

Abstract
The nucleotide state of actin (ATP, ADP-Pi, or ADP) is known to impact its interactions with other actin molecules upon polymerization as well as with multiple actin binding proteins both in the monomeric and filamentous states of actin. Recently, molecular dynamics simulations predicted that a sequence located at the interface of subdomains 1 and 3 (W-loop; residues 165–172) changes from an unstructured loop to a β-turn conformation upon ATP hydrolysis (Zheng, X., Diraviyam, K., and Sept, D. (2007) Biophys. J. 93, 1277–1283). This region participates directly in the binding to other subunits in F-actin as well as to cofilin, profilin, and WH2 domain proteins and, therefore, could contribute to the nucleotide sensitivity of these interactions. The present study demonstrates a reciprocal communication between the W-loop region and the nucleotide binding cleft on actin. Point mutagenesis of residues 167, 169, and 170 and their site-specific labeling significantly affect the nucleotide release from the cleft region, whereas the ATP/ADP switch alters the fluorescence of probes located in the W-loop. In the ADP-Pi state, the W-loop adopts a conformation similar to that in the ATP state but different from the ADP state. Binding of latrunculin A to the nucleotide cleft favors the ATP-like conformation of the W-loop, whereas ADP-ribosylation of Arg-177 forces the W-loop into a conformation distinct from those in the ADP and ATP-states. Overall, our experimental data suggest that the W-loop of actin is a nucleotide sensor, which may contribute to the nucleotide state-dependent changes in F-actin and nucleotide state-modulated interactions of both G- and F-actin with actin-binding proteins.
Keywords: Actin, ADP, ATP, Cytoskeleton, Nucleotide, Protein Conformation, Protein Structure, Kabiramide C, SpvB, WH2 Domain
Introduction
Actin, one of the most abundant and conserved eukaryotic proteins, is involved in a variety of cellular functions, from cell division and migration to intracellular transport and endo- and exocytosis. Under physiological conditions, monomeric actin (G-actin)3 and filamentous actin (F-actin) are in a dynamic equilibrium, strongly favoring the latter. Very low basal ATPase activity of G-actin is amplified strongly upon its polymerization, and the release of inorganic Pi in general follows the polymerization with some delay (2, 3). Consequently, under equilibrium conditions, a typical actin filament contains ATP protomers at the growing end followed by an ADP-Pi-enriched region and then by an extended ADP region in the rest of the filament. The nature of the bound nucleotide influences the properties of G- and F-actin and controls their interactions with a number of actin-binding proteins (ABP), which play key regulatory and structural roles in the dynamics and maintenance of the actin cytoskeleton. Nucleotide-sensitive ABPs regulate nucleotide exchange rates and the polymerization of G-actin, the nucleation of new filaments, and disassembly of mature filaments. Thus, preferential binding of the Arp2/3 nucleating complex to ATP and ADP-Pi F-actin, cofilin to ADP-F-actin, and profilin to ATP-G-actin leads to the acceleration of directed actin filament treadmilling by orders of magnitude (for a recent review, see Ref. 4). Such selective recognition of ABPs implies that actin undergoes conformational changes upon ATP hydrolysis and Pi release; however, the detailed structural basis of such changes is still debated.
Molecular events, presumably implicated in the nucleotide-dependent conformational transitions of actin upon ATP hydrolysis, can be classified as global and local depending on the amplitude of the conformational transition. Global conformational changes suggest rotation of entire subdomains, resulting in an open-closed transition of the nucleotide binding cleft (5). In contrast, local conformational changes are restricted to relatively small regions on the actin molecule (Fig. 1A), such as the direct sensors of ATP hydrolysis the H-loop (residues 70–78) and the S-loop (residues 11–16) (6–8) and the distant sensors, the C terminus (9, 10), and the DNase I binding loop (D-loop) (6, 11).
FIGURE 1.

The W-loop region of actin in different nucleotide states. A, shown are nucleotide sensitive regions of actin. The S-loop (residues 11–16, blue), H-loop (residues 70–78, magenta), and G-loop (residues 154–161, yellow) are involved in a direct interaction with a nucleotide (ATP, ADP-Pi, or ADP), whereas the D-loop (residues 38–52, green), W-loop (residues 165–172, red), and the C terminus (349–375, cyan) are putative distant nucleotide sensors. The Arg-177 residue, ADP-ribosylated by SpvB enzyme, is represented as a stick model and colored in apricot. B, shown is a schematic representation of secondary structure of the W-loop region in the ATP, ADP-Pi, and ADP nucleotide states. C, superposition of the W-loop region of α-skeletal actin in ATP, ADP-Pi, and ADP states from the molecular dynamics simulations (1) were kindly provided by Dr. Sept. Residues 166–171 are colored as shown in C. Color intensity increases in the following order: ATP, ADP-Pi, ADP actin states. Distances between the carboxyl group of Tyr-166 and an amine group of Tyr-169 are shown by yellow (ATP state), cyan (ADP-Pi state), and black (ADP state) dashed lines. D, sequence alignment of the W-loop residues of mammalian α-skeletal and S. cerevisiae actins is shown. Residues are colored as in C.
Opening of the nucleotide cleft of actin upon ATP hydrolysis and Pi release has been initially suggested through an analogy with other members of the hexokinase family ATPases to which actin belongs (12). To date, this cleft opening hypothesis remains to be confirmed. Solution data on the cleft opening has been viewed as inconclusive and open to alternative interpretations (10). Furthermore, most crystal structures of ADP-actin as well as ATP-actin are in the closed state, with a single exception of the β-actin-profilin complex, which was solved in the open conformation (13). A recent report utilizing dynamic fluorescence quenching supports the opening and closing of the cleft upon profilin and cofilin binding, respectively (14), whereas the results of several molecular dynamic simulation studies are inconclusive (1, 15–18). Thus, it appears that additional experiments involving direct measurements of intercleft distance are required to resolve this ambiguity.
Recently, nucleotide-dependent conformational changes on actin have been examined by Sept and co-workers (1) using multiple, long molecular dynamics simulations. Although the opening of the nucleotide cleft was not observed in this study, several sites on actin were found to respond specifically to the ATP, ADP-Pi, or ADP state of the nucleotide binding cleft (Fig. 1A). First, it has been found that the H-loop, previously denoted as a sensor loop (6), loses most of the hydrogen-bonding networks responsible for the formation of a β-sheet in the ATP and ADP-Pi states and transits to an unstructured coil in the ADP state. Second, in agreement with crystallography studies of Dominguez and co-workers (6, 7), it has been suggested that the D-loop forms a stable α-helix in the ADP state but adopts a coil conformation in both the ATP and ADP-Pi states. Third, the position of the C-terminal region (residues 349–375; Fig. 1A) differed by 5 Å between the average ATP and ADP states. Finally, it was noted (1) that the W-loop (residues 165–172) shifted from a coil state in ATP-actin to a classical β-turn conformation in the ADP- and ADP-Pi-actin due to the formation of backbone hydrogen bonds between Tyr-166 and Tyr-169 (Tyr-166 and Phe-169 in yeast; Fig. 1, B and C). The last observation merits special attention for the following reasons; (i) the W-loop is a prime interaction site for the WH2 domain proteins and also participates in the binding of profilin (5), cofilin (19), vitamin D binding protein (20–22), and MAL (23), most of which are known to have different affinities for ATP- and ADP-actin; (ii) the W-loop is a part of the F-actin interface in most filament models published to date (24–26); (iii) in contrast to all other nucleotide-sensitive areas mentioned above, the W-loop has not been tested experimentally for nucleotide-dependent conformational rearrangements on actin and evaded prior scrutiny.
