

Abstract
Centrobin/NIP2 is a centrosomal protein that is required for centrosome duplication. It is also critical for microtubule organization in both interphase and mitotic cells. In the present study, we observed that centrobin is phosphorylated in a cell cycle stage-specific manner, reaching its maximum at M phase. PLK1 is a kinase that is responsible for M phase-specific phosphorylation of centrobin. The microtubule forming activity of centrobin was enhanced by PLK1 phosphorylation. Furthermore, mitotic spindles were not assembled properly with the phospho-resistant mutant of centrobin. Based on these results, we propose that centrobin functions as a microtubule stabilizing factor and PLK1 enhances centrobin activity for proper spindle formation during mitosis.
Keywords: Cell Cycle, Centrosome, Microtubules, Mitosis, Serine-threonine Protein Kinase, NEK2, PLK1, Centrobin
Introduction
Microtubules are assembled and disassembled spontaneously depending on the local concentration of tubulin dimers, and this phenomenon is known as dynamic instability of microtubules (1). Microtubules in a physiological cytoplasmic environment polymerize faster and transit more frequently between polymerization and depolymerization (2). A number of protein factors are known to control in vivo microtubule dynamics in diverse manners. For example, MAP4 promotes microtubule formation by enhancing the rescue frequency without decreasing the catastrophe frequency (3). XMAP215 increases the polymerization rate of pure tubulin by catalyzing the addition of up to 25 tubulin dimers to the growing plus end (4). In addition, microtubules also exhibit a high catastrophe frequency caused by destabilizers such as XKCM1 (5).
A global change in microtubule dynamics is observed in cells entering M phase (6, 7). A large array of stable microtubules at interphase is replaced by short, highly dynamic microtubules at M phase (8). Total microtubule levels drop and the turnover rate increases in cells approaching mitosis. Microtubule polymerization recovers during prometaphase and returns to interphase levels by the end of metaphase. Protein kinases such as CDK1 and MAP4/microtubule affinity-regulating kinase are known to regulate microtubule dynamics during this period. For example, CDK1 phosphorylates MAP4, which reduces microtubule polymerization activity (9, 10). CDK1 may directly phosphorylate tubulins, prohibiting their incorporation into growing microtubules (11). MAP/microtubule affinity-regulating kinase phosphorylates MAP2c, MAP4, and Tau, causing increased microtubule dynamics (12, 13). Nonetheless, it remains to be investigated what proteins are involved in microtubule dynamics and how their activity is controlled during the cell cycle.
Centrobin/NIP2 was initially identified as a centrosome protein required for centriole duplication (14). Centrobin is expressed abundantly in tissues with high proliferation activities (14); however, it was also detected in the centrosomes of resting cells, suggesting that centrobin is a core centrosome protein required for basic functions of the centrosome (15). Furthermore, centrobin is involved in the microtubule organizing activity of the cell (16). Knockdown of centrobin causes cell shrinkage, defects in spindle assembly, and abnormal nuclear morphology (16). Mitotic defects with abnormal spindle formation were also observed in centrobin-suppressed early mouse embryos (17). It was reported that centrobin is a substrate of NEK2 and its centrosomal localization is controlled by NEK2 (16). NEK2 activity is cell cycle stage-specific and is induced at S phase and maintained high through the G2 phase, but eventually degraded in early mitosis (18). This finding suggests that another kinase may be critical for controlling centrobin functions such as proper spindle assembly during mitosis.
In the present study, we showed that PLK1 can phosphorylate centrobin. Furthermore, PLK1 enhanced the microtubule formation activity of centrobin. Based on these results, we propose that PLK1 phosphorylation is required for centrobin function in spindle assembly during mitosis.
EXPERIMENTAL PROCEDURES
Antibodies
Antibodies against β-tubulin and γ-tubulin were purchased from Sigma. Antibodies against Myc, HA, and GFP epitopes were purchased from Cell Signaling, Babco, and Santa Cruz, respectively. The monoclonal and polyclonal antibodies against PLK1 and phospho-histone H3 were purchased from Zymed Laboratories Inc. and Upstate, respectively. The rabbit polyclonal centrobin/NIP2 antibody was used as previously described (16). All purchased antibodies were diluted to 1:1000 for immunoblotting and 1:100 for immunocytochemistry. The secondary antibody conjugated with mouse or rabbit horseradish peroxidase (Sigma) was diluted to 1:10,000 for immunoblotting.
Cell Culture and Treatment
HeLa, U2OS, 293T, and 293T 17 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml). The cells were treated with 4 mm hydroxyurea for S phase arrest, 100 μm monastrol and 0.33 μm nocodazole for prometaphase arrest, and 10 μm MG132 for metaphase arrest.
RNA Interference
siRNA targeted to centrobin (AAG GAT GGT TCT AAG CAT ATC), PLK1 (AAG CGG GAC TTC CGC ACA TAC), and control siRNA (AAG TAG CCG AGC TTC GAT TGC) were transfected into HeLa or U2OS cells using Lipofectamine 2000 and Oligofectamine reagents (Invitrogen) according to the manufacturer's instructions. Cells (2.5 × 104) were seeded into 4-well dishes with 500 μl of medium without antibiotics. One day after seeding, 50 pmol of siRNA was mixed with 50 μl of Opti-MEM (Invitrogen) in one tube and 2 μl of Oligofectamine was mixed with 13 μl of Opti-MEM in another tube. After 5 min, two tubes were combined and incubated for a further 20 min. After incubation, the siRNA mixture was added into the cells. A day after incubation, the medium was exchanged with fresh medium.
