decreased membrane cholesterol content chronically by using lipoprotein-free media or acutely by treatment with methyl-β-cyclodextrin (34). Interestingly, cholesterol depletion also significantly blocks efficient internalization of GLUT4, presumably leading to its delayed degradation in lysosomes (41). However, the detailed mechanisms on how membrane lipid mobility mediates internalization and intracellular trafficking of GLUT4 and IR are not known. Tumor necrosis factor α-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a decrease of GLUT4 levels (42). Thus, we cannot exclude the possibility that a decrease of GLUT4 level in FA2H knockdown adipocytes may be partially due to the loss of IR protein.
We have shown effects of FA2H production of 2-OH FA on mobility of raft-associated lipids, but it is likely that other effects of these FA also occur in adipocytes. It has been well documented that chemical structures of lipid species are major determinants of physical properties of lipid membranes and their intracellular trafficking. For example, lipid-mimetic dialkylindocarbocyanine derivatives with short or unsaturated hydrocarbon chains enter the endocytic recycling compartment efficiently, whereas lipid analogs with long, saturated tails are sorted out of this pathway and targeted to the late endosomes/lysosomes (43, 44). Similarly, short chain sphingomyelins recycle to the PM more effectively, whereas long chain sphingomyelins are preferentially routed to the late endocytic pathway (45).
How chemical modifications of lipid acyl moieties (e.g. hydroxylation) influence vesicular trafficking is less clear. FA2H catalyzes the 2-hydroxylation of straight chain FAs and is responsible for the formation of 2-hydroxylated sphingolipids (14, 40). Saccharomyces cerevisiae yeast strains with defective SCS7/FA2H are resistant to the antifungal syringomycin E (30) and the antitumor compound PM02734 (46), suggesting that 2-hydroxylated sphingolipids could influence the physical properties of some plasma membrane microdomains/lipid rafts and their internalization. This is supported by biophysical studies of membranes in reconstituted systems (47, 48). In this study, we provide direct evidence that FA2H depletion modulates diffusional mobility of raft-associated lipids and co-localization of GLUT4 with lysosomes, leading to decreased GLUT4 level in a mammalian adipocyte model system. Moreover, FA2H was also required for epidermal lamellar membrane formation, suggesting a role in the exocytic pathway (15). How FA2H mediates translocation of membrane proteins (e.g. GLUT4 and IR) to lipid rafts and regulates distinct intracellular trafficking pathways warrants investigation. Detailed proteomic and lipidomic analysis of raft-associated fractions in the presence and absence of FA2H will be greatly helpful to understand the underlying biochemical mechanisms.
Differentiation of pre-adipocytes into adipocytes requires the coordination of a complex network of transcription factors, cofactors, and signaling molecules (49, 50). Hormone-induced differentiation of 3T3-L1 adipocytes is a commonly used model to recapitulate the biochemical, molecular biological, and ultrastructural characteristics of differentiated mammalian adipocytes. Our data showed the inhibition of adipocyte differentiation by two independent siRNAs recognizing FA2H, suggesting FA2H activity is required for this process. A study using lentivirus-mediated shRNA show that GLUT4 is not required for adipogenic differentiation but is necessary for full lipogenic capacity of differentiated adipocytes (51). Similar results were presented using caveolin-1 knockdown 3T3-L1 cells (16). In this respect, accelerated degradation of GLUT4 and IR cannot fully account for the finding that knockdown of FA2H results in inhibition of differentiation. One intriguing possibility is that FA2H may mediate some signaling pathways or generate some lipid ligands for activation of transcription factors necessary for adipocyte differentiation. Moreover, whether FA2H contributes to the commitment of a mesenchymal stem cell to the adipocyte lineage is of great interest for future study.