In this study we explored the W-loop sensitivity to nucleotide-dependent transitions in actin by site-directed mutagenesis and specific fluorescent labeling of several residues within this loop. Our results provide the first experimental evidence for nucleotide-dependent conformational changes in this region. Future research should examine the interactions of different ABPs with the W-loop region on actin in different nucleotide states.
EXPERIMENTAL PROCEDURES
Chemical Reagents
Analytical grade chemicals were purchased from Sigma unless specified otherwise. ATP and HEPES were from Merck. Tetramethylrhodamine-5-maleimide, pyrene maleimide, and fluorescein maleimide were purchased from Invitrogen. Kabiramide C (KabC) and the SpvB-expressing plasmid were generous gifts from Dr. Gerard Marriott (University of Wisconsin) and Dr. C. Eric Stebbins (Rockefeller University), respectively.
Site-directed Mutagenesis
The QuikChange® XL site-directed mutagenesis kit from Stratagene was used to introduce mutations into the yeast actin coding sequence. The following primers and their reverse complements were used to introduce the desired mutations: A167C, 5′-CGTCGTTCCAATTTACTGTGGTTTCTCTCTACCTC-3′; G168C, 5′-CGTTCCAATTTACGCTTGTTTCTCTCTACCTC-3′; F169C, 5′-CCAATTTACGCTGGTTGTTCTCTACCTCACGCC-3′; S170C, 5′-CAATTTACGCTGGTTTCTGTCTACCTCACGCCATTTTG.
A derivative of pRS314 plasmid (27) carrying the TRP1 gene, C374A mutant actin coding sequence and the promoter region, was used as a template for mutagenesis (28). In each case the introduced mutations were confirmed by sequencing. The mutant plasmids were transformed into a trp1, ura3-52 haploid cell in which the chromosomal ACT1 gene had been disrupted by replacement of the coding sequence with the LEU2 gene. Wild-type actin was expressed in these cells from another centromeric plasmid, pCENWT, containing the URA3 gene. Transformants were selected on tryptophan-deficient medium and then subjected to plasmid shuffling to eliminate the WT actin gene. The mutant plasmids were rescued from the trp+, ura− cells, amplified in Escherichia coli cells and sequenced to verify that the mutations were intact.
Yeast Strain Growth
The effect of mutations on the growth characteristics of yeast strains was examined as described (29). Briefly, WT and mutant strains from fresh overnight cultures were diluted to 0.1 A600 and grown in YPD liquid media at 30 °C with agitation. Cell density was monitored by measuring the optical density of 1-ml aliquots removed at selected time points. Growth curves were plotted as A600 versus time. Temperature sensitivity of actin mutants and their growth on solid media were examined as described previously (29) with minor modifications. Briefly, overnight cultures of WT and mutant actin strains were diluted to an A600 of 0.1, and serial dilutions of 1, 1/10, 1/100, 1/1000, and 1/10000 were made in phosphate-buffered saline. 3 μl of each dilution was spotted onto YPD, YPD + 0.5 m NaCl, and YPG plates (2% yeast peptone, 2% glycerol, and 1% yeast extract). The last two conditions were used to test for hyperosmolar sensitivity and for mitochondrial defects, respectively. Growth of colonies on the plates was examined after 72 h.
Actin Preparation
Yeast actin was purified as described (30, 31). G-actin was stored on ice in G buffer (0.2 mm Ca2+, 0.2 mm ATP, 1 mm dithiothreitol, 10 mm HEPES, pH 7.5) and used within 10 days from the purification. G-actin was converted to the Mg-G-actin form with the addition of 0.1 mm MgCl2 and 0.2 mm EGTA. ATP-G-actin, in the presence of 0.5 mm ATP and 1 mm glucose, was converted to ADP-G-actin with hexokinase (3 units/ml). Actin concentration was determined by using A290 nm (1%) = 11.5 cm−1 (in the presence of 0.5 m NaOH) and a molecular mass of 42.3 kDa.
Actin Polymerization and Pi Release Measurements
Actin polymerization in the presence or absence of phalloidin was induced with 3 mm MgCl2 and was monitored via light scattering with the PTI spectrofluorometer (Photon Technology International, South Brunswick, NJ) set at 325 nm for both the excitation and emission wavelengths. The release of inorganic phosphate was monitored in parallel experiments under identical conditions via color reaction with the EnzChek phosphate assay kit (Invitrogen). The accumulation of the chromophoric reporter of Pi was monitored at 360 nm in a Hewlett Packard 8453 spectrophotometer. Both types of measurements were initiated within 1 h of each other and performed under carefully controlled temperature, at 24 °C.
Nucleotide Exchange and Fluorescence Measurements
The rate of nucleotide exchange in G-actin (1–2 μm) was monitored via the change in ϵ-ATP fluorescence as described before (10). All measurements were carried out in a G buffer at 22 °C. Monitoring ϵ-ATP release from actin was initiated with the addition of 100 μm ATP. The decrease in ϵ-ATP fluorescence upon its release from actin was followed in the PTI fluorometer with the excitation set at 350 nm and the emission at 410 nm. The rates of nucleotide exchange were determined by fitting the data to a single exponential decay expression. Binding of cofilin to the WT- and F-169C G-actin and binding of latrunculin A to Mg-ADP rabbit skeletal actin were measured via nucleotide release inhibition (32). Cofilin binding to F-actin was evaluated by densitometry of SDS gels of F-actin and cofilin co-pelleted in an OPTIMA-TLX120 ultracentrifuge at 300,000 × g for 30 min at 24 °C. The fluorescence of pyrene, rhodamine, and fluorescein was monitored with the excitation set at 344, 520, and 475 nm and the emission wavelengths set at 377, 575, and 515 nm, respectively.
A titration of actin with inorganic phosphate was performed as follows. Aliquots of inorganic phosphate from a 200 mm stock solution, pH 7.5, were added to a sample containing 5 μm pyrene-labeled (95%) F169C G-actin, and the Pi-dependent decrease in pyrene fluorescence was recorded in a PTI fluorescence spectrometer with the excitation and emission wavelengths set at 344 and 376 nm, respectively. Experimental data were fitted to the quadratic binding isotherm,
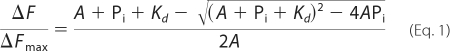 |
where A and Pi are the concentrations of G-actin and inorganic phosphate, respectively, and Kd is the observed dissociation constant. ΔF is the observed fluorescence change of the Cys-169-pyrene actin after correction for dilution. Because the final ATP addition coincided well with a saturation point of the titration, an assumption was made that Pi saturation brings the pyrene fluorescence of ADP-actin to the level of ATP-actin. Therefore, ΔFmax is the fluorescence change upon the addition of ATP to ADP-actin.
Critical Concentration Measurements
To determine the critical concentration (Cc) for actin polymerization, each of the W-loop mutants of actin (20 μm) was polymerized in G buffer supplemented with 3 mm MgCl2 (F buffer) for 2 h. Then, actin was diluted with the polymerization buffer to 15 different concentrations within the range 0.1–15 μm and incubated for 4 h at room temperature. Light scattering from each sample was measured in a firmly immobilized cuvette, and the resulting values were plotted versus total actin concentration. The Cc was determined from the intersection of two linear regression lines with distinctly different slopes. The Cc of ADP-actin was determined similarly to that of ATP-actin, except that ATP in polymerized actin was converted to ADP by adding hexokinase and glucose. After 1 h, samples were diluted with ADP-containing buffer to 15 different concentrations within the range 0.1–25 μm and incubated for 5 h at room temperature.