Transfection and Immunocytochemistry
Transient transfection into 293T, HeLa, and 293T 17 cells were carried out using Lipofectamine Plus (Invitrogen) following the manufacturer's instructions. For immunocytochemistry, 4 × 104 cells were seeded into a 4-well dish and transfected 24 h later. One or 2 days after transfection, the cells were fixed either with cold methanol for 10 min at −20 °C or with 3.7% paraformaldehyde for 10 min at room temperature. The paraformaldehyde-fixed cells were permeabilized with 0.5% PBST (phosphate-buffered saline with 0.5% Triton X-100); however, the methanol-fixed cells were not permeabilized. The fixed cells were blocked with 10% normal goat serum in 0.1% PBST (PBS with 0.1% Triton X-100) for 10 min, incubated with primary antibodies for 1 h, washed with 0.1% PBST three times, and incubated with either fluorescein isothiocyanate- or TRITC-conjugated secondary antibody (Jackson ImmunoResearch) for 30 min. Next, the cells were washed three times with 0.1% PBST, incubated with 4′,6-diamidino-2-phenylindole solution, and observed using a fluorescence microscope with a CCD (Qicam Fast 1394; Qimaging) camera. The data were quantified with Image-pro software and statistically analyzed with Sigma plot.
Immunoprecipitation and Immunoblot Analysis
Immunoprecipitation and immunoblot analyses were carried out as previously described (19). In brief, 2 × 106 cells were seeded into 60-mm dishes and transfected 24 h later. After 24 h, the cells were lysed with RIPA (50 mm Tris-HCl, pH 7.3, 150 mm NaCl, 0.1% SDS, 0.5% deoxycholate, 1% Nonidet P-40, and 1 mm EDTA) or Nonidet P-40 buffer (50 mm Tris-HCl, pH 7.3, 150 mm NaCl, 1% Nonidet P-40, 1 mm EDTA) with protease inhibitors for 20 min on ice and centrifuged at 15,000 × g for 15 min at 4 °C. For immunoprecipitation, the supernatant was incubated with specific antibodies for 2 h and then incubated with protein A-Sepharose (Amersham Biosciences) for 1 h at 4 °C. The immunoprecipitates were immunoblotted with the indicated antibodies as described.
In Vivo Labeling Assay
The in vivo labeling assay was conducted following Mayor et al. (20). In brief, HeLa cells were incubated for 3 h in a phosphate-free Dulbecco's modified Eagle's medium supplemented with 10% dialyzed fetal bovine serum and 250 μCi/ml of [32P]orthophosphate. Cells were collected and lysed with RIPA buffer. The lysates were subjected to immunoprecipitation followed by immunoblotting and autoradiography.
In Vitro Kinase Assay
The PLK1 kinase assay was carried out as previously described (21). In brief, 293T cells transfected with FLAG-tagged wild-type PLK1 (pFlagPLK1WT), kinase-defect PLK1 (pFlagPLK1KD), or constitutively active PLK1 (pFlagPLK1CA) expression vectors were lysed with Nonidet P-40 lysis buffer and subjected to immunoprecipitation with an antibody against the FLAG tag. The immunoprecipitates were washed three times with lysis buffer and once with kinase buffer (50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 1 mm Na3VO4, 1 g/ml of heparin). Kinase reactions were conducted for 20 min at 30 °C in kinase buffer supplemented with 5 μm ATP, 1 mm dithiothreitol, and 5 μCi of [γ-32P]ATP in a total volume of 20 μl. The centrobin substrates were prepared from a bacterially expressed fusion protein or immunoprecipitates of 293T cells with ectopically expressed centrobin. The reactions were stopped by the addition of the 2× SDS sample buffer and heated for 5 min at 95 °C. Protein samples were resolved with 10% SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was exposed to the x-ray film.
Microtubule Fractionation
Microtubule fractionation was performed as previously described (22). Under MT-depolymerizing conditions, cellular MTs were depolymerized with 33 μm nocodazole for 1 h. The cells were then washed twice with PBS and lysed with TNE buffer (50 mm Tris-HCl, pH 7.5, containing 150 mm NaCl, 1% Nonidet P-40, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml of aprotinin) for 3 min at 4 °C. The cells were then centrifuged at 16,000 × g for 5 min at 4 °C. Under the MT-stabilizing condition, cells were incubated with 20 μm Taxol for 1 h, washed twice with PEM (80 mm Pipes, pH 6.6, 1 mm EGTA, 1 mm MgSO4), and lysed for 1 min at room temperature with PEM containing 1% Nonidet P-40, 20 μm Taxol, 1 mm GTP, 1 mm phenylmethylsulfonyl fluoride, and 10 μg/ml of aprotinin. The lysate was then centrifuged at 15,000 × g for 5 min at room temperature. The supernatant obtained was clarified by centrifugation at 100,000 × g for 60 min at 4 °C. The precipitates were washed twice with lysis buffer, and these fractions were then subjected to immunoblot analysis.
Microtubule Pull-down Assay
Full-length centrobin was generated by transfection of a vector into 293T 17 cells. The expression vector was created by inserting GST and the centrobin sequence into the pCMV-Tag3B vector. Twenty-four hours after transfection, the cells were lysed with RIPA buffer with protease inhibitors for 20 min on ice, sonicated, and centrifuged at 15,000 × g for 15 min at 4 °C. GST was bacterially expressed in the BL21 LysE strain of Escherichia coli. Proteins were purified using glutathione-Sepharose, and after purification, proteins were dialyzed against PEM buffer (80 mm Pipes, pH 6.9, 1 mm MgSO4, 1 mm EGTA). The fusion proteins were centrifuged at 35,000 × g at 20 °C for 30 min. The supernatants were mixed with 10 μg of microtubules that were polymerized from purified tubulins with 10 μm Taxol according to the manufacturer's instruction (Cytoskeleton). The microtubule and protein complexes were then incubated for 30 min at 37 °C and centrifuged at 35,000 × g at 20 °C for 30 min. The supernatant was carefully removed and mixed with an equal volume of the 2× protein sample buffer. The pellet was then carefully washed with PEM buffer before resuspension in the 1× protein sample buffer.
