

Abstract
Histamine dehydrogenase (HADH) isolated from Nocardioides simplex catalyzes the oxidative deamination of histamine to imidazole acetaldehyde. HADH is highly specific for histamine, and we are interested in understanding the recognition mode of histamine in its active site. We describe the first crystal structure of a recombinant form of HADH (HADH) to 2.7-Å resolution. HADH is a homodimer, where each 76-kDa subunit contains an iron-sulfur cluster ([4Fe-4S]2+) and a 6-S-cysteinyl flavin mononucleotide (6-S-Cys-FMN) as redox cofactors. The overall structure of HADH is very similar to that of trimethylamine dehydrogenase (TMADH) from Methylotrophus methylophilus (bacterium W3A1). However, some distinct differences between the structure of HADH and TMADH have been found. Tyr60, Trp264, and Trp355 provide the framework for the “aromatic bowl” that serves as a trimethylamine-binding site in TMADH is comprised of Gln65, Trp267, and Asp358, respectively, in HADH. The surface Tyr442 that is essential in transferring electrons to electron-transfer flavoprotein (ETF) in TMADH is not conserved in HADH. We use this structure to propose the binding mode for histamine in the active site of HADH through molecular modeling and to compare the interactions to those observed for other histamine-binding proteins whose structures are known.
Keywords: Bacteria, Crystal Structure, Dehydrogenase, Enzyme Catalysis, Enzyme Mechanisms, Enzyme Structure, Flavoproteins, Iron-Sulfur Protein, Post Translational Modification, Protein Structure
Introduction
Histamine is an essential biogenic amine present in prokaryotes and tissues of animals and plants. In humans, histamine acts as a neurotransmitter, mediates allergic reactions, plays a role in cell proliferation, and is important in signaling the release of gastric acid into the stomach (1). Histamine receptors are the targets of drugs that treat allergies and stomach acidity, but there is very little structural information on the histamine-binding sites of these proteins. There is interest in isolating histamine-metabolizing enzymes that could be used as biosensors, and such enzymes may also prove useful for understanding histamine recognition (2). Although histamine oxidase from Arthrobacter globiformis (a topaquinone-containing copper amine oxidase) has been used as a histamine sensor, this enzyme was found to be more sensitive for dopamine and tyramine than for histamine (2). Trimethylamine dehydrogenase (TMADH)5 from Methylophilus methylotrophus (3) and the quinoprotein methylamine dehydrogenase from Paracoccus denitrificans (4) have been used in general amine-sensing electrodes without the complication of O2 chemistry, but neither are as effective as histamine sensors.
Histamine dehydrogenase (HADH) can be isolated from cultures of Nocardiodes simplex grown on histamine as the sole nitrogen source (5). HADH is a homodimer of ∼76-kDa subunits and catalyzes the oxidative deamination of histamine to give imidazole acetaldehyde (Scheme 1) where the enzyme is remarkably selective for histamine (5, 6) thereby showing great potential for use in a biosensor. HADH was originally classified as a quinone-containing amine dehydrogenase (5). Subsequent work by our group (6) and others (7) showed HADH to be a homologue of TMADH and the closely related dimethylamine dehydrogenase (DMADH) from M. methylotrophus, sharing ∼40% sequence identity and 56% similarity with both proteins. The cofactors in this small family of proteins are the uncommon 6-S-cysteinyl-FMN, or 6-S-Cys-FMN (Fig. 1) and [4Fe-4S]. The oxidized form of HADH displays a UV-visual spectrum with a λmax at 440 nm and a shoulder at 340 nm characteristic of the oxidized flavin, 6-S-Cys-FMNox, as does TMADH (6). TMADH binds one molecule of ADP per subunit whose function is not known (8). The conserved ADP binding domain is also found in HADH (Lys392–Leu423) (6) and DMADH and aligns with the FAD binding domain of other flavoproteins, such as Escherichia coli 2,4-dienoyl-CoA reductase (9).
SCHEME 1.

FIGURE 1.
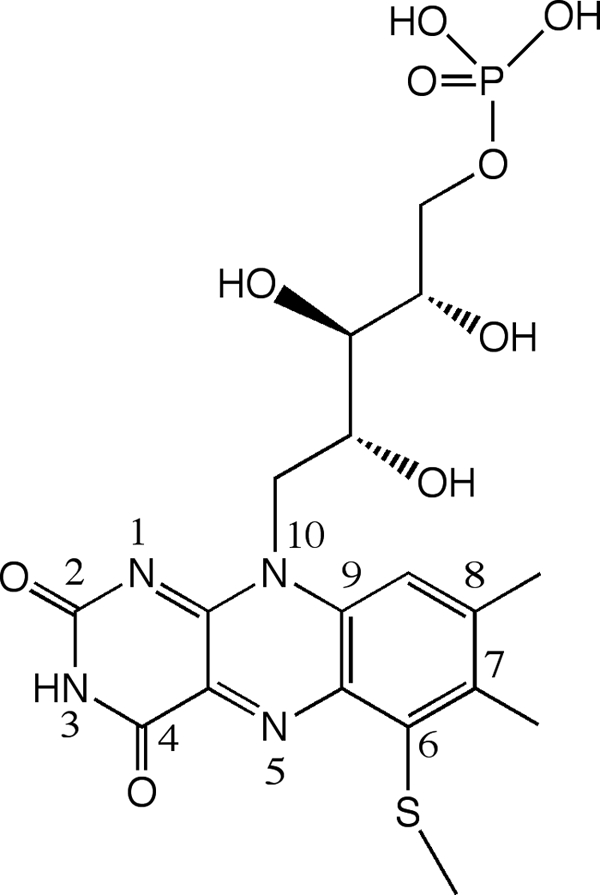
Structure of 6-S-Cys-FMN cofactor.