Electron Microscopy
For actin filament imaging, 10 μm actin was polymerized in the presence of 3 mm MgCl2 for 2 h. Immediately before loading on the EM grids (Ted Pella Inc., CA), actin was diluted 4-fold with the F buffer. The samples were deposited on the EM grids, incubated for 1 min, and then negatively stained with 1% uranyl acetate (w/v) for 0.75–1 min. Grids were examined in a JEM1200EX electron microscope at an accelerating voltage of 80 keV and a nominal magnification in the 100,000–150,000× range.
Actin Labeling
Actin was supplemented with 10 mm dithiothreitol and incubated for 1 h to ensure efficient reduction of sulfhydryl groups. Before labeling, dithiothreitol was removed from G-actin solutions over disposable PD-10 desalting columns (GE Healthcare) equilibrated with G or TG buffer (5 mm Tris, pH 8, 0.2 mm CaCl2, 0.3 mm ATP). Next, G-actin was incubated with an equimolar amount of the label for 1–4 h on ice, and the reaction was stopped with 1 mm dithiothreitol. The unreacted label was removed from the samples over a Sephadex G-50 gel filtration column. C374A actin, used as a negative control, showed very low levels of labeling and no nucleotide-specific changes.
RESULTS
Effect of Mutations on Yeast Cell Growth
To examine the conformational dynamics of the W-loop in actin by fluorescent probes, yeast actin mutants A167C, F169C, and S170C were generated in a C374A background to yield a single reactive cysteine in the W-loop for each mutant. In the utilized system each particular mutant actin was expressed as the only actin in yeast. We were not able to replace WT actin with the G168C/C374A actin mutant, suggesting that this actin mutation is lethal to yeast.
Because actin in yeast is essential for cell cytokinesis, endocytosis, and organelle transport, comparative growth rates of yeast cells provide general characterization of the functional performance of actin mutants in vivo. Yeast cells with C374A actin as well as A167C and S170C actins in the C374A background showed similar growth rates and reached a plateau at ∼87% that for cells expressing WT actin (Fig. 2A). Cells with F169C/C374A actin showed a slightly but reproducibly slower growth rate and reached plateau at the ∼77% level as compared with WT yeast. This correlates with an overall lower colony density of the F169C yeast on YPD plates under most conditions tested. Although all W-loop mutants showed temperature sensitivity when grown at 37 °C, growth inhibition was most dramatic for the F169C actin, indicating a lower thermal stability of either monomers or filaments (or both) of this mutant (Fig. 2B). To detect whether the introduced actin mutations cause defects in the mitochondria function, we assessed the ability of the mutants to grow on plates containing glycerol as a sole carbon source. Both F169C and A167C showed low viability on glycerol plates, whereas S170C actin was able to utilize glycerol similarly to the WT and C374A controls (Fig. 2, low panel). In addition, F169C and A167C cells showed growth retardation under hyperosmolar conditions (0.5 m NaCl), suggesting that these actin mutants do not satisfy fully the requirements for a compensatory restructuring of the actin cytoskeleton (33).
FIGURE 2.

Characterization of S. cerevisiae A167C, F169C, and S170C mutant actins. A, growth of yeast with the W-loop actin mutants in a liquid YPD medium (A) and on YPD agar plates at different temperatures (B; top panel) in the presence of 0.5 m NaCl and on YPG agar plate with glycerol as the only carbon source (B, bottom panel) was carried out as described under “Experimental Procedures.” Compared with the WT actin strain, most of the mutant strains showed only mild growth changes in liquid culture at 30 °C and on plates at 24 and 30 °C. However, the growth of A167C and S170C strains was significantly inhibited at 37 °C, whereas the growth of the F169C strain was abolished under the same conditions.
Polymerization Properties of A167C, F169C, and S170C Actin Mutants
All three W-loop actin mutants polymerized in the presence of 3 mm MgCl2, albeit at different rates and to significantly different extents, as monitored by light scattering and confirmed by EM (Figs. 3 and 4). Filaments of all mutants but F169C appeared normal upon EM examination. The F169C mutant actin formed short and curved abnormal filaments with a tendency to aggregate. Filament structures are stabilized through the binding of phalloidin at the interface of three subunits in F-actin, and thus, the assembly of polymerization-impaired actins can be frequently rescued by this reagent. Similarly, cofilin can also rescue the polymerization of some assembly impaired actin mutants and chemically or enzymatically modified actins by bridging adjacent subunits and remodeling the intersubunit interface in the filament (34, 35). However, for the F169C actin mutant, only phalloidin (but not cofilin) efficiently improved both the filaments morphology and polymerization extent (Figs. 3 and 4). Notably, the F169C mutation did not significantly affect affinity of yeast cofilin to ATP-G-actin (Kd 0.16 ± 0.05 μm (31) and 0.17 ± 0.06 μm for WT actin and F169C actin, respectively) and preserved cofilin binding to F-actin (Kd ∼ 0.05 (36) and 0.21 ± 0.05 μm for WT and F169C actins, respectively).
FIGURE 3.

EM images of WT-actin and A167C, F169C, and S170C mutant actins. All actins but the F169C mutant form long helical filaments in the presence of 3 mm MgCl2. The F169C mutant actin alone and in the presence of cofilin forms short crooked filaments and filament aggregates, but long filaments of normal morphology are formed in the presence of phalloidin.
FIGURE 4.

Polymerization of W-loop mutant actins and Pi release. The polymerization of A167C and S170C (A) and F169C (B) actins (10 μm) was monitored by light scattering (black traces) and the accompanying Pi release (gray traces) was detected in parallel, enzyme coupled reactions by measuring the absorbance increase at 480 nm (EnzCheck phosphate assay kit, Invitrogen). The traces for polymerization and Pi release by the A167C and WT actin were virtually superimposable (for clarity, only A167C traces are shown). The polymerization and Pi release followed the same kinetics for WT, A167C, and S170C actins but not for the F169C mutant actin. Unless stabilized by phalloidin (F169C+phalloidin), the polymerization of 10 μm F169C actin was accompanied by a notably lower increase of light scattering, consistent with the higher critical concentration and shorter filament size for this mutant. Pi release (shown for various concentrations of F169C actin) followed the light scattering increase only initially (explanation in the text under the “Results” section). C, monitoring of ϵ-ATP release from 1 μm Ca2+-ATP WT and mutant actins via fluorescence decay was initiated with adding 100 μm ATP to the solution. Experimental curves (black and gray symbols) were fitted to a single exponential decay equation (solid lines), and the nucleotide release rates are given in Table 1. Notably, experimental curves corresponding to ϵ-ATP release from WT-actin (gray symbols) and Cys-167-actin (black symbols) are virtually superimposable.