In Vitro Tubulin Polymerization Assay
GST-doublecortin and GST-EB1 were bacterially expressed in the BL21 LysE strain of E. coli and dialyzed against PEM buffer. Purified tubulin was diluted in PEM buffer supplemented with 1 mm GTP to a final concentration of 16 μm. The tubulin solution was kept on ice prior to beginning the polymerization reaction. The fusion proteins were added to the tubulin solutions, mixed well, and immediately transferred into pre-warmed 96-well plates. Reaction volumes were adjusted to 100 μl. Absorbance was measured at 340 nm once every 30 s for 50 min in the Spectramax M5 (Molecular Devices) at 37 °C. For the in vitro microtubule stability assay, 5 μg/μl of tubulin was used, and at this concentration, tubulin polymerizes spontaneously. The polymerization rate of 5 μg/μl of tubulin was observed with or without 3 μm colcemid. Microtubule seeds were created by the sonication of preassembled tubulins and 70 μg of protein/reaction.
RESULTS
Augmented Phosphorylation of Centrobin at M Phase
We performed in vivo labeling assays to determine the cell cycle stage-specific phosphorylation of centrobin (Fig. 1A). HeLa cells were cultured in the presence of hydroxyurea and nocodazole to arrest the cell cycle at S and M phases, respectively. As reported previously, the endogenous centrobin protein remained constant irrespective of the cell cycle stage (Fig. 1A) (16). However, the phosphorylation level of centrobin was augmented in cells treated with nocodazole (Fig. 1A). This finding suggests that centrobin phosphorylation oscillates during the cell cycle and increases at M phase.
FIGURE 1.

PLK1 phosphorylation of centrobin at G2/M phase. A, HeLa cells were treated with 4 mm hydroxyurea (HU) or 0.33 μm nocodazole (NZ) for 24 h. During the last 3 h, the cells were cultured in the presence of [32P]orthophosphate and then subjected to immunoprecipitation followed by immunoblot analysis with the centrobin antibody. The amount of 32P-labeled centrobin was determined with autoradiography and measured densitometrically. B, the siPLK1-transfected HeLa cells were treated with 0.33 μm nocodazole for 16 h. During the last 3 h, the cells were cultured in the presence of [32P]orthophosphate. The endogenous centrobin and PLK1 proteins were immunoprecipitated (IP) with the specific antibodies and subjected to immunoblotting and autoradiography. The relative amount of [32P]centrobin was measured densitometrically.
We previously reported that centrobin is a substrate of NEK2 (16). It is known that NEK2 activity is low in early G1 phase cells, increases during cell cycle progression, but is degraded during early mitosis (18). This NEK2 activity cycle does not correspond to the M phase-specific phosphorylation of centrobin shown in Fig. 1A. This discrepancy suggests that an additional kinase that phosphorylates centrobin is present during M phase.
We speculated that PLK1 may phosphorylate centrobin because PLK1 activity is highest at the M phase (23). In fact, PLK1 was detected at the centrosome during the G2/M phase along with centrobin (Fig. 2A) (23). Microscopic observation as well as fluorescence-activated cell sorter analysis revealed an increase in mitotic cell population in both the PLK1- and centrobin-suppressed cells, suggesting that both PLK1 and centrobin are required for mitosis (Fig. 2, B–E). Defects in spindle assembly were common in both the PLK1- and centrobin-suppressed cells (Fig. 2F) (16, 24). To test whether centrobin is a substrate of PLK1 at M phase, we performed in vivo labeling assays in the PLK1-suppressed cells whose cell cycle was arrested at M phase with nocodazole. The results showed that centrobin phosphorylation was reduced in PLK1-suppressed cells; however, centrobin protein levels remained constant, suggesting that PLK1 may be responsible for phosphorylation of centrobin at the M phase (Fig. 1B).
FIGURE 2.

Centrobin and PLK1 suppression interfered with spindle assembly. A, HeLa cells were transfected with pGFP-PLK1. Twenty-four hours later, the cells at interphase and M phase were co-immunostained with antibodies specific to centrobin (red) and GFP (green). The insets are magnified views of the centrosome signals. DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 10 μm. B, the U2OS cells were transfected with control (siCTL) or centrobin (siCBN) siRNAs. Twenty-four hours later, the cells were subjected to immunoblot analysis with antibodies specific to centrobin, PLK1, and phospho-histone H3 (pHH3). C, the HeLa cells were transfected with varying amounts of PLK1 siRNA (siPLK1). Forty-eight hours after transfection, the cells were subjected to immunoblot analysis with antibodies specific to PLK1 and β-tubulin. D, 48 h after transfection with siCTL or siCBN, the cells were photographed under a phase-contrast microscope. E, fluorescence-activated cell sorter analysis was performed with the U2OS cells transfected with siCTL, siCBN, or siPLK1. The cells were gated with the DNA contents and pHH3 antibody. The proportion of the double positive cells is indicated. F, the centrobin- and PLK1-suppressed cells at M phase were co-immunostained with antibodies specific to centrobin (red) and β-tubulin (green). DNA was stained with 4′,6-diamidino-2-phenylindole (blue).