The oxidation of histamine by HADH leaves the flavin in its reduced form, 6-S-Cys-FMNred, which is reoxidized by a stepwise electron transfer reaction through the [4Fe-4S] to either a mediator, such as phenazinemethosulfate (6) or, in TMADH, an electron transferring flavoprotein (ETF) (10). If either HADH or TMADH are reduced under single turnover conditions, a disproportionation reaction occurs between 6-S-Cys-FMNred and [4Fe-4S]2+ to produce a semiquinone, 6-S-Cys-FMNsq, and [4Fe-4S]+ (Scheme 2) (6). In TMADH, the two spins form a triplet that is detected by EPR as a strong half-field signal (11–13). Under steady-state conditions at high substrate concentrations, the 6-S-Cys-FMNsq is stabilized by the binding of a substrate cation and can no longer be oxidized by [4Fe-4S], leading to substrate inhibition (6, 14, 15).
SCHEME 2.

Although HADH is a homolog of TMADH, there are many differences that show that HADH is a functionally distinct enzyme. The substrate specificity of HADH differs significantly from both TMADH and DMADH, as secondary and tertiary amines are not substrates for HADH. HADH can only oxidize histamine (Km = 31 μm, kcat/Km = 2.1 × 105 m−1 s−1), agmatine (Km = 37 μm, kcat/Km = 6.0 × 104 m−1 s−1), and putrescine (Km = 1280 μm, kcat/Km = 1500 m−1 s−1) (6). The trimethylamine-binding site of TMADH is well characterized, showing that the (CH3)3NH+ is held in place by π-cation interactions with Tyr60, Trp265, and Trp365 (16). Two additional residues, Ser74 and Trp105, along with Tyr60, have been shown to control the selectivity for trimethylamine over dimethylamine. Analogous to TMADH, monoamine oxidase and polyamine oxidase use aromatic amino acids to form π-cation interactions with their amine substrates (17, 18). The equivalent positions in HADH from an amino acid sequence alignment are Gln59, Glu79, Ala110, Trp264, and Asp364 (6). These differences suggest that the histamine-binding site of HADH is significantly different from other flavin-dependent amine metabolizing enzymes. DMADH seems to provide a bridge between TMADH and HADH, containing residues Gln59, Thr74, Phe105, Trp271, and Trp357. Although sequence alignment suggests that the active sites of TMADH and HADH are very different, EPR shows that the magnetic interactions between the 6-S-Cys-FMN and [4Fe-4S] also differ between TMADH and HADH. In contrast to TMADH, the cofactors are not strongly spin-coupled in HADH suggesting that there are structural or conformational differences between the two enzymes (6).
The pH dependence of the steady-state kinetic parameters of histamine oxidation by HADH yielded two pKa values (pKa1 = 5.6 ± 0.3 and pKa2 = 5.4 ± 0.4) (6). The imidazole group of histamine must be neutral when it binds to the active site but the protonation state of the primary amino group of histamine was not conclusive as we were not able to determine the kinetics above pH 9 (6). The second pKa may represent the ionization of an active site residue, where an anionic residue in the active site could be responsible for stabilizing the monoprotonated histamine (6). HADH exhibits a significant primary kinetic deuterium isotope effect using α,α,β,β-d4-histamine on kcat/Km (>7.0) that is absent in kcat (6). This suggests that C-H bond cleavage, and therefore 6-S-Cys-FMN reduction, is rate-limiting at subsaturating concentrations of histamine but that the subsequent steps, possibly electron transfer, define kcat. This contrasts with TMADH, where the effect of using (CD3)3NH+ can only be examined by stopped-flow methods, and there is no isotope effect in the steady state (19).
Taken together, these studies strongly suggest that HADH is unique compared with TMADH and that they do not share a common mechanism. Detailed structure/function studies on HADH are needed to understand the substrate specificity and the fundamental chemistry underlying catalysis in this uncommon amine oxidizing flavoprotein. Toward this goal, we report the first three-dimensional structure of a histamine dehydrogenase.
MATERIALS AND METHODS
Expression and Purification of HADH
A recombinant form of HADH was expressed and purified as described previously (20). Briefly, HADH was expressed in E. coli Rosetta 2 (DE3) cells (Novagen) using a pET-21b vector (Novagen). HADH was purified by Toyopearl DEAE, Toyopearl Butyl-650 (Tosoh Bioscience), and HiLoad Superdex 200 16/60 sizing column (GE Healthcare) chromatography. Full flavinylation of HADH was confirmed by the ratio of A444/A382 = 1.38 (7). The activity of HADH against histamine was determined using the standard assay (5). The selenomethionine-substituted HADH (SeMet-HADH) was prepared and purified as described previously (20) and the substitution of 10 of the 13 Met with SeMet was confirmed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry at the University of Kansas Analytical Proteomic Laboratory.