The rates and extent of polymerization of the W-loop mutants decreased in the order A167C > S170C > F169C. Accordingly, the critical concentrations for ATP-actin polymerization (Cc) increased in the same order: 0.3 ± 0.05, 1.5 ± 0.1, and 3.2 ± 0.2 μm for A167C, S170C, and F169C actins, respectively (data not shown). The Cc for polymerization of these mutants in the ADP state also followed the same order: 3.8 ± 0.8, 10.4 ± 0.9, and 12.8 ± 0.5 μm for A167C, S170C, and F169C actins, respectively (compared with 4.1 ± 0.9 μm for WT-actin). Notably, the Cc values for the mutants S170C and F169C are significantly higher than those for WT and A167C actins. This agrees well with predictions from F-actin models (24–26), which suggest that the W-loop plays an important role in establishing the F-actin interface and, therefore, in actin polymerization.
Because the W-loop area is a part of the interprotomer interface in F-actin (24–26), it can serve potentially as an allosteric activator or a contributor to the polymerization enhanced ATPase. Thus, we tested whether mutations in this region affect the Pi release upon actin polymerization. The Pi release correlates well with the increase in light scattering upon polymerization of the A167C and S170C mutants (Fig. 4A) and the WT actin (data not shown). In contrast to that, the Pi release upon polymerization of the F169C mutant correlates with light scattering only at the beginning of the polymerization but accelerates strongly as the polymerization approaches a plateau (Fig. 4B). We hypothesized that this accelerated ATP hydrolysis and Pi release was not an intrinsic property of monomeric F169C but the result of filament instability and accelerated cycling of monomeric actin through short and unstable filaments and/or oligomers. To test this, the Pi release was monitored at various concentrations of F169C actin upon its polymerization in the presence of 3 mm MgCl2. F169C actin produces negligible amounts of Pi at close to or below its critical concentration (3.2 μm). At higher concentrations of actin, the Pi release increases in a nonlinear, hyperstoichiometric manner (Fig. 4B; gray traces), suggesting that the polymerization is a primary reason for the high level of Pi production. Because only Pi and not ADP can be released by actin protomers in the filament, fresh ATP can be hydrolyzed only upon filament disassembly and reassembly, i.e. upon treadmilling (37). Because short, unstable filaments treadmill significantly faster than stable ones, these results agree with the EM data and confirm the instability of F169C actin filaments.
Nucleotide Exchange
If the molecular dynamics predictions of the W-loop sensitivity to the nucleotide state of actin (1) are correct, we can expect a reciprocal effect of structural changes in the W-loop on the properties of the nucleotide binding cleft. To test for that, nucleotide exchange rates were monitored for the W-loop mutants and the WT actin. The A167C substitution did not affect the nucleotide exchange rate as compared with the WT actin (Fig. 4C). In contrast, mutations of 169 and 170 residues to cysteine slowed the nucleotide exchange rates by a factor of ∼2 and ∼8, respectively (Fig. 4C and Table 1). Moreover, under some conditions, site-specific labeling of cysteine 169 with either tetramethylrhodamine-5-maleimide or fluorescein probes decreased further the nucleotide exchange rate (Table 1), suggesting that structural changes in the W-loop can indeed affect the nucleotide binding cleft on actin.
TABLE 1.
Nucleotide exchange rates of the W-loop cysteine mutants of yeast actin
All rates are the mean values of at least three determinations. S.D. are given next to the rate values. Under identical conditions, the nucleotide release rates of rabbit skeletal actin were 0.0011 ± 0.0002 and 0.0053 ± 0.0005 s−1 in the presence of Ca2+ and Mg2+ cations, respectively. TMR, tetramethylrhodamine-5-maleimide.
| Divalent cation |
K-ϵ-ATP |
|||||
|---|---|---|---|---|---|---|
| WT | A167C | S170C | F169C | F169C-TMR | F169C- Fluor | |
| s−1 | ||||||
| 0.2 mm Ca2+ | 0.013 ± 0.002 | 0.012 ± 0.006 | 0.0017 ± 0.0004 | 0.006 ± 0.001 | 0.006 ± 0.002- | 0.005 ± 0.002 |
| 0.1 mm Mg2+ | 0.121 ± 0.003 | 0.07 ± 0.01 | 0.013 ± 0.004 | 0.014 ± 0.006 | ||
ATP Hydrolysis Affects the Properties of Fluorescent Probes Attached to the W-loop Residues
To further test the connections between the nucleotide cleft and the W-loop, cysteine residues of the W-loop mutants were labeled with different fluorescent probes. Among the three mutants, F169C actin labeled with pyrene maleimide showed the most prominent changes upon conversion of ATP to ADP. In the presence of glucose and Mg2+ cations, hexokinase converts ATP to ADP and results in ATP substitution by ADP in the nucleotide cleft of actin. This event is accompanied by a fast and steep increase in the Cys-169-pyrene fluorescence (47 ± 6%; n = 10; Table 2) followed by a slow fluorescence rise. Importantly, the fast component of fluorescence increase is completely reversible upon the addition of extra ATP (Fig. 5A), indicating a specific, nucleotide state-dependent conformational perturbation. In contrast, the slow component could not be reversed by ATP, reflecting apparently slow and irreversible conformational changes on actin due to its intrinsic instability in the ADP/apo states (38).
TABLE 2.
Maximum fluorescence intensities of the Cys-169-pyrene actin under different conditions
Mean values of fluorescence intensity maxima from at least three measurements are given with their S.D.
| Actin | MgATP | MgADP |
|---|---|---|
| % | % | |
| C169-pyrene | 100 | 147 ± 6 |
| C169-pyrene+LatA | 99 ± 3 | 105 ± 2 |
| C169-pyrene+KabC | 298 ± 6 | 300 ± 8 |
| C169-pyrene+SpvB | 119 ± 1 | 113 ± 1 |
| C169-pyrene F-actin | 136 ± 6 |
FIGURE 5.

A pyrene probe at Cys-169 reports nucleotide and ligand specific changes. A, the switch from the ATP to ADP state of actin (upon the addition of hexokinase; “Experimental Procedures”) increases the fluorescence of a pyrene probe at Cys-169 in the absence (red and blue traces) but not in the presence of LatA (black trace). The addition of LatA (arrowhead) or extra ATP (open arrow) to the ADP-actin decreases the fluorescence close to that of ATP-actin. A.U., absorbance unit. B, the addition of KabC to ATP-actin (black trace), ADP-actin (red trace), or ADP-actin in the presence of LatA (blue trace) increases the fluorescence of Cys-169-attached pyrene (∼3×). C, ADP-ribosylation of Arg-177 on ATP-actin by SpvB in the presence of NAD+ increases the Cys-169-pyrene fluorescence (black and red traces) by 17 ± 2%. A switch from ATP- to ADP-state in the ADP-ribosylated actin (black arrow) results in a fluorescence decay, which is reversed with extra ATP (open black arrow). D, fluorescence spectra of Cys-169-pyrene actin in different states are shown. Black line, Ca2+-ATP actin and Mg2+-ATP actin (identical spectra); red line, Mg2+-ADP-actin; green line, phalloidin stabilized F-actin in the presence of 3 mm MgCl2.
Low amplitude but reproducible nucleotide-dependent effects were observed also with acrylodan and pyrene maleimide probes at the position Cys-167 (7–10% change upon ATP to ADP transition) and with a fluorescein maleimide probe at positions Cys-169 (∼10% change upon ATP to ADP transition) and Cys-170 (∼25% change; data not shown). In contrast to nucleotide effects, no significant changes were observed with any probe or mutant upon the switch from Ca2+- to Mg2+-ATP actin.