PLK1 Phosphorylates Centrobin in Vitro
We performed in vitro kinase assays to examine PLK1 phosphorylation of centrobin in detail (Fig. 3A). Centrobin fusion proteins (GST-CBN) were used as substrates, and PLK1 was prepared from 293T cells in which wild-type (WT), kinase-dead (KD), or constitutively active (CA) FLAG-PLK1 proteins were ectopically expressed (25). Results showed that the GST-CBN fusion protein was phosphorylated efficiently by the wild-type and constitutively active FLAG-PLK1, but not by the kinase-dead FLAG-PLK1 (Fig. 3A). FLAG-PLK1 selectively phosphorylated GST-CBN1–445, but not GST-CBN445–903, suggesting that the PLK1 phosphorylation sites reside at the N terminus of centrobin (Fig. 3A). The PLK1 phosphorylation sites were further defined using a different set of centrobin-truncated fusion proteins and were limited to 1–193 residues of the protein (Fig. 3B). For further definition of the PLK1 phosphorylation sites within GST-CBN1–193, we used bacterially expressed PLK1CA and PLK1KD as enzymes and found that GST-CBN1–29 was the smallest substrate fragment (Fig. 3, C and D). There are 8 serines and threonines within the 1–29 residues of centrobin. We prepared the phospho-resistant GST-CBN1–29 proteins in which each serine or threonine was substituted with an alanine and these mutated proteins were used as substrates. However, PLK1 phosphorylated all single base-substituted CBN1–29 proteins, suggesting that multiple residues are phosphorylated by PLK1 (data not shown). In fact, we observed no PLK1 phosphorylation in the 4-residue substituted CBN1–29 (GST-CBN1–29,T3A,S4,21,22A; Fig. 3E). Therefore, PLK1 phosphorylation sites were minimally limited to the threonine 3, serine 4, serine 21, and serine 22 residues of the centrobin protein.
FIGURE 3.

In vitro kinase assay of centrobin by PLK1. A, WT, KD, or CA forms of the FLAG-PLK1 proteins were ectopically expressed in 293T cells and immunoprecipitated with the FLAG antibody. Bacterially expressed centrobin fusion proteins (GST, GST-CBN, GST-CBN1–445, and GST-CBN445–903) were used as substrates. Amounts of the enzymes and substrates were determined with immunoblot analyses and the kinase activity was determined via autoradiography. Asterisks in the immunoblot data indicate the IgG heavy chain bands. B, the in vitro kinase assay was performed with the WT and KD FLAG-PLK1 proteins. As substrates, bacterially expressed GST, GST-CBN1–193, GST-CBN194–560, and GST-CBN561–903 were used. Arrowheads indicate the GST fusion proteins. C–E, the in vitro kinase assay was performed with the bacterially expressed kinase-active (CA) or kinase-dead (KD) PLK1 protein. C, as substrates, GST-CBN1–193 was truncated into three fragments (GST-CBN1–56, GST-CBN57–119, and GST-CBN120–193). Casein was used as a control substrate. D, as substrates, GST-CBN1–56 was truncated into two fragments (GST-CBN1–29 and GST-CBN30–56). The autophosphorylated PLK1 and phosphorylated GST-CBN proteins were marked with asterisks. E, the GST-CBN1–29 fusion proteins in which 4 specific serine or threonine residues were mutated into alanines (GST-CBN1–29, GST-CBN1–29,T3A,S4A, GST-CBN1–29,S21,22A, GST-CBN1–29,T3A,S4,21,22A) were used as substrates. The phosphorylation activity was determined by autoradiography, and the amount of the substrates was determined by Coomassie Blue staining.
PLK1 Phosphorylates Centrobin in Vivo
We examined in vivo phosphorylation of centrobin in FLAG-PLK1 overexpressing 293T cells (Fig. 4A). Immunoblot analysis revealed the presence of a slow migrating MycCBN band in the wild-type FLAG-PLK1-expressing cells that was not present in the kinase-dead FLAG-PLK1-expressing cells (Fig. 3A). The slow migrating MycCBN band appeared more prominently in the constitutively active FLAG-PLK1-expressing cells (Fig. 4A). Co-expression of GFP-NEK2 also produced a slow migrating MycCBN band (Fig. 4B). The slow migrating band disappeared when immunoprecipitated MycCBN was treated with calf intestine alkaline phosphatase, indicating that it is a phosphorylated form of MycCBN (Fig. 4B).
FIGURE 4.

Centrobin was phosphorylated by PLK1 in vivo. A, 293T cells were cotransfected with pMycCBN and pFlagPLK1 (WT, KD, CA). Twenty-four hours later, the cells were subjected to immunoblot analysis with antibodies specific to the Myc and FLAG tags. The natural and mobility-shifted MycCBN bands are marked with arrows. B, 293T cells were cotransfected with pMycCBN and pGFP-NEK2 or pGFP-PLK1. Twenty-four hours later, the MycCBN proteins were immunoprecipitated with the Myc antibody, treated with calf intestine phosphatase (CIP), and subjected to immunoblot analysis. The natural and mobility-shifted MycCBN bands are marked with arrows. C, the 293T cells were cotransfected with pMycCBN and the forms of pHaNEK2 and/or pFlagPLK1 plasmids are indicated (WT, KD, and CA). The expressed ectopic MycCBN, HaNEK2, and FLAG-PLK1 proteins were determined by immunoblot analyses. The natural and mobility-shifted MycCBN bands are marked with arrows.
NLP, a centrosomal protein involved in microtubule nucleation in interphase cells, was phosphorylated by both NEK2 and PLK1 (26). Interestingly, NLP may be phosphorylated by PLK1 efficiently only when it had been primed by NEK2 in advance (26). To determine whether NEK2 also functions as a priming kinase in centrobin phosphorylation, we overexpressed MycCBN along with the HaNEK2 and/or FLAG-PLK1 proteins. As previously observed, the MycCBN band was retarded when kinase-active HaNEK2 or FLAG-PLK1 were expressed (Fig. 4C). The phosphorylated form of MycCBN was detected in cells with kinase-dead NEK2 and kinase-active PLK1, suggesting that PLK1 phosphorylation of centrobin does not require the priming kinase activity of NEK2 (Fig. 4C).