Crystallization and Data Collection
The crystallization of HADH and SeMet-HADH, subsequent data collection, and data processing were carried out as described previously (20). In short, crystals of HADH were obtained by equilibration versus 0.1 m HEPES, pH 7.4, 2.0 m ammonium sulfate, 2% polyethylene glycol 400, and 4% glycerol using hanging-drop vapor diffusion. Glycerol (25%) was used as a cryoprotectant and data were collected from cryocooled crystals. A data set was collected to 2.7-Å resolution from crystals grown from SeMet-HADH at beamline BL9-2 at the Stanford Synchrotron Radiation Laboratory (SSRL) (21). The estimated maximal error is 0.165 Å and the average coordinate error estimated from the Luzzati plot is 0.294 Å. Data collection was performed at 100 K. Data integration and scaling were performed with MOSFLM (22) and SCALA (23).
Structure Solution, Refinement, Rebuilding, and Model Analysis
As described previously (20), structure determination by SOLVE/RESOLVE (24, 25) using the complete three-wavelength multiwavelength anomalous dispersion data set was not immediately successful. However, using only the single-wavelength 0.98-Å data, we were able to determine the structure by molecular replacement using the program Phaser (26). The search model was the polypeptide monomer of TMADH (PDB accession number 1DJN, 2.2-Å molecule A only) (8), which had nonconserved residues pruned back to the last common atom. A log likelihood gain value of 247 and a translation function Z-score of 18.1 were obtained in Phaser (26) for two solutions without packing clashes. The x-ray diffraction data used to solve the HADH structure was collected to a resolution of 2.7 Å with 99.7% completeness and an Rmerge of 16%. The Ramachandran plot showed 88.4% of residues in the most favored region, 11.1% in the additional allowed regions, 0.5% in the generously allowed regions, and no residue was found in the disallowed region. The crystal belongs to an orthorhombic space group P212121, with unit cell parameters a = 101.14, b = 107.03, c = 153, 35 Å. Each asymmetric unit contained two molecules of HADH. The Matthew's coefficient was 2.79 Å3 Da−1, giving a solvent content of 55.96% (27). NCS restraints were imposed during the early phases of refinement and relaxed during later phases of refinement. In the final rounds of refinement, NCS restraints were removed. The refinement statistics are summarized in Table 1. The initial model of HADH was iteratively refined using the data set of 2.7-Å resolution with REFMAC (28) and built manually using COOT (29). The model was validated using PROCHECK (30) and WHAT IF (31). Superimposition of structures of HADH and TMADH and all molecular figures were generated by PyMOL (32). The PDB deposition code is 3K30.
TABLE 1.
Refinement statistics
| Refinement | HADH (PDB code 3K30) |
|---|---|
| Resolution (Å)a | 84.43-2.70 (2.77-2.70) |
| No. reflectionsa | 43,913 (3,143) |
| Completenessa | 99.6% (98.6%) |
| Rwork/Rfree (5.1%) | 18.1/24.2 |
| No. atoms | |
| Protein | 10,544 |
| 6-S-Cys-FMN | 62 |
| [4Fe-4S] | 16 |
| ADP | 54 |
| Water | 8 |
| B-factors | |
| Protein | 21.5 |
| 6-S-Cys-FMN | 19.0 |
| [4Fe-4S] | 21.3 |
| ADP | 17.6 |
| Water | 8.6 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.017 |
| Bond angles (°) | 1.753 |
| DPIb | 0.314 |
a Highest resolution shell is shown in parentheses.
b Diffraction-component precision indicator based on R-factor.
Modeling Study
Structural models for Arg444 (HADH) and Tyr442 (TMADH) were obtained by extracting the relevant amino acids directly from their corresponding crystal structure using SYBYL 8.0 (Tripos International, St. Louis, MO). The models were then protonated in SYBYL according to the following specifications: Arg444 was assumed to be cationic, whereas Tyr442 was assumed to be neutral; both amino acids were assigned neutral backbones with amine and aldehyde termination at the N and carbonyl ends, respectively. The HOMO-LUMO gaps for each were computed in Gaussian 03 (Gaussian, Inc., Wallingford, CT) at a B3-LYP level of representation (33, 34) using the 6-31G* basis set (35).
Docking Study
The ligands (monoprotonated and neutral forms of histamine and trimethylamine) were sketched in SYBYL 8.0 (Tripos International) and refined via molecular mechanics optimization using the Tripos Force Field (36) and Gasteiger-Marsili electrostatics (37). The two protonated states of histamine ligands were docked into the HADH active site (receptor) by FlexX (38), retaining for explicit consideration all residues and crystallographically resolved heteroatomic moieties within 8.0 Å of the receptor center. All other docking controls were left at default settings. Three ligands (trimethylamine and two protonated states of histamine) were docked into the TMADH active site (receptor) by Surflex (39), as guided by a receptor-based protomol file defined by the position of the 6-S-Cys-FMN cofactor molecule (i.e. ligand binding modes that interact with this molecule are preferentially sought). Ligands via space-filling spheres were scaled according to van der Waals radii. All other docking controls were left at default settings.