The polymerization of F169C pyrene actin with 3 mm MgCl2 and phalloidin also resulted in a fluorescence increase (36 ± 6%; n = 3; Fig. 5D), which was not affected significantly by additions of Pi or its analog BeFx (data not shown). Therefore, the fluorescence increase upon F169C-pyrene actin polymerization may be brought primarily by burying the pyrene moiety in the adjacent actin protomers.
Effects of Actin Binding Ligands on the F169C-pyrene Fluorescence
KabC, a marine macrolide toxin of the trisoxazole family, binds to actin with its macrolide ring region located in the hydrophobic patch between SD1 and -3 (residues 341–349), whereas its long aliphatic “tail” forms hydrophobic contacts with residues Gly-168 and Tyr/Phe-169 of the W-loop (39).4 Therefore, we checked whether KabC affects the fluorescence of the pyrene probe at Cys-169. The addition of KabC to either ATP or ADP actin increased strongly and to the same final level the fluorescence of Cys-169- pyrene (∼3-fold raise over the initial MgATP actin level; Fig. 5B; Table 2). The addition of KabC eliminates any nucleotide-dependent changes in pyrene fluorescence, indicating that local effects of KabC on the structure of the W-loop prevail over those caused by ATP hydrolysis.
In contrast to KabC, latrunculin A (LatA) binds to actin ∼30 Å away from residue 169, inside the nucleotide-binding cleft. Therefore, it cannot affect directly the fluorescence of the pyrene probe at Cys-169. Using a nucleotide exchange inhibition assay (32), we found that LatA binds to ADP-actin with Kd = 1.02 ± 0.14, which is ∼5 times weaker than its affinity to ATP-actin (Kd∼ 0.2 μm (40, 41)). LatA had no effect on the fluorescence of Cys-169-pyrene actin in the ATP state, but in each test it decreased the fluorescence of ADP-actin to that of ATP-actin (Fig. 5A). Subsequent addition of ATP to the LatA-ADP-actin complex does not anymore affect the fluorescence of pyrene, suggesting that both compounds shift the probe at Cys-169 to a similar conformational state. Moreover, the addition of LatA to ATP-actin blocks completely the ADP-induced transition from a low to high pyrene fluorescence level even over a much longer time scale (Fig. 5A) needed to ensure the efficient substitution of ATP to ADP in the presence of LatA (40). Therefore, our data suggest strongly that LatA allosterically affects the WH2 loop of actin by bringing it to the ATP-like state.
Effects of Inorganic Phosphate
It has been predicted that the W-loop adopts similar β-turn conformations in the ADP and ADP-Pi states and a loop conformation in the ATP-state (1). This is rather unexpected because the ADP-Pi and ATP states are known to be functionally and structurally similar in F-actin, whereas the ADP state is characterized by a higher Cc, greater flexibility, and lower stability of the filaments (42, 43). To test whether the observed conformational changes in the W-loop are due mainly to ATP hydrolysis or Pi release, we monitored the effect of Pi on the fluorescence of the Cys-169-pyrene actin. Because yeast actin polymerization is inhibited in the presence of monovalent cations (Na+ and K+ (44)) and the F169C mutant has a high Cc (3.2 μm in 3 mm MgCl2), we could use high concentrations of inorganic phosphate (up to 54 mm) without causing actin polymerization (as monitored by light scattering; data not shown). Also, pyrene excimer, which can accompany the formation of antiparallel dimers and oligomers at early stages of actin polymerization (30, 45, 46), has never been detected for Cys-169-pyrene actin under all the conditions tested.
We found that the fluorescence of MgATP Cys-169-pyrene actin was not sensitive to Pi additions, whereas that of MgADP actin decreased significantly in a dose-dependent manner (Fig. 6A). As with LatA, the effects of Pi and ATP complemented each other, i.e. ATP decreased the fluorescence of MgADP-Cys-169 pyrene actin to the same final level, and the amplitudes of the ATP-induced shifts decreased with higher concentrations of Pi. A plot of the fluorescence decay of MgADP actin as a function of the Pi added could be fitted well to a curve with a Kd = 12.7 ± 0.5 mm (Fig. 6B). To confirm specific binding of Pi to actin rather than a nonspecific collision quenching of fluorescence, MgADP Cys-169-pyrene actin was titrated also with Na2SO4 and NaCl. Both salts produced only a minor loss of pyrene fluorescence compared with that caused by inorganic phosphate (Fig. 6B; only the titration with Na2SO4 that caused stronger effects, is shown). Overall, as measured by the fluorescence of the pyrene probe at Cys-169, inorganic phosphate shifts the W-loop to an ATP-like state.
FIGURE 6.

Pi affects the fluorescence of the Cys-169-pyrene actin in the ADP but not in the ATP state of Cys-169-pyrene actin. A, titrations of the ATP- (gray trace) and ADP (black trace)- Cys-169-pyrene actin with inorganic phosphate. The additions of Pi are indicated by arrowheads; additions of ATP and hexokinase are denoted by open and closed arrows, respectively. A.U., absorbance unit. B, titration of Cys-169-pyrene actin in the ADP state with phosphate and Na2SO4 are represented by black and white symbols, respectively. Gray symbols represent a titration of Cys-169-pyrene actin in the ATP state with phosphate. The Pi titration of the Cys-169-pyrene ADP-actin was fitted to a quadratic binding equation as described under “Experimental Procedures.” Bars represent S.D. of three independent experiments.
Effects of Arg-177 ADP-ribosylation with SpvB Toxin
Residue Arg-177 is connected either directly or via a water molecule to His-73 (1), the residue reported to be critical in delaying Pi release after hydrolysis (47). Moreover, Arg-177 has been suggested to have a shuttle function, facilitating Pi transport along the backdoor release pathway, from the nucleotide binding site to the periphery of actin (47). Although Arg-177 is not a part of the actin catalytic site (48), its ADP-ribosylation by bacterial toxins (such as botulinum C2 and Salmonella SpvB toxins) facilitates nucleotide exchange and strongly inhibits ATP hydrolysis in G-actin and upon its polymerization (49). Because Arg-177 is located relatively close to the W-loop (residues 165–172), we hypothesized that conformational changes from and to the nucleotide binding cleft of actin could be transmitted through a pathway that includes this residue. To test for this possibility, the F169C-pyrene fluorescence was monitored upon Arg-177 ADP-ribosylation by SpvB. The addition of SpvB increased the fluorescence of MgATP F169C-pyrene by 17 ± 2% (n = 3), mimicking to some extent the effect of ADP. In addition, ADP-ribosylation reversed to a limited degree the effect of ADP on F169C-pyrene actin. The addition of hexokinase to Arg-177-ADP-ribosylated actin decreased the fluorescence by ∼5% in a reversible manner (Fig. 5C). Therefore, ADP-ribosylation of Arg-177 indeed affects the conformation of the W-loop, shifting it to a position that is different from that in ATP or ADP actin.
DISCUSSION
Nucleotide-dependent conformational changes in actin are a subject of considerable interest. Although ATP hydrolysis per se is not required for actin polymerization (50, 51), it defines the properties of actin filaments and provides the energy for their predominant disassembly at the pointed ends and predominant assembly at the barbed ends i.e. the treadmilling of filaments under equilibrium conditions. Many ABPs interact preferentially with the ATP, ADP-Pi, or ADP states of both G- and F-actin. Profilin, thymosin-β4, vitamin D-binding protein, Arp2/3 complex, and WH2 domain proteins have higher affinity to ATP-actin. Other proteins, such as gelsolin and cofilin, bind predominantly to ADP-actin (52–54). However, the detailed nature of conformational rearrangements on actin underlying the ABP binding selectivity is unknown. Many of these proteins bind to actin at the interface between SDs 1 and 3, where the W-loop, a recently predicted nucleotide sensor (1), is located.