Centrobin Associates with Microtubules
Centrobin associates with a stable microtubule network in the cytoplasm as well as daughter centrioles (14, 16). We biochemically examined the physical association of centrobin with microtubules. Microtubules in HeLa cells were stabilized with Taxol treatment or destabilized with nocodazole treatment and fractionated by centrifugation. The majority of endogenous centrobin was co-precipitated with microtubules in the Taxol-treated cells (Fig. 5A). Alternatively, a significant amount of centrobin was detected in the supernatant of nocodazole-treated cells. A similar distribution pattern was also observed with PLK1 and NEK2 (Fig. 5A). An in vitro microtubule-binding assay revealed that GST-MycCBN was precipitated only in the presence of microtubules (Fig. 5B). These results indicate that centrobin is directly associated with microtubules both in vivo and in vitro.
FIGURE 5.
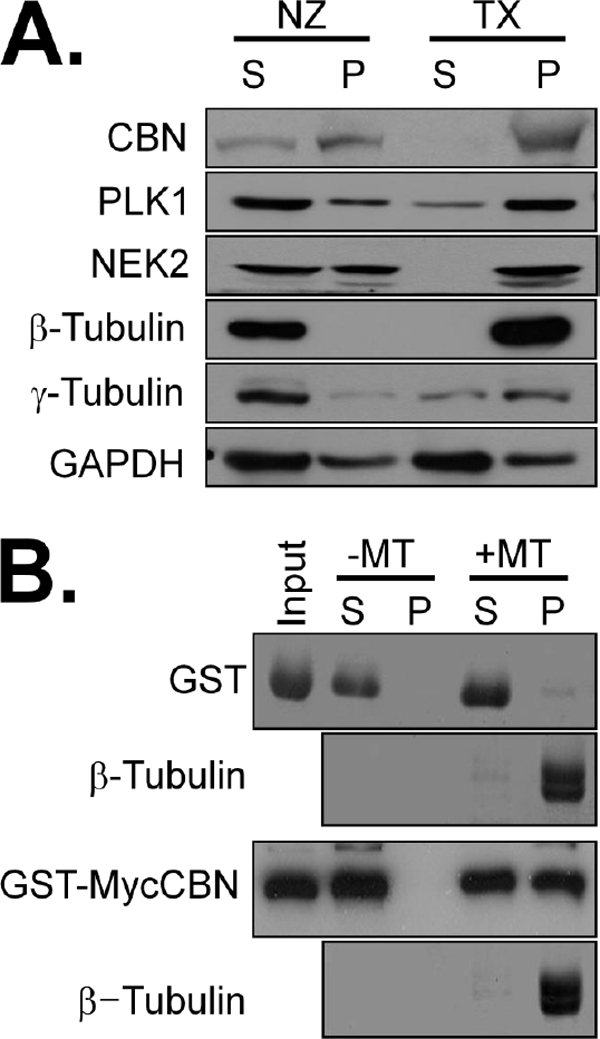
Centrobin association with microtubules in vivo and in vitro. A, HeLa cells were treated with 33 μm nocodazole (NZ) to disrupt microtubules or 20 μm paclitaxel (TX) to stabilize them. The cell lysates were fractionated into supernatant (S) and precipitates (P) by ultracentrifugation and subjected to immunoblot analysis with antibodies specific to centrobin (CBN), PLK1, NEK2, β-tubulin, γ-tubulin, and GAPDH. B, the tubulin monomers were polymerized with 10 μm paclitaxel. Ectopically expressed GST and GST-MycCBN proteins were purified from 293T cells. The purified proteins and microtubules were incubated for 30 min and centrifuged at 35,000 × g to separate microtubules (P) from tubulin monomers (S). β-Tubulin was detected with Coomassie Blue staining and GST fusion proteins were detected with the GST antibody.
Centrobin Promotes Microtubule Formation in Vitro
To determine the effects of centrobin on microtubule dynamics, we performed an in vitro tubulin polymerization assay. The GST-MycCBN protein was ectopically expressed in 293T cells and purified. The amount and purity of the GST-MycCBN protein was determined by Coomassie Blue staining of the gel (Fig. 6A). The concentration of tubulin in the reaction mixture was suboptimal for spontaneous polymerization in vitro. Addition of GST-MycCBN to the reaction mixture induced microtubule polymerization in a concentration-dependent manner, as shown by the measurement of the absorbance at 340 nm (Fig. 6B). These results suggest that centrobin promotes microtubule formation in vitro.
FIGURE 6.

Centrobin-promoted microtubule formation in vitro. A, the GST-MycCBN fusion protein was ectopically expressed in 293T cells and purified as described under “Experimental Procedures.” The purity and amount of the centrobin fusion protein were determined with Coomassie Blue staining. Bovine serum albumin (BSA) was used as a loading control. B, various amounts of GST-MycCBN (0, 10, 30, and 90 nm) were added into the reaction mixture of the in vitro tubulin polymerization assay. For this experiment and the following experiments, the amount of polymerized microtubule was monitored every 30 s for 50 min. The experiments were repeated twice and the results are indicated as mean ± S.E. C, after the polymerization assay in B, the reaction mixture was centrifuged. The supernatant (S) and pellet (P) of the reaction mixture was subjected to immunoblot analysis with the β–tubulin antibody.
To confirm that an increase in absorbance at 340 nm was due to polymerized microtubules rather than aggregation of the centrobin protein, we determined tubulin distribution in the reaction mixture. The reaction mixture was centrifuged to separate polymerized microtubules from unpolymerized tubulins. Immunoblot analysis revealed that tubulin was polymerized in the reaction mixture and centrobin-enhanced tubulin polymerization (Fig. 6C).