The effect of FA2H on lipid rafts might reflect the fact that this enzyme localizes to the ER where sphingolipids are made. FA2H contains a putative ER targeting signal in its C terminus and is co-localized with the ER marker calnexin (9). 2-OH FAs generated by FA2H are activated to CoA thioesters and utilized for sphingolipid synthesis (52). Alternatively, the 2-OH fatty acyl-CoA could be utilized by peroxisomal 2-hydroxyphytanoyl-CoA lyase via the α-oxidation pathway, leading to the generation of odd chain length fatty acids (1). Many details regarding the generation site or the fate of 2-OH FA are still lacking. It is possible that 2-OH FA is generated in the ER prior to delivery to the peroxisome for further degradation by α-oxidation. The role of peroxisomal phytanoyl-CoA hydroxylase, initially thought to generate 2-OH FA is now unclear (10–12). Moreover, the presence of another FA 2-hydroxylase in peroxisome cannot be excluded. 2-Hydroxylation generates a chiral carbon in fatty acids, and both enantiomers are detected in brain samples (53), suggesting the presence of multiple 2-hydroxylases with opposite stereospecificities. In this respect, it would be informative to gain a better understanding of the stereospecificity of FA2H. Our results showed the ability of racemic 2-OH FA to partially reverse the inhibitory effects of FA2H depletion on lipogenesis. It is important to resolve the enantiomers of 2-OH FA and examine the different roles of the two enantiomers in adipocyte lipogenesis. Some other explanation why 2-OH palmitic acid cannot totally reverse the effects of FA2H depletion may exist. It is not clear whether unsaturated FAs (e.g. oleic acid) are good substrates for FA2H. Moreover, FA2H may have some hydroxylation-independent effects.
Collectively, this study provides multiple lines of evidence to support the regulation of adipocyte differentiation and lipogenesis by FA2H. This study might open a door to explore potential roles of an underappreciated fatty acid catabolic pathway in adipocyte physiology.
Supplementary Material
Acknowledgments
We thank Dr. Pin Yue for help with statistical analyses and Yingqiu Liu for help with culture of 3T3-L1 adipocytes. We thank Drs. John Cooper and Olivia Mooren for help with TIRF microscopy. We also thank Drs. Nada Abumrad, Phil Stahl, and Elliot Elson for critical reviews of the manuscript and for helpful suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grant R21DK082951 (to X. S.). This work was also supported by Scientist Development Grant 835140N from the American Heart Association (to X. S.) and by the core services of the Nutrition Obesity Research Center Grant P30DK56341 at Washington University School of Medicine.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S5.
- FA
- fatty acids
- FA2H
- fatty acid 2-hydroxylase
- 2-OH FA
- 2-hydroxy FA
- IR
- insulin receptor
- TAG
- triacylglycerol
- siRNA
- small interfering RNA
- DMEM
- Dulbecco's modified Eagle's medium
- FRAP
- fluorescence recovery after photobleaching
- LAMP1
- lysosome associated membrane protein 1
- PM
- plasma membrane
- TIRF
- total internal reflection fluorescence
- PC
- phosphatidylcholine
- CTxB
- cholera toxin subunit B
- FAS
- fatty-acid synthase
- RT-PCR
- real time-PCR
- ROI
- region of interest
- ER
- endoplasmic reticulum
- Mf
- mobile fraction.