Cysteine replacements in the W-loop affect to a different extent the properties of actin. Surprisingly, the eight-amino acid residues of the W-loop of mammalian actins are identical to those of Schizosaccharomyces pombe actin but share only 50% identity with the corresponding sequence of Saccharomyces cerevisiae actin (Fig. 1D). One substitution (E167A) is not conserved, whereas others (Y169F, A170S, L171I) are conserved or semi-conserved. Our attempts to substitute a highly conserved Gly-168 with cysteine failed due to the lethality of this mutant. This suggests strong structural constraints for a residue at this position. Gly-168 is located at the tip of the W-loop and is likely to be critical for the formation of a proper turn. In contrast, the properties of A167C mutant actin were virtually indistinguishable from those of WT actin, at least in terms of their polymerization and nucleotide exchange. The mildness of this phenotype can be justified partly by the relative insignificance of this residue in filament formation and the semi-conserved nature of the substitution (small-to-small residue). However, even a more conserved S170C substitution resulted in a more severe phenotype, with a slower polymerization rate, higher Cc, and, most importantly, a significantly (∼8 times) slower nucleotide exchange rate. This implies that residue 170 is involved in actin polymerization and affects the nucleotide cleft properties of G-actin. Interestingly, residue 170 is Ala in a vast majority of actin isoforms (including S. pombe actin), but it is Ser in S. cerevisiae actin. Analysis of crystal structures of various actins with Protein Interaction Calculator software (55) indicates that Ala-170 can be involved in hydrophobic interactions (within 5 Å) with 5–6 other vicinal residues, namely, amino acids 136, 139, 163, 165, 172, and 175. In striking contrast, the polar side chain of Ser-170 in S. cerevisiae actin does not form any contacts (PDB codes 1VYN and 1YAG) with nearby residues. Interestingly, the nucleotide release rate of skeletal actin is about an order of magnitude slower than that of yeast WT actin under identical conditions (0.0011 versus 0.013 s−1), whereas the S170C substitution makes the nucleotide release properties of both actins comparable. Although Cys and Ser are both polar, non-charged residues, Cys has a positive hydropathy index (+2.5 compared with −0.8 for Ser and, in fact, is even higher than that of Ala (56)), indicating a higher tendency of Cys to seek hydrophobic environment. The significant difference in nucleotide release rates of the two actin isoforms and the dramatic effect of Cys-170 on this rate prompts the speculation that hydrophobic interactions of Ala (in skeletal actin) or Cys (in S170C yeast actin) residues might restrict the dynamic fluctuations of the W-loop and the nucleotide cleft area, resulting in slower nucleotide release and impaired polymerization properties of this mutant. However, additional molecular simulation and mutagenesis studies are required to test this hypothesis.
Although the nucleotide exchange properties of the F169C mutant with a non-conserved F-to-C substitution were affected to a lesser degree (2-fold inhibition compared with WT actin), its polymerization phenotype was more severe. The polymerization impairment due to the F169C substitution is consistent with a model-predicted participation of this residue in the formation of intersubunit contacts in F-actin (24–26, 57). For example, in a recent high resolution model of F-actin, the aromatic ring of Tyr-169 in skeletal actin (corresponding to Phe-169 in yeast actin) makes abundant contacts with several conserved, nonpolar/hydrophobic residues (Val-43, Met-44, and Val-45) in the D-loop of the longitudinally adjacent protomer (26). Given that both the D-loop and the W-loop are nucleotide-sensitive areas, it is tempting to hypothesize that a putative W-loop/D-loop contact may play a critical role in weakening the SD1–3/SD2 longitudinal contacts upon ATP-hydrolysis and Pi release (58). Yet the high Cc of F169C ADP-actin (∼13 μm compared with ∼4 μm for that of WT and Cys-167 ADP-actins) suggests that the W-loop might participate in the stabilization of filament contacts also in ADP-F-actin.
Both Ala-167 and Ser-170 (Glu-167 and Ala-170 in human actins, respectively) also play important roles in filament contact formation, as follows both from the current study and another recent work (59). However, these residues are less conserved, and their role in filament stabilization is more difficult to explain based on the existing filament models. Thus, A167R and A167E mutations have been recently shown to severely impair the polymerization of S. cerevisiae yeast actin (59), which is quite unexpected given that Glu-167 is present in most other actins (Fig. 1D), including actin from another yeast organism, S. pombe. Therefore, WT-like behavior of the A167C mutant observed in our assays is likely due to a relatively conserved nature of the substitution (Ala-to-Cys) and not the insignificance of this residue to actin polymerization.
The addition of phalloidin rescued the polymerization of F169C actin, whereas cofilin failed to significantly improve its polymerization and the structure of the filaments. This contrasts with the behavior of several other actin species with impaired polymerization, namely T203C yeast mutant actin, tetramethylrhodamine-5-maleimide-modified α-skeletal actin, and enzymatically cross-linked actin oligomers, for which the polymerization can be rescued with cofilin (34, 35). Cofilin rescues actin polymerization by bridging neighboring subunits and changing the actin-actin interface (34). It follows from the failure of cofilin to improve the F169C filaments that a bulky ring of the Tyr/Phe-169 residue may be involved in filament contacts both in the cofilin-free and cofilin-decorated F-actin.
Nucleotide Effects on the Properties of the W-loop
Fluorescent probes attached to any of the tested W-loop cysteine mutants responded to different extents to the substitution of ATP with ADP at the nucleotide cleft. The most responsive mutant was F169C, and the most sensitive probe for this mutant was pyrene maleimide. Pyrene fluorescence increases reversibly with the transition of G-actin to the ADP-state. Molecular dynamics simulations performed by Zheng et al. (1) showed that the W-loop forms a β-turn in the ADP and ADP-Pi states and adopts a loop conformation in the ATP-state, i.e. the ADP- and ADP-Pi states of the W-loop are similar to each other, but different from the ATP state. Yet numerous studies indicate that the properties of F-actin in ATP and ADP-Pi states are similar to each other but differ from those of ADP-F-actin (for review, see Ref. 60). The latter is in agreement with our experiments on G-actin, indicating that the W-loop properties are indeed similar in the ATP and ADP-Pi actin but different in the ADP-actin. In support of that, the addition of inorganic phosphate to F169C-pyrene actin caused no significant changes in the fluorescence of ATP-actin but brought the fluorescence of ADP-actin close to that of ATP-actin (Fig. 6). Several factors may potentially account for the apparent discrepancy between the molecular dynamic simulation data and our results, including isoform specificity (α-skeletal actin in Zheng et al. (1) versus S. cerevisiae actin in the present study), the effect of mutation, and/or labeling. An alternative explanation could be that the observed W-loop properties are defined by the side chain rather than the backbone. Indeed, in the molecular dynamic simulation study (1), the Tyr-169 side chain occupies a similar position in ATP and ADP-Pi states but swings out significantly in the ADP-state (Fig. 1C).
Pi titration of ADP-actin in the presence of KabC did not influence fluorescence intensity, indicating that the strong local effects of KabC overcome the allosteric effects of Pi. This is not surprising as KabC makes direct contacts with residues 168 and 169. The Pi affinity to F169C-G-actin was estimated at ∼13 mm, which is about 2.5–7 times stronger than measured previously for skeletal α-actin (∼30–90 mm (61). Although we do not have a direct explanation for this discrepancy, it may stem from the difference between actin isoforms or the labeling of the W-loop with pyrene maleimide.