Centrobin Is Critical for Microtubule Stabilization in Vitro
Microtubule formation may be divided into three phases: nucleation, polymerization, and dynamic instability. To determine at which level centrobin enhances microtubule formation, we compared centrobin with other proteins whose roles in microtubule formation have been well documented. The purity and amount of the control proteins such as doublecortin and EB1 were determined by Coomassie Blue staining (Fig. 7A).
FIGURE 7.

Centrobin enhanced nucleation and stabilization of microtubules formed in vitro. A, the centrobin fusion protein (GST-MycCBN) was purified from the ectopically expressed 293T cells, whereas the doublecortin (GST-DCX) and GST-EB1 fusion proteins were purified from bacteria. The purity and amount of the proteins were determined with Coomassie Blue staining. B, the in vitro tubulin polymerization assay was performed in the presence of centrobin and doublecortin fusion proteins. C, in vitro tubulin polymerization assays were performed in the presence or absence of the microtubule seed. Effects of centrobin and EB1 fusion proteins on microtubule formation were compared. D, the in vitro tubulin polymerization assay was conducted in the presence of GST-MycCBN. The amount of tubulin in the reaction mixture was sufficient to induce spontaneous polymerization without a protein factor. Colcemid was added to destabilize the microtubules formed in vitro. The experiments were repeated twice and the results indicated as mean ± S.E.
Doublecortin is a microtubule stabilizer and is able to nucleate tubulin polymerization in vitro (27). As expected, addition of doublecortin to the tubulin mixture induced tubulin polymerization in a similar pattern as centrobin (Fig. 7B). This finding suggests that centrobin and doublecortin have a similar molecular property and that they both act as a microtubule nucleating and/or stabilizing factor. EB1 is known as a polymerizing factor that can induce microtubule formation only in the presence of the microtubule seed (28, 29). We tested microtubule forming activity of centrobin in the presence or absence of the seed. As expected, EB1 enhanced tubulin polymerization only in the presence of the microtubule seeds (Fig. 7C). Alternatively, centrobin was able to form microtubules irrespective of the seeds in the reaction mixture (Fig. 7C). These results suggest that centrobin has a microtubule nucleation activity rather than a polymerizing activity.
To test if centrobin is a microtubule stabilizer, we added colcemid, a microtubule-destabilizing agent, to the reaction mixture and observed microtubule formation activity in vitro (30). We used a sufficient amount of tubulin to induce spontaneous polymerization without a protein factor (Fig. 7D). In the presence of 3 μm colcemid, the microtubule assembly rate was reduced by 50% of the control (Fig. 7D). However, centrobin significantly limited the microtubule disassembly activity of colcemid (Fig. 7D). These results suggest the centrobin functions as a microtubule stabilizer.
PLK1 Phosphorylation Enhanced the Microtubule Formation Activity of Centrobin
We examined the effects of PLK1 phosphorylation on microtubule nucleation and stabilization activity of centrobin in vitro. We used GST-MycCBN isolated from cells co-expressing pGFP-PLK1CA or pGFP-PLK1KD (Fig. 8A). The results showed that the microtubule formation activity of centrobin was significantly enhanced with PLK1CA but not with PLK1KD (Fig. 8A). Next, we performed an in vitro tubulin polymerization assay using the phospho-resistant centrobin protein in which PLK1 phosphorylation sites were substituted with alanines (GST-MycCBNT3A,S4,21,22A; Fig. 8B). The results showed that the microtubule formation activity of the phospho-resistant centrobin was not enhanced by PLK1 (Fig. 8B). These results suggest that PLK1 phosphorylation enhances the microtubule formation activity of centrobin.
FIGURE 8.

PLK1 enhanced the microtubule-polymerizing activity of centrobin in vitro. A, 293T cells were cotransfected with pGST-MycCBN and the constitutively active (pGFP-PLK1CA) or kinase-dead (pGFP-PLK1KD) forms of the PLK1 plasmids. Twenty-four hours after transfection, GST-MycCBN was purified and added to the reaction mixture of the in vitro tubulin polymerization assay. B, the wild type (GST-MycCBN) and phospho-resistant (GST-MycCBNT3A,S4,21,22A) centrobin proteins were expressed in 293T cells in the presence or absence of the constitutively active PLK1 (GFP-PLK1CA). The purified GST-MycCBN protein was added to the reaction mixture of the in vitro tubulin polymerization assay. The experiments were repeated twice and the results indicated as mean ± S.E.
Mitotic Spindles Were Not Assembled Properly with the Phospho-resistant Centrobin
Defects in spindle assembly were observed in centrobin-suppressed cells (Fig. 2F) (16, 24). We examined whether the spindle assembly defect was rescued by the phospho-resistant mutant of centrobin or not. HeLa cells were transfected with siCBN and subsequently with the wild-type (pGST-MycCBN) or phospho-resistant (pGST-MycCBNT3A,S4,21,22A) centrobin plasmids. Knockdown and ectopic expression of centrobin was confirmed by immunoblot analysis (Fig. 9A). The cells were arrested at prometaphase with monastrol for 6 h and synchronously released to metaphase in the presence of MG132. As expected, most of the centrobin-depleted cells revealed defects in spindle formation (Fig. 9B) (16). The wild-type centrobin (GST-MycCBN) rescued the spindle assembly defects successfully. However, the phospho-resistant mutant of centrobin (GST-MycCBNT3A,S4,21,22A) did not (Fig. 9B). These results suggest that PLK1 phosphorylation at specific sites of centrobine is critical for spindle assembly during mitosis.
FIGURE 9.