RESULTS AND DISCUSSION
Primary Characterization of HADH
The recombinant HADH is fully flavinylated judging from the ratio of A444/A382 = 1.38, and exhibits identical UV-visual and EPR spectroscopic properties as the native HADH (6) (data not shown). The kinetic parameters of histamine oxidation by recombinant HADH and the native HADH are in good agreement, and furthermore, like the native form, histamine shows substrate inhibition with recombinant HADH (supplemental Table S1). These results show that recombinant HADH can be used in place of native HADH in the structure determination.
The Overall Structure of HADH
Recombinant HADH was crystallized as a homodimer with two molecules per asymmetric unit (Fig. 2a). The overall structure is very similar to that of the native TMADH (40) (supplemental Fig. S1). Secondary structure matching reveals a core root mean square deviation of 1.29 Å between the two proteins. Full-length HADH contains 690 residues. Both molecule A and molecule B were modeled with residues 7–690. Each molecule contains 6-S-Cys-FMN and [4Fe-4S] as redox active cofactors, as predicted from the previous study, and also a single ADP per molecule (6). The root mean square deviation for Cα atoms of molecules A and B of HADH is 0.34 Å. With the high structural similarity between subunits A and B, analysis was focused on subunit A.
FIGURE 2.

a, overview of recombinant HADH structure. Monomer B is shown in pale blue. Monomer A is colored by domains. Domain 1 is shown in green, domain 2 in pink, and domain 3 in yellow. 6-S-Cys-FMN and [4Fe-4S] are shown in red and ADP is shown in blue. Subsequent views are rotated by 90 degrees. b, 6-S-Cys-FMN in HADH, electron density map around 6-S-Cys-FMN. c, stereoview of 6-S-Cys-FMN from HADH both with (carbon shown in magenta) and without restraints (carbon shown in green) on the planarity of the isoalloxazine ring system compared with that in TMADH (carbon shown in cyan).
Each subunit is comprised of three domains (Fig. 2a and supplemental Fig. S1), a large domain (residues 7–385), a medium domain (residues 386–491 and residues 622–690), and a small domain (residues 492–621). The large domain contains an N-terminal triose-phosphate isomerase barrel, the most common tertiary fold observed in protein crystal structures (41). The 6-S-Cys-FMN is located at the opening of the barrel and surrounded by α helices and large excursions at the end of β strands of the barrel that serve to cover and bury the 6-S-Cys-FMN. The 6-S-Cys-FMN is covalently linked to Cys35 positioned after the first parallel β-strand of the barrel. The [4Fe-4S] cluster is positioned outside the barrel and connected to the end of an α-helix close to the medium domain. The [4Fe-4S] cluster is coordinated to four Cys residues (Cys348, Cys351, Cys354, and Cys366). As predicted from the sequence alignment (6), one molecule of ADP is bound per protein molecule, located in the medium domain. ADP is exposed to the surface, next to the interface of the medium and small domains, but is not covalently linked, as is observed for TMADH (8, 42).
The Active Site of HADH
The electron density for the 6-S-Cys-FMN is very clear (Fig. 2b), consistent with a fully flavinylated enzyme. By comparison, crystals of recombinant TMADH lacked much of the electron density at the flavin site. The estimated average occupancy for the FMN in the recombinant TMADH (PDB code 1DJN) was reported as 0.55 (43). The electron density suggested that the isoalloxazine ring of the 6-S-Cys-FMN in recombinant HADH is not planar but in a “butterfly bend” conformation centered at the N5 and N10 positions as seen in the recombinant TMADH (43). An overlap is shown of FMN from TMADH (cyan carbons), FMN from HADH without planar restraints (green carbons) and with planar restraints (magenta carbons) on the isoalloxazine ring system in Fig. 2c. When the FMN in HADH is refined without planarity restraints on the isoalloxazine ring, the deviation from planarity is ∼15°, which is slightly less than that in TMADH (20°) (43).
Although the factors contributing to the butterfly bend in TMADH have not been defined yet, the cysteinyl cross-link formation has been shown not to cause the bend as a similar bend was seen in the x-ray structure of the C35A mutant of TMADH (39). We have looked into the active site of HADH surrounding the benzene ring of FMN where the bend has been seen (Fig. 2c) in comparison to the corresponding area in TMADH, but we have not been able to find particular interactions that contribute to the extent of butterfly bend. Further study using higher resolution crystals and site-directed mutagenesis is necessary.
The redox potentials of HADH have been determined to be +34 mV (fully oxidized form to one electron-reduced form) and +30 mV (one electron-reduced form to fully reduced form), respectively (44), whereas the reported values of TMADH are +44 and +36 mV, respectively (45). Theoretical studies on lumiflavin and C6-methyl-sulfanyllumiflavin suggested that the butterfly bend raises the two-electron reduction potential, making the flavin more reactive to substrate oxidation (46, 47). The changes in the redox potential of the 6-S-Cys-FMN cofactors in HADH and TMADH are relatively small, yet support the theoretical study where the redox potential of the former is slightly more negative than the latter.