Effects of Drugs and Modifications
LatA is a marine drug that binds to G-actin in the nucleotide binding cleft region and blocks filament formation most likely by interfering with specific conformational changes necessary for polymerization (62). Although LatA does not affect the fluorescence of the probe in the ATP-state of F169C-pyrene actin, it quenches efficiently the fluorescence of ADP-actin to the level of ATP-actin (Fig. 5A). Therefore, our data suggest that LatA forces the W-loop of actin to the ATP-like conformation. This is not unexpected, because LatA binds with higher affinity to ATP-actin (Ref. 40 and our data) and apparently stabilizes that conformation. Our result correlates with the observations that the binding of LatA to actin is affected by profilin and cofilin, proteins that interact with actin in the cleft between SD1 and -3, at a significant distance from the LatA binding site. Binding of LatA to actin is allosterically inhibited by cofilin (63) (which has higher affinity for ADP-actin) but improved slightly by profilin (has higher affinity for ATP-actin) (40). Future studies should clarify whether the W-loop is involved in the regulation of nucleotide sensitive interactions of actin with these and other proteins.
Although it is not clear to date how conformational changes are transmitted from the nucleotide cleft region to the W-loop, our data support a view that Arg-177 might be involved in this process. In the primary sequence of actin, Arg-177 is the closest residue to the W-loop that is known to be directly implicated in the regulation of nucleotide cleft properties. Thus, Arg-177 is one of the key residues in the backdoor mechanism of Pi release upon hydrolysis (47). We found that ADP-ribosylation of Arg-177 by SpvB changes the environment of pyrene at Cys-169. This modification both increases the fluorescence signal in the ATP state of actin and inverses the changes caused by a transition from the ATP to ADP state. Therefore, ADP-ribosylation does not block, but modifies the conformational signal transmitted from the nucleotide binding cleft to the W-loop.
Comparison of S. cerevisiae and Mammalian Actins
The W-loop region of S. cerevisiae actin has a lower level of conservation than most other actins (Fig. 1D). This raises a potential concern that the observed nucleotide sensitivity can be attributed to the yeast actin but not be a universal property of all actins. However, several lines of evidence suggest that this is unlikely to be the case. First, the nucleotide sensitive hydrogen bond is formed between a fairly conserved Tyr-166-Tyr/Phe-169 pair of residues. Second, three antiparallel β-strands connecting the W-loop with the nucleotide binding region (Fig. 1A) are also well conserved among different actin species. Third, the nucleotide sensitivity of the W-loop was predicted initially based on the molecular dynamics simulation of skeletal actin (1) but has been confirmed now for yeast actin, suggesting its conservation across species. Finally, the nucleotide-dependent regulation of actin polymerization as well as actin interaction with several key ABPs is highly conserved through evolution, and therefore, the underlying allosteric interconnections on actin should be also conserved.
In conclusion, we demonstrate experimentally that the W-loop of actin (residues 165–172) undergoes nucleotide-dependent conformational changes and, thus, together with the C terminus and the DNase binding loop, is a nucleotide sensitive region on actin. Given the importance of this and the surrounding sites of actin to protein-protein interactions (64) and to the formation of F-actin interface, the W-loop region is a prime candidate for contributing to the nucleotide sensitivity of these interactions.
Acknowledgments
We thank Dr. Eric Stebbins (Rockefeller University) for the kind donation of SpvB encoding plasmid, Dr. Gerard Marriott (University of Wisconsin-Madison) for the gift of KabC, and Melissa McKane (University of Iowa) for production of the yeast strain expressing A167C actin.
This work was supported, in whole or in part, by National Institutes of Health Grants GM077190 (to E. R.) and GM33689 (to P. A. R.). This work was also supported by National Science Foundation Grant MCB0316269 (to E. R.).
D. S. Kudryashov, M. R. Sawaya, T. O. Yeates, and E. Reisler, unpublished structure of KabC with yeast actin.
- G-actin
- monomeric actin
- F-actin
- filamentous actin
- ABP
- actin binding proteins
- A167C
- actin mutant A167C/C374A
- F169C
- actin mutant F169C/C374A
- S170C
- actin mutant S170C/C374A
- EM
- electron microscopy
- KabC
- kabiramide C
- LatA
- latrunculin A
- SD
- subdomain
- SpvB
- Salmonella enterica virulence-associated protein B
- WT
- wild type
- YPD
- yeast extract/peptone/dextrose
- Cc
- critical concentration.
REFERENCES
- 1.Zheng X., Diraviyam K., Sept D. (2007) Biophys. J. 93, 1277–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korn E. D., Carlier M. F., Pantaloni D. (1987) Science 238, 638–644 [DOI] [PubMed] [Google Scholar]
- 3.Carlier M. F. (1987) Biochem. Biophys. Res. Commun. 143, 1069–1075 [DOI] [PubMed] [Google Scholar]
- 4.Bugyi B., Le Clainche C., Romet-Lemonne G., Carlier M. F. (2008) FEBS Lett. 582, 2086–2092 [DOI] [PubMed] [Google Scholar]
- 5.Schutt C. E., Myslik J. C., Rozycki M. D., Goonesekere N. C., Lindberg U. (1993) Nature 365, 810–816 [DOI] [PubMed] [Google Scholar]
- 6.Graceffa P., Dominguez R. (2003) J. Biol. Chem. 278, 34172–34180 [DOI] [PubMed] [Google Scholar]
- 7.Otterbein L. R., Graceffa P., Dominguez R. (2001) Science 293, 708–711 [DOI] [PubMed] [Google Scholar]
- 8.Rould M., Wan Q., Joel P. B., Lowey S., Trybus K. M. (2006) J. Biol. Chem. 281, 31909–31919 [DOI] [PubMed] [Google Scholar]
- 9.Crosbie R. H., Miller C., Cheung P., Goodnight T., Muhlrad A., Reisler E. (1994) Biophys. J. 67, 1957–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kudryashov D. S., Reisler E. (2003) Biophys. J. 85, 2466–2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strzelecka-Gołaszewska H., Moraczewska J., Khaitlina S. Y., Mossakowska M. (1993) Eur. J. Biochem. 211, 731–742 [DOI] [PubMed] [Google Scholar]
- 12.Bork P., Sander C., Valencia A. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 7290–7294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chik J. K., Lindberg U., Schutt C. E. (1996) J. Mol. Biol. 263, 607–623 [DOI] [PubMed] [Google Scholar]