REFERENCES
- 1.Foulon V., Sniekers M., Huysmans E., Asselberghs S., Mahieu V., Mannaerts G. P., Van Veldhoven P. P., Casteels M. (2005) J. Biol. Chem. 280, 9802–9812 [DOI] [PubMed] [Google Scholar]
- 2.Wanders R. J. (2004) Mol. Genet. Metab. 83, 16–27 [DOI] [PubMed] [Google Scholar]
- 3.Su X., Han X., Yang J., Mancuso D. J., Chen J., Bickel P. E., Gross R. W. (2004) Biochemistry 43, 5033–5044 [DOI] [PubMed] [Google Scholar]
- 4.Roberts L. D., Virtue S., Vidal-Puig A., Nicholls A. W., Griffin J. L. (2009) Physiol. Genomics 39, 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levis G. M., Mead J. F. (1964) J. Biol. Chem. 239, 77–80 [PubMed] [Google Scholar]
- 6.Hama H. (2010) Biochim. Biophys. Acta 1801, 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wanders R. J., Jansen G. A., Lloyd M. D. (2003) Biochim. Biophys. Acta 1631, 119–135 [DOI] [PubMed] [Google Scholar]
- 8.Alderson N. L., Rembiesa B. M., Walla M. D., Bielawska A., Bielawski J., Hama H. (2004) J. Biol. Chem. 279, 48562–48568 [DOI] [PubMed] [Google Scholar]
- 9.Eckhardt M., Yaghootfam A., Fewou S. N., Zöller I., Gieselmann V. (2005) Biochem. J. 388, 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukherji M., Kershaw N. J., Schofield C. J., Wierzbicki A. S., Lloyd M. D. (2002) Chem. Biol. 9, 597–605 [DOI] [PubMed] [Google Scholar]
- 11.Foulon V., Asselberghs S., Geens W., Mannaerts G. P., Casteels M., Van Veldhoven P. P. (2003) J. Lipid Res. 44, 2349–2355 [DOI] [PubMed] [Google Scholar]
- 12.Croes K., Foulon V., Casteels M., Van Veldhoven P. P., Mannaerts G. P. (2000) J. Lipid Res. 41, 629–636 [PubMed] [Google Scholar]
- 13.Lingwood D., Simons K. (2010) Science 327, 46–50 [DOI] [PubMed] [Google Scholar]
- 14.Zöller I., Meixner M., Hartmann D., Büssow H., Meyer R., Gieselmann V., Eckhardt M. (2008) J. Neurosci. 28, 9741–9754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uchida Y., Hama H., Alderson N. L., Douangpanya S., Wang Y., Crumrine D. A., Elias P. M., Holleran W. M. (2007) J. Biol. Chem. 282, 13211–13219 [DOI] [PubMed] [Google Scholar]
- 16.González-Muñoz E., López-Iglesias C., Calvo M., Palacín M., Zorzano A., Camps M. (2009) Endocrinology 150, 3493–3502 [DOI] [PubMed] [Google Scholar]
- 17.Su X., Mancuso D. J., Bickel P. E., Jenkins C. M., Gross R. W. (2004) J. Biol. Chem. 279, 21740–21748 [DOI] [PubMed] [Google Scholar]
- 18.Liu Y., Zhou D., Abumrad N. A., Su X. (2010) Am. J. Physiol. Cell Physiol. 298, C921–C928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lazar D. F., Wiese R. J., Brady M. J., Mastick C. C., Waters S. B., Yamauchi K., Pessin J. E., Cuatrecasas P., Saltiel A. R. (1995) J. Biol. Chem. 270, 20801–20807 [DOI] [PubMed] [Google Scholar]
- 20.Yang C. Z., Mueckler M. (1999) J. Biol. Chem. 274, 25297–25300 [DOI] [PubMed] [Google Scholar]
- 21.Lippincott-Schwartz J., Snapp E., Kenworthy A. (2001) Nat. Rev. Mol. Cell Biol. 2, 444–456 [DOI] [PubMed] [Google Scholar]
- 22.Su X., Kong C., Stahl P. D. (2007) J. Biol. Chem. 282, 21278–21284 [DOI] [PubMed] [Google Scholar]
- 23.Bolte S., Cordelières F. P. (2006) J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 24.Manders E. M., Verbeek F. J., Aten J. A. (1993) J. Microsc. 169, 375–382 [DOI] [PubMed] [Google Scholar]
- 25.Lopez J. A., Burchfield J. G., Blair D. H., Mele K., Ng Y., Vallotton P., James D. E., Hughes W. E. (2009) Mol. Biol. Cell 20, 3918–3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nahlé Z., Hsieh M., Pietka T., Coburn C. T., Grimaldi P. A., Zhang M. Q., Das D., Abumrad N. A. (2008) J. Biol. Chem. 283, 14317–14326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villena J. A., Kim K. H., Sul H. S. (2002) Horm. Metab. Res. 34, 664–670 [DOI] [PubMed] [Google Scholar]