Mitotic spindles were not assembled properly with the phospho-resistant mutant of centrobin. A, HeLa cells were transfected with siCBN and subsequently with the wild type (pGST-MycCBN) or phospho-resistant (pGST-MycCBNT3A,S4,21,22A) centrobin plasmids. The endogenous and ectopic (WT, GST-MycCBN, and MU, GST-MycCBNT3A,S4,21,22A) centrobin protein levels were determined by immunoblot analysis with the centrobin antibody. β-Tubulin was detected as a loading control. B, 48 h after transfection, the cells were treated with monastrol (100 μm) for 6 h and released for 1.5 h in the presence of MG132 (10 μm). The cells were coimmunostained with antibodies specific to Myc and γ-tubulin. DNA was stained with 4′,6-diamidino-2-phenylindole. The spindle morphology was classified into bipole, abnormal bipole, and monopole. The experiments were repeated five times and 100 cells per experimental group were analyzed. The results are indicated as mean ± S.D.
DISCUSSION
Centrobin was previously reported as a substrate of NEK2 (16). Here, we report that centrobin is also a substrate of PLK1. Substrate targeting of PLK1 is directed by the polo-box domain, which recognizes a specific phosphopeptide within the substrate protein (31). The priming phosphorylation may be carried out by PLK1 itself (32, 33) or other kinases such as CDK1 (31, 34). NEK2 was also considered one of the priming kinases for PLK1 action (26). Regarding centrobin phosphorylation, PLK1 does not need NEK2 as a priming kinase because centrobin is efficiently phosphorylated by PLK1 alone or in the presence of kinase-dead NEK2. Rather, these two kinases are likely to phosphorylate centrobin at different cell cycle stages for distinct and specific cellular functions. We previously observed that the centrosomal centrobin level was enhanced in the NEK2-suppressed interphase cells (16). Alternatively, the centrosomal centrobin level was hardly affected in PLK1-suppressed mitotic cells (Fig. 2F).
The microtubule-stabilizing activity of centrobin is comparable with that of doublecortin. Both proteins promote tubulin polymerization in vitro in the absence of seed (35) (Fig. 7B). Overexpression of doublecortin or centrobin induced microtubule bundling in vivo (16, 35). Furthermore, centrobin created microtubules resistant to colcemid, suggesting that centrobin binds to microtubules and stabilizes them (Fig. 7D). Centrobin was detected to be stable at the daughter centriole and was observed transiently at a newly formed microtubule network in interphase cells and mitotic spindles (16). Taken together, we propose that centrobin may be required for stabilizing newly formed microtubules within as well as outside the centrosome.
There is a range of evidence that PLK1 is implicated in spindle assembly during mitosis. The microinjection of PLK1 antibody caused a reduction in γ-tubulin recruitment to centrosomes followed by defects in spindle assembly (36). The PLK1 knockdown experiments using RNA interference also showed similar phenotypes (24, 37). A few substrates have been reported to be responsible for PLK1 regulation on proper spindle assembly. Stathmin/Op18 is a negative regulator of microtubule nucleation, and its affinity for tubulin was reduced by PLK1 phosphorylation (38). PLK1 phosphorylates and activates the abnormal spindle protein, Asp, to facilitate the nucleation of minus ends of microtubules at the centrosome (39). In contrast, there exists an opposite view in which PLK1 phosphorylation decreases the microtubule-stabilizing activity of TCTP and promotes an increase in microtubule dynamics that occurs after metaphase (40). In this study, we discovered centrobin as a microtubule stabilizer whose activity is enhanced by PLK1 phosphorylation. Centrobin may be critical for stabilizing the newly formed spindle during mitosis and its activity may be enhanced by PLK1 phosphorylation. It remains to be investigated how centrobin is linked to other factors for proper mitotic spindle formation in mammalian cells.
Acknowledgment
We thank Joonhyun Park (Seoul National University) for critical support for this work.
This work was supported in part by grants from the BioImaging Research Center at Gwangju Institute of Science and Technology, Basic Research Program Grant R01-2007-000-20116-0, and Science Research Center Program Grant R11-2005-009-03005-0.
- MAP
- microtubule-associated protein
- CBN
- centrobin
- WT
- wild-type
- KD
- kinase-dead
- CA
- constitutively active
- GFP
- green fluorescent protein; Pipes,1,4-piperazinediethanesulfonic acid
- TRITC
- tetramethylrhodamine isothiocyanate
- PBS
- phosphate-buffered saline
- GST
- glutathione S-transferase
- siRNA
- small interfering RNA.