- 14.Kardos R., Pozsonyi K., Nevalainen E., Lappalainen P., Nyitrai M., Hild G. (2009) Biophys. J. 96, 2335–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfaendtner J., Branduardi D., Parrinello M., Pollard T. D., Voth G. A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 12723–12728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu J. W., Voth G. A. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13111–13116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalhaimer P., Pollard T. D., Nolen B. J. (2008) J. Mol. Biol. 376, 166–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Splettstoesser T., Noe F., Oda T., Smith J. C. (2009) Proteins 76, 353–364 [DOI] [PubMed] [Google Scholar]
- 19.Paavilainen V. O., Oksanen E., Goldman A., Lappalainen P. (2008) J. Cell Biol. 182, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otterbein L. R., Cosio C., Graceffa P., Dominguez R. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 8003–8008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verboven C., Bogaerts I., Waelkens E., Rabijns A., Van Baelen H., Bouillon R., De Ranter C. (2003) Acta Crystallogr. D Biol. Crystallogr. 59, 263–273 [DOI] [PubMed] [Google Scholar]
- 22.Head J. F., Swamy N., Ray R. (2002) Biochemistry 41, 9015–9020 [DOI] [PubMed] [Google Scholar]
- 23.Mouilleron S., Guettler S., Langer C. A., Treisman R., McDonald N. Q. (2008) EMBO J. 27, 3198–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holmes K. C., Popp D., Gebhard W., Kabsch W. (1990) Nature 347, 44–49 [DOI] [PubMed] [Google Scholar]
- 25.Holmes K. C., Angert I., Kull F. J., Jahn W., Schröder R. R. (2003) Nature 425, 423–427 [DOI] [PubMed] [Google Scholar]
- 26.Oda T., Iwasa M., Aihara T., Maéda Y., Narita A. (2009) Nature 457, 441–445 [DOI] [PubMed] [Google Scholar]
- 27.Sikorski R. S., Hieter P. (1989) Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng L., Kim E., Lee W. L., Miller C. J., Kuang B., Reisler E., Rubenstein P. A. (1997) J. Biol. Chem. 272, 16829–16837 [DOI] [PubMed] [Google Scholar]
- 29.McKane M., Wen K. K., Boldogh I. R., Ramcharan S., Pon L. A., Rubenstein P. A. (2005) J. Biol. Chem. 280, 36494–36501 [DOI] [PubMed] [Google Scholar]
- 30.Kim E., Reisler E. (2000) Biophys. Chem. 86, 191–201 [DOI] [PubMed] [Google Scholar]
- 31.Grintsevich E. E., Benchaar S. A., Warshaviak D., Boontheung P., Halgand F., Whitelegge J. P., Faull K. F., Loo R. R., Sept D., Loo J. A., Reisler E. (2008) J. Mol. Biol. 377, 395–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hertzog M., Carlier M. F. (2005) Curr. Protoc. Cell Biol. Chapter 13, Unit 13.6 [DOI] [PubMed] [Google Scholar]
- 33.Chowdhury S., Smith K. W., Gustin M. C. (1992) J. Cell Biol. 118, 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kudryashov D. S., Galkin V. E., Orlova A., Phan M., Egelman E. H., Reisler E. (2006) J. Mol. Biol. 358, 785–797 [DOI] [PubMed] [Google Scholar]
- 35.Kudryashov D. S., Durer Z. A., Ytterberg A. J., Sawaya M. R., Pashkov I., Prochazkova K., Yeates T. O., Loo R. R., Loo J. A., Satchell K. J., Reisler E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 18537–18542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bobkov A. A., Muhlrad A., Kokabi K., Vorobiev S., Almo S. C., Reisler E. (2002) J. Mol. Biol. 323, 739–750 [DOI] [PubMed] [Google Scholar]
- 37.Pollard T. D., Goldberg I., Schwarz W. H. (1992) J. Biol. Chem. 267, 20339–20345 [PubMed] [Google Scholar]
- 38.De La Cruz E. M., Pollard T. D. (1995) Biochemistry 34, 5452–5461 [DOI] [PubMed] [Google Scholar]
- 39.Klenchin V. A., Allingham J. S., King R., Tanaka J., Marriott G., Rayment I. (2003) Nat. Struct. Biol. 10, 1058–1063 [DOI] [PubMed] [Google Scholar]
- 40.Yarmola E. G., Somasundaram T., Boring T. A., Spector I., Bubb M. R. (2000) J. Biol. Chem. 275, 28120–28127 [DOI] [PubMed] [Google Scholar]
- 41.Coué M., Brenner S. L., Spector I., Korn E. D. (1987) FEBS Lett. 213, 316–318 [DOI] [PubMed] [Google Scholar]
- 42.Carlier M. F., Pantaloni D. (1988) J. Biol. Chem. 263, 817–825 [PubMed] [Google Scholar]
- 43.Lal A. A., Brenner S. L., Korn E. D. (1984) J. Biol. Chem. 259, 13061–13065 [PubMed] [Google Scholar]
- 44.Kim E., Miller C. J., Reisler E. (1996) Biochemistry 35, 16566–16572 [DOI] [PubMed] [Google Scholar]
- 45.Bubb M. R., Govindasamy L., Yarmola E. G., Vorobiev S. M., Almo S. C., Somasundaram T., Chapman M. S., Agbandje-McKenna M., McKenna R. (2002) J. Biol. Chem. 277, 20999–21006 [DOI] [PubMed] [Google Scholar]
- 46.Wen K. K., Rubenstein P. A. (2009) J. Biol. Chem. 284, 16776–16783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wriggers W., Schulten K. (1999) Proteins Struct. Funct. Genet. 35, 262–273 [PubMed] [Google Scholar]
- 48.Schüler H., Nyåkern M., Schutt C. E., Lindberg U., Karlsson R. (2000) Eur. J. Biochem. 267, 4054–4062 [PubMed] [Google Scholar]
- 49.Geipel U., Just I., Schering B., Haas D., Aktories K. (1989) Eur. J. Biochem. 179, 229–232 [DOI] [PubMed] [Google Scholar]
- 50.De La Cruz E. M., Mandinova A., Steinmetz M. O., Stoffler D., Aebi U., Pollard T. D. (2000) J. Mol. Biol. 295, 517–526 [DOI] [PubMed] [Google Scholar]
- 51.Cooke R. (1975) Biochemistry 14, 3250–3256 [DOI] [PubMed] [Google Scholar]
- 52.Laham L. E., Way M., Yin H. L., Janmey P. A. (1995) Eur. J. Biochem. 234, 1–7 [DOI] [PubMed] [Google Scholar]
- 53.Blanchoin L., Pollard T. D. (1998) J. Biol. Chem. 273, 25106–25111 [DOI] [PubMed] [Google Scholar]
- 54.Carlier M. F., Laurent V., Santolini J., Melki R., Didry D., Xia G. X., Hong Y., Chua N. H., Pantaloni D. (1997) J. Cell Biol. 136, 1307–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tina K. G., Bhadra R., Srinivasan N. (2007) Nucleic Acids Res. 35, W473–W476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kyte J., Doolittle R. F. (1982) J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 57.Cong Y., Topf M., Sali A., Matsudaira P., Dougherty M., Chiu W., Schmid M. F. (2008) J. Mol. Biol. 375, 331–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Orlova A., Egelman E. H. (1992) J. Mol. Biol. 227, 1043–1053 [DOI] [PubMed] [Google Scholar]
- 59.Stokasimov E., McKane M., Rubenstein P. A. (2008) J. Biol. Chem. 283, 34844–34854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pollard T. D., Borisy G. G. (2003) Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 61.Fujiwara I., Vavylonis D., Pollard T. D. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 8827–8832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morton W. M., Ayscough K. R., McLaughlin P. J. (2000) Nat. Cell Biol. 2, 376–378 [DOI] [PubMed] [Google Scholar]
- 63.Bernstein B. W., Chen H., Boyle J. A., Bamburg J. R. (2006) Am. J. Physiol. Cell Physiol. 291, C828–C839 [DOI] [PubMed] [Google Scholar]
- 64.Dominguez R. (2004) Trends Biochem. Sci. 29, 572–578 [DOI] [PubMed] [Google Scholar]