The electron density for the [4Fe-4S] cluster is also very clear (supplemental Fig. S2). The spatial arrangement of 6-S-Cys-FMN and the [4Fe-4S] cluster in HADH is very similar to that in TMADH (8) (Fig. 3). The distance between the 8α-methyl carbon of 6-S-Cys-FMN and the closest iron in [4Fe-4S] is 5.7 Å, and to the closest Cys ligand (Cys354) of the cluster is 4.1 Å, respectively. It has been proposed that the electrons from substrate-reduced flavin pass through the 8α-methyl to the closest Cys ligand upon reoxidation in TMADH (8). The oxidation of 6-S-Cys-FMNred in HADH may go through a similar electron transfer mechanism.
FIGURE 3.

A stereoview of the active site of HADH (green) and TMADH (cyan). The superimposition was done on the whole molecule. Residue numbers in black are for HADH and those in blue correspond to TMADH.
Fig. 3 shows the superimposed stereoview of the FMN-binding site. In both structures, the Tyr-His-Asp triad comprising Tyr169, His172, and Asp267 in TMADH and Tyr176, His179, and Asp270 in HADH is located close to the pyrimidine moiety of the isoalloxazine ring of 6-S-Cys-FMN. Tyr169 in TMADH is proposed to stabilize the semiquinone of 6-S-Cys-FMN and [4Fe-4S]+ (48). To support this idea, the Y169F mutant of TMADH showed the loss of the spin coupling of the semiquinone and [4Fe-4S]+ (48). It was proposed that the charge repulsion of the negative charge on the hydroxyl group of Tyr169 pushes the unpaired electron density on the 6-S-Cys-FMN toward [4Fe-4S] and mediates the spin-spin interaction. His172 in TMADH is proposed to be neutral to achieve the maximum rate of 6-S-Cys-FMN reduction (49).
In the native HADH, the spin coupling of the semiquinone of 6-S-Cys-FMN and [4Fe-4S]+ are not as strong as that observed in TMADH (6). We previously proposed that the spatial arrangement of the two cofactors or the surrounding residues could affect the spin coupling of the two cofactors (6). However, the active site structure of HADH revealed that the distances between the two cofactors are almost identical in HADH and TMADH (Fig. 3). We saw small differences in the location of two of the triad residues in HADH. The hydroxyl of Tyr176 resides 3.6 Å away from the O2 carbonyl of 6-S-Cys FMN, which is ∼1 Å further away than the hydroxyl of Tyr169 in TMADH. His179 is 0.5 Å closer to the pyrimidine moiety of 6-S-Cys-FMN than His172 in TMADH. Arg222 in TMADH is 2.6 Å from the O2 carbonyl of 6-S-Cys-FMN and is proposed to stabilize the negative charge building up on the N1 and C2 of the isoalloxazine ring during the 6-S-Cys-FMN biogenesis (50). Arg230 in HADH is spatially conserved with Arg222 in TMADH and is 3.6 Å away from the O2 carbonyl of the 6-S-Cys-FMN. Further studies are necessary to define whether the small differences of Tyr176 and His179 are the origin of the weak spin-spin coupling observed in HADH. We expect that F176Y exhibits spin-spin coupling to the same extent as the wild-type but W176Y may exhibit stronger coupling only if the mutation does not disrupt the rest of the active site environment.
In the proposed catalytic mechanism of HADH and TMADH, a water molecule plays a catalytic role, i.e. to produce corresponding aldehydes from the imine products. However, no water molecule has been found in the active site of HADH, probably due to 2.7-Å resolution, but this could also be due to high mobility of water molecules as no water molecule has been detected in the active site of TMADH (2.2 Å) either.
Electron Transfer Pathway
X-ray crystal structures of TMADH-ETF complex (2.0 and 3.7 Å) revealed that ETF interacts with a shallow surface groove of TMADH, comprised of nine amino acids, namely Ser391, Leu393, Thr414, His416, Gln462, Ala464, Tyr478, Gly479, and Ala480 (Fig. 4a and supplemental Fig. S3), through its proposed recognition peptide sequence (51). Leu194 in the β subunit of ETF is proposed to act as an anchor and positions ETF into the groove of TMADH via van der Waals interactions with Leu393 and Tyr478.
FIGURE 4.

a, surface grooves of TMADH (cyan) involved in electron transfer to ETF (51). The groove in light blue interacts with a recognition loop of the β subunit of ETF through van der Waals interactions between Leu393 and Tyr478 and Leu198 of ETF. The groove colored light yellow is involved in the electron-transferring reaction. Tyr442 is proposed to interact with Arg237 in the α subunit of ETF to facilitate the electron transfer reaction to FAD in ETF. Val344 interacts with ferricenium, an artificial electron acceptor. b, the corresponding surface grooves of HADH (see text for residue numbers) are shown in blue and orange, respectively.
The presence of an ETF-like protein has not yet been determined in N. simplex. By inspection of the crystal structure of HADH, there seems to be a similar shallow groove on the surface (Fig. 4b). From the sequence alignment (supplemental Fig. S3), the corresponding residues in HADH are Arg393, Leu395, Asp416, Val418, Tyr462, Arg463, Phe475, Gly476, and Phe477. Leu395 and Gly476 are conserved and Phe475 is homologous to Tyr478 in TMADH. The hydrophobic patch comprising Leu393, Tyr478, Gly479, and Ala480 in the groove that interacts with Leu194 of the recognition loop in ETF also exists in HADH (Leu395, Phe475, Gly476, and Phe477). However, three other residues in HADH are charged (Arg393, Asp416, and Arg463) when compared with the corresponding residues in TMADH (Ser391, Thr414, and Ala464).
The surface residue Tyr442 in TMADH has been shown to play a key role in the electron-transfer reaction between the [4Fe-4S] center and ETF (52–54). Tyr442 resides in the surface groove (Fig. 4a and supplemental Fig. S4a) different from that discussed earlier. Mutations of Tyr442 to Phe, Leu, Cys, and Gly lead to perturbation of the interaction between TMADH and ETF, resulting in retardation of the electron-transfer reaction (52). It has been proposed that Tyr442 transiently pairs with Arg237 in the α subunit of ETF to stabilize productive electron-transfer configurations. The residue corresponding to Tyr442 is Arg444 in HADH. Molecular orbital calculations suggest that the Arg444 residue in HADH may be better suited to stabilize the charge transfer complex than the Tyr442 in TMADH. Specifically, the HOMO-LUMO gap of Arg444 is 4.49 eV, whereas the HOMO-LUMO gap of Tyr442 is 4.91 eV, thus Arg444 requires less energy to accept a negative charge than Tyr442 would.
In TMADH, Val344 resides at the bottom of the surface groove where Tyr442 resides and engages in van der Waals interactions with Cys345 and Tyr442 (Figs. 3 and 4a). Val344 has been shown to participate in the electron transfer to ferricenium, an artificial electron acceptor (52). From the molecular modeling study, it has been proposed that ferricenium binds in this hydrophobic surface groove and can be in close contact with Val344. Mutations of Val344 to amino acids with smaller side chains (Ala, Gly, or Cys) resulted in increasing the rate of electron transfer and it was proposed that shortening the electron-transfer pathway and/or changes in packing density accelerates the reaction. Mutation of Val344 to bulkier side chains (Ile or Tyr) to reduce the size of the groove resulted in an impaired electron transfer reaction. In HADH, the corresponding residue to Val344 is Glu347 making this groove hydrophilic rather than hydrophobic (supplemental Fig. S4). Taken together, we found that the electronic environment of these surface grooves involved in ETF binding and electron transfer reaction in TMADH is not strictly conserved in HADH, where both grooves in the latter contain charged residues.
Two possible electron-transfer pathways from the [4Fe-4S] to the surface Tyr442 are proposed for TMADH, namely Cys345 → Glu439 → Tyr442 or Cys345 → Val344 (48) (supplemental Fig. 5a). The first path is used to transfer electrons to ETF and the second path is used to transfer electrons to ferricenium. Interestingly, HADH has a very analogous pair of choices, namely Cys348 → Glu347 → Arg444 or Cys348 → Ala441. These pathways are geometrically comparable with the two pathways in TMADH, except the positions of the possible intermediates (Glu437 in HADH and Gly439 in TMADH) are reversed (supplemental Fig. 5b). Although we have not identified an ETF-like protein in N. simplex, it is possible that HADH utilizes the Cys348 → Glu347 → Arg444 pathway to its electron acceptor protein.
Substrate (Histamine)-binding Site
Although the overall three-dimensional fold of TMADH and HADH are very similar, there are some significant differences between the two active sites. In TMADH, substrate binds to the “aromatic bowl” created by Tyr60, Trp264, and Trp355 (55). The binding of trimethylamine in TMADH involves cation-π interaction of the methylammonium ion and the aromatic bowl. The corresponding residues in HADH are Gln65, Trp267, and Asp358 as predicted from the sequence alignment (6), as shown in Fig. 3.
Modeling of Histamine into the Active Site of HADH
To gain insight into the histamine selectivity of HADH, histamine was modeled into the active site in both neutral and monoprotonated forms. The modeling was chosen over crystal structure determination of the histamine-bound form of HADH as we have not found the optimal condition to prevent oxidation of histamine to the corresponding aldehyde in the active site of HADH yet. Both forms bound in the same fashion, with the latter predicted to be more energetically favored than the former (−21.5 kcal/mol versus −14.4 kcal/mol). Histamine is positioned over the si-face of the isoalloxazine ring of 6-S-Cys-FMN (Fig. 5). The positively charged amino group of histamine forms salt bridges or hydrogen bonding interactions with the carboxylate groups of Asp358 and Glu79. The carbonyl oxygen of the Asn115 side chain is in hydrogen bond interaction with the NE2 atom of the imidazole ring. The amido group of Gln65 is in hydrogen bonding distance from the CD2 proton of the imidazole ring. In addition to the hydrogen bonding residues, a number of hydrophobic side chains (Ile74, Phe77, Tyr181, and Trp267, respectively) line the binding pocket and have van der Waals contact with histamine. Among them, the aromatic ring of Tyr181 is in π-stacking interaction with the neutral imidazole ring of histamine and Phe77 is packed edge-to-face against the imidazole ring. The closest approach of the plane of the ring is 3.6 and 3.7 Å for Tyr181 and Phe77, respectively. This position places the protonated amino group of histamine close to the isoalloxazine ring, as would be required for catalysis to the imidazole acetaldehyde. The histamine-binding site is located at the center of the dimer interface and a channel connects both binding sites to the surface (Fig. 6).
FIGURE 5.

Stereoview of the proposed histamine-binding site in the active site of HADH. Histamine (green C atoms) was modeled in as the monoprotonated form. The neutral imidazole ring is stabilized by a π-stacking interaction with Tyr181, an end-on interaction with Phe77, and a hydrogen bond with Asn115. The positively charged amino group is stabilized by a salt bridge with Glu79 and Asp358. Hydrophobic residues surrounding histamine are shown in blue. 6-S-Cys-FMN is shown in magenta.
FIGURE 6.

The proposed substrate-entry channels in HADH. There are two channels connecting the surface to each active site (only one channel is shown). The bound histamine predicted from the docking study is shown in magenta. a, a top view of the channel. b, a side view of the channel.
Modeling of Histamine into the Active Site of TMADH
To gain further insight into histamine selectivity of HADH, we docked trimethylamine and monoprotonated and neutral forms of histamine into the active site of TMADH. Trimethylamine successfully docked into the proposed aromatic bowl in the active site, comprising Tyr60, Trp264, and Trp355 (supplemental Fig. 6a) (56). However, both forms of histamine ligands failed to dock into the active site, as docking efforts consistently placed histamine outside of the active site. When manually superimposing histamine onto the aromatic bowl, where trimethylamine binds, we found that histamine is too bulky to fit into the aromatic bowl (supplemental Fig. 6b) leading to insurmountable steric clashes. These results indicate that the substrate-binding site of HADH and TMADH are evolved to best accommodate each substrate by specific interactions with amino acid side chains as well as the size of the cavity.
Comparison of Histamine Binding Motifs in Histamine-binding Proteins
The histamine binding motif in the active site of HADH (Fig. 7a) from the modeling study correlates closely with the x-ray crystallographically determined binding sites of histamine N-methyltransferase from human (HNMT) (Fig. 7b) (57) and histamine-binding proteins (HBPs) from the saliva of blood sucking insects, such as the ticks Rhipicephalaus appendiculatus (Fig. 7c) (58) and Argas monolakensis (supplemental Fig. 7a) (59), and the malaria mosquito, Anopheles gambiae (supplemental Fig. 7b) (60). HBPs bind histamine that is released by the host during blood feeding to suppress inflammatory response of the host. HNMT inactivates histamine by transferring a methyl group from S-adenosyl-l-methionine to the imidazole ring. In the HBPs, the negatively charged residues such as Asp and Glu stabilize the positively charged amino group of histamine, whereas π-stacking interactions with Tyr, Phe, and Trp help to stabilize the imidazole ring of histamine (58–60). The selective binding of histamine in HNMT is achieved by ionic or hydrogen bonding interactions of Glu, Gln, and Asp with the positively charged amino group and π-stacking interaction of Tyr and Trp with the imidazole ring (57). Even though the function and mechanism of HBPs, HNMT, and HADH differ, the histamine-binding motif is clearly conserved and this contributes to their high histamine selectivity. HNMT and HBPs are not sequentially homologous to HADH, suggesting that the similarities in histamine-binding motifs have arisen through convergent evolution.
FIGURE 7.

Histamine-binding sites in HADH in comparison to other structurally characterized histamine-binding proteins. a, HADH (PDB code 3K30); b, HNMT (PDB code 1JQD); c, a female-specific HBP from the soft tick A. monolakensis (PDB code 3BU1). Hydrogen bond interactions are shown in orange dashed lines. Hydrophobic residues are shown in blue. Water molecules are shown as small red spheres.
The sequences of the human histamine receptors (HHRs) are known (61–65), but the crystal structures have yet to be determined. Molecular modeling studies of HHRs have provided insights into the binding mode of histamine into these receptors (66–69). In all four HHRs, a salt bridge between a conserved Asp and the positively charged amino group of histamine is proposed to be essential for histamine binding. HHR1 and HHR4 both contain an Asn residue that is predicted to be in hydrogen bond interaction to the imidazole nitrogen of histamine. In HHR1, a Tyr residue is predicted to stabilize histamine binding by forming a π-stacking interaction with the imidazole ring (70). The binding motif of HADH demonstrates significant similarities with HHRs, and especially the binding motif of HHR1.
Conclusion
We have solved the crystal structure of a recombinant form of HADH to 2.7-Å resolution. The overall structure, domain organization, and the spatial arrangement of 6-S-Cys-FMN and [4Fe-4S] are very similar to those of TMADH but we found significant differences at the substrate-binding site as well as on the surface proposed to be from contact with electron acceptors like ETF in TMADH. From the docking study, we predict that monoprotonated histamine preferentially binds to the active site in the following manner. The anionic residues (Glu79 and Asp358) stabilize the protonated amino group of histamine by salt bridge, whereas the aromatic residues (Phe77 and Tyr181) stabilize the imidazole ring via a π-stacking interaction. This binding mode of histamine is very similar to those found in the structures of other histamine-binding proteins and consists of a model for understanding the selectivity of HHRs, in particular HHR1. The similar binding mode of histamine into HADH and HHR1 could make the HADH substrate recognition site a potential model for HHR1.
Supplementary Material
Acknowledgments
We thank Scott Schreiber for assistance in protein purification. We thank reviewers for their constructive and valuable critiques to improve our manuscript. We also thank Masao Ikeda-Saito and Kunio Miki for critical reading and helpful discussion in revising the manuscript. Crystals were grown and initially screened using the facilities of the Protein Structure Laboratory core facility at The University of Kansas (supported by National Institutes of Health Grant RR-017708 RPH). Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the United States Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
This work was supported, in whole or in part, by National Institutes of Health Grants 5P20 RR17708 (to J. L., COBRE Center in Protein Structure and Function) and GM079446 (to J. L. and M. M.), National Science Foundation MCB-0747377 (to M. M.), and the University of Kansas Center for Research (to J. L. and M. M.).
The atomic coordinates and structure factors (code 3K30) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7 and Table S1.
- TMADH
- trimethylamine dehydrogenase
- HADH
- histamine dehydrogenase
- DMADH
- dimethylamine dehydrogenase
- ETF
- electron transferring flavoprotein
- HHR
- human histamine receptor
- SeMet
- selenomethionine
- HNMT
- histamine N-methyltransferase
- HBP
- histamine-binding protein
- PDB
- Protein data bank.
REFERENCES
- 1.Thurmond R. L., Gelfand E. W., Dunford P. J. (2008) Nat. Rev. Drug Discov. 7, 41–53 [DOI] [PubMed] [Google Scholar]
- 2.Iwaki S., Ogasawara M., Kurita R., Niwa O., Tanizawa K., Ohashi Y., Maeyama K. (2002) Anal. Biochem. 304, 236–243 [DOI] [PubMed] [Google Scholar]
- 3.Loechel C., Basran A., Basran J., Scrutton N. S., Hall E. A. (2003) Analyst 128, 166–172 [DOI] [PubMed] [Google Scholar]
- 4.Bao L., Sun D., Tachikawa H., Davidson V. L. (2002) Anal Chem. 74, 1144–1148 [DOI] [PubMed] [Google Scholar]
- 5.Siddiqui J. A., Shoeb S. M., Takayama S., Shimizu E., Yorifuji T. (2000) FEMS Microbiol. Lett. 189, 183–187 [DOI] [PubMed] [Google Scholar]
- 6.Limburg J., Mure M., Klinman J. P. (2005) Arch. Biochem. Biophys. 436, 8–22 [DOI] [PubMed] [Google Scholar]
- 7.Fujieda N., Satoh A., Tsuse N., Kano K., Ikeda T. (2004) Biochemistry 43, 10800–10808 [DOI] [PubMed] [Google Scholar]
- 8.Lim L. W., Shamala N., Mathews F. S., Steenkamp D. J., Hamlin R., Xuong N. H. (1986) J. Biol. Chem. 261, 15140–15146 [PubMed] [Google Scholar]
- 9.Hubbard P. A., Liang X., Schulz H., Kim J. J. (2003) J. Biol. Chem. 278, 37553–37560 [DOI] [PubMed] [Google Scholar]
- 10.Huang L., Rohlfs R. J., Hille R. (1995) J. Biol. Chem. 270, 23958–23965 [DOI] [PubMed] [Google Scholar]
- 11.Steenkamp D. J., Beinert H. (1982) Biochem. J. 207, 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fournel A., Gambarelli S., Guigliarelli B., More C., Asso M., Chouteau G., Hille R., Bertrand P. (1998) J. Chem. Phys. 109, 10905–10913 [Google Scholar]
- 13.Jang M. H., Basran J., Scrutton N. S., Hille R. (1999) J. Biol. Chem. 274, 13147–13154 [DOI] [PubMed] [Google Scholar]
- 14.Rohlfs R. J., Huang L., Hille R. (1995) J. Biol. Chem. 270, 22196–22207 [DOI] [PubMed] [Google Scholar]
- 15.Roberts P., Basran J., Wilson E. K., Hille R., Scrutton N. S. (1999) Biochemistry 38, 14927–14940 [DOI] [PubMed] [Google Scholar]
- 16.Basran J., Mewies M., Mathews F. S., Scrutton N. S. (1997) Biochemistry 36, 1989–1998 [DOI] [PubMed] [Google Scholar]
- 17.Scrutton N. S., Raine A. R. (1996) Biochem. J. 319, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Binda C., Mattevi A., Edmondson D. E. (2002) J. Biol. Chem. 277, 23973–23976 [DOI] [PubMed] [Google Scholar]
- 19.Basran J., Sutcliffe M. J., Scrutton N. S. (2001) J. Biol. Chem. 276, 24581–24587 [DOI] [PubMed] [Google Scholar]
- 20.Reed T. M., Hirakawa H., Mure M., Scott E. E., Limburg J. (2008) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64, 785–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen A. E., Ellis P. J., Miller M. D., Deacon A. M., Phizackerley R. P. (2002) J. Appl. Cryst. 35, 720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leslie A. G. W. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 23.Collaborative Computational Project, N. (1994) Acta Crystallogr. D. Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 24.Terwilliger T. C., Berendzen J. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terwilliger T. C. (2000) Acta Crystallogr. D Biol. Crystallogr. 56, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matthews B. W. (