REFERENCES
- 1.Desai A., Mitchison T. J. (1997) Annu. Rev. Cell Dev. Biol. 13, 83–117 [DOI] [PubMed] [Google Scholar]
- 2.Kinoshita K., Arnal I., Desai A., Drechsel D. N., Hyman A. A. (2001) Science 294, 1340–1343 [DOI] [PubMed] [Google Scholar]
- 3.Ookata K., Hisanaga S., Bulinski J. C., Murofushi H., Aizawa H., Itoh T. J., Hotani H., Okumura E., Tachibana K., Kishimoto T. (1995) J. Cell Biol. 128, 849–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brouhard G. J., Stear J. H., Noetzel T. L., Al-Bassam J., Kinoshita K., Harrison S. C., Howard J., Hyman A. A. (2008) Cell 132, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walczak C. E., Mitchison T. J., Desai A. (1996) Cell. 84, 37–47 [DOI] [PubMed] [Google Scholar]
- 6.Cassimeris L. (1999) Curr. Opin. Cell Biol. 11, 134–141 [DOI] [PubMed] [Google Scholar]
- 7.Andersen S. S. (2000) Trends Cell Biol. 10, 261–267 [DOI] [PubMed] [Google Scholar]
- 8.Rusan N. M., Tulu U. S., Fagerstrom C., Wadsworth P. (2002) J. Cell Biol. 158, 997–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitazawa H., Iida J., Uchida A., Haino-Fukushima K., Itoh T. J., Hotani H., Ookata K., Murofushi H., Bulinski J. C., Kishimoto T., Hisanaga S. (2000) Cell Struct. Funct. 25, 33–39 [DOI] [PubMed] [Google Scholar]
- 10.Ookata K., Hisanaga S., Sugita M., Okuyama A., Murofushi H., Kitazawa H., Chari S., Bulinski J. C., Kishimoto T. (1997) Biochemistry 36, 15873–15883 [DOI] [PubMed] [Google Scholar]
- 11.Fourest-Lieuvin A., Peris L., Gache V., Garcia-Saez I., Juillan-Binard C., Lantez V., Job D. (2006) Mol. Biol. Cell 17, 1041–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drewes G., Ebneth A., Preuss U., Mandelkow E. M., Mandelkow E. (1997) Cell 89, 297–308 [DOI] [PubMed] [Google Scholar]
- 13.Ebneth A., Drewes G., Mandelkow E. M., Mandelkow E. (1999) Cell Motil. Cytoskeleton 44, 209–224 [DOI] [PubMed] [Google Scholar]
- 14.Zou C., Li J., Bai Y., Gunning W. T., Wazer D. E., Band V., Gao Q. (2005) J. Cell Biol. 171, 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee J., Kim S., Jeong Y., Rhee K. (2009) Mol. Cells 28, 31–36 [DOI] [PubMed] [Google Scholar]
- 16.Jeong Y., Lee J., Kim K., Yoo J. C., Rhee K. (2007) J. Cell Sci. 120, 2106–2116 [DOI] [PubMed] [Google Scholar]
- 17.Sonn S., Jeong Y., Rhee K. (2009) Mol. Reprod. Dev. 76, 587–592 [DOI] [PubMed] [Google Scholar]
- 18.Fry A. M., Schultz S. J., Bartek J., Nigg E. A. (1995) J. Biol. Chem. 270, 12899–12905 [DOI] [PubMed] [Google Scholar]
- 19.Yoo J. C., Chang J. R., Kim S. H., Jang S. K., Wolgemuth D. J., Kim K., Rhee K. (2004) Exp. Cell Res. 292, 393–402 [DOI] [PubMed] [Google Scholar]
- 20.Mayor T., Hacker U., Stierhof Y. D., Nigg E. A. (2002) J. Cell Sci. 115, 3275–3284 [DOI] [PubMed] [Google Scholar]
- 21.Lee K. S., Erikson R. L. (1997) Mol. Cell. Biol. 17, 3408–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nadano D., Nakayama J., Matsuzawa S., Sato T. A., Matsuda T., Fukuda M. N. (2002) Biochem. J. 364, 669–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golsteyn R. M., Mundt K. E., Fry A. M., Nigg E. A. (1995) J. Cell Biol. 129, 1617–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sumara I., Giménez-Abián J. F., Gerlich D., Hirota T., Kraft C., de la Torre C., Ellenberg J., Peters J. M. (2004) Curr. Biol. 14, 1712–1722 [DOI] [PubMed] [Google Scholar]
- 25.Jang Y. J., Ma S., Terada Y., Erikson R. L. (2002) J. Biol. Chem. 277, 44115–44120 [DOI] [PubMed] [Google Scholar]
- 26.Rapley J., Baxter J. E., Blot J., Wattam S. L., Casenghi M., Meraldi P., Nigg E. A., Fry A. M. (2005) Mol. Cell. Biol. 25, 1309–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moores C. A., Perderiset M., Kappeler C., Kain S., Drummond D., Perkins S. J., Chelly J., Cross R., Houdusse A., Francis F. (2006) EMBO J. 25, 4448–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakamura M., Zhou X. Z., Lu K. P. (2001) Curr. Biol. 11, 1062–1067 [DOI] [PubMed] [Google Scholar]
- 29.Ligon L. A., Shelly S. S., Tokito M., Holzbaur E. L. (2003) Mol. Biol. Cell 14, 1405–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bondallaz P., Barbier A., Soehrman S., Grenningloh G., Riederer B. M. (2006) Cell. Motil. Cytoskeleton 63, 681–695 [DOI] [PubMed] [Google Scholar]
- 31.Elia A. E., Cantley L. C., Yaffe M. B. (2003) Science 299, 1228–1231 [DOI] [PubMed] [Google Scholar]
- 32.Lee K. S., Park J. E., Kang Y. H., Zimmerman W., Soung N. K., Seong Y. S., Kwak S. J., Erikson R. L. (2008) Cell Cycle 7, 141–145 [DOI] [PubMed] [Google Scholar]
- 33.Kang Y. H., Park J. E., Yu L. R., Soung N. K., Yun S. M., Bang J. K., Seong Y. S., Yu H., Garfield S., Veenstra T. D., Lee K. S. (2006) Mol. Cell 24, 409–422 [DOI] [PubMed] [Google Scholar]
- 34.Baumann C., Körner R., Hofmann K., Nigg E. A. (2007) Cell 128, 101–114 [DOI] [PubMed] [Google Scholar]
- 35.Gleeson J. G., Lin P. T., Flanagan L. A., Walsh C. A. (1999) Neuron 23, 257–271 [DOI] [PubMed] [Google Scholar]
- 36.Lane H. A., Nigg E. A. (1996) J. Cell Biol. 135, 1701–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Vugt M. A., van de Weerdt B. C., Vader G., Janssen H., Calafat J., Klompmaker R., Wolthuis R. M., Medema R. H. (2004) J. Biol. Chem. 279, 36841–36854 [DOI] [PubMed] [Google Scholar]
- 38.Budde P. P., Kumagai A., Dunphy W. G., Heald R. (2001) J. Cell Biol. 153, 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.do Carmo Avides M., Tavares A., Glover D. M. (2001) Nat. Cell Biol. 3, 421–424 [DOI] [PubMed] [Google Scholar]
- 40.Yarm F. R. (2002) Mol. Cell. Biol. 22, 6209–6221 [DOI] [PMC free article] [PubMed] [Google Scholar]