- 28.Foufelle F., Gouhot B., Pégorier J. P., Perdereau D., Girard J., Ferré P. (1992) J. Biol. Chem. 267, 20543–20546 [PubMed] [Google Scholar]
- 29.Letexier D., Pinteur C., Large V., Fréring V., Beylot M. (2003) J. Lipid Res. 44, 2127–2134 [DOI] [PubMed] [Google Scholar]
- 30.Hama H., Young D. A., Radding J. A., Ma D., Tang J., Stock S. D., Takemoto J. Y. (2000) FEBS Lett. 478, 26–28 [DOI] [PubMed] [Google Scholar]
- 31.Löfgren H., Pascher I. (1977) Chem. Phys. Lipids 20, 273–284 [DOI] [PubMed] [Google Scholar]
- 32.Singh R. D., Puri V., Valiyaveettil J. T., Marks D. L., Bittman R., Pagano R. E. (2003) Mol. Biol. Cell 14, 3254–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bacia K., Majoul I. V., Schwille P. (2002) Biophys. J. 83, 1184–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwik J., Boyle S., Fooksman D., Margolis L., Sheetz M. P., Edidin M. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 13964–13969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palacios S., Lalioti V., Martinez-Arca S., Chattopadhyay S., Sandoval I. V. (2001) J. Biol. Chem. 276, 3371–3383 [DOI] [PubMed] [Google Scholar]
- 36.Hanzal-Bayer M. F., Hancock J. F. (2007) FEBS Lett. 581, 2098–2104 [DOI] [PubMed] [Google Scholar]
- 37.Laude A. J., Prior I. A. (2004) Mol. Membr. Biol. 21, 193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bickel P. E. (2002) Am. J. Physiol. Endocrinol. Metab. 282, E1–E10 [DOI] [PubMed] [Google Scholar]
- 39.Roettger B. F., Pinon D. I., Burghardt T. P., Miller L. J. (1999) Am. J. Physiol. 276, C539–C547 [DOI] [PubMed] [Google Scholar]
- 40.Maldonado E. N., Alderson N. L., Monje P. V., Wood P. M., Hama H. (2008) J. Lipid Res. 49, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shigematsu S., Watson R. T., Khan A. H., Pessin J. E. (2003) J. Biol. Chem. 278, 10683–10690 [DOI] [PubMed] [Google Scholar]
- 42.Stephens J. M., Lee J., Pilch P. F. (1997) J. Biol. Chem. 272, 971–976 [DOI] [PubMed] [Google Scholar]
- 43.Mukherjee S., Soe T. T., Maxfield F. R. (1999) J. Cell Biol. 144, 1271–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hao M., Mukherjee S., Sun Y., Maxfield F. R. (2004) J. Biol. Chem. 279, 14171–14178 [DOI] [PubMed] [Google Scholar]
- 45.Koivusalo M., Jansen M., Somerharju P., Ikonen E. (2007) Mol. Biol. Cell 18, 5113–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herrero A. B., Astudillo A. M., Balboa M. A., Cuevas C., Balsinde J., Moreno S. (2008) Cancer Res. 68, 9779–9787 [DOI] [PubMed] [Google Scholar]
- 47.Jenske R., Lindström F., Gröbner G., Vetter W. (2008) Chem. Phys. Lipids 154, 26–32 [DOI] [PubMed] [Google Scholar]
- 48.Shah J., Atienza J. M., Rawlings A. V., Shipley G. G. (1995) J. Lipid Res. 36, 1945–1955 [PubMed] [Google Scholar]
- 49.MacDougald O. A., Lane M. D. (1995) Annu. Rev. Biochem. 64, 345–373 [DOI] [PubMed] [Google Scholar]
- 50.Rosen E. D., MacDougald O. A. (2006) Nat. Rev. Mol. Cell Biol. 7, 885–896 [DOI] [PubMed] [Google Scholar]
- 51.Liao W., Nguyen M. T., Imamura T., Singer O., Verma I. M., Olefsky J. M. (2006) Endocrinology 147, 2245–2252 [DOI] [PubMed] [Google Scholar]
- 52.Mizutani Y., Kihara A., Chiba H., Tojo H., Igarashi Y. (2008) J. Lipid Res. 49, 2356–2364 [DOI] [PubMed] [Google Scholar]
- 53.Jenske R., Vetter W. (2008) J. Agric. Food Chem. 56, 11578–11583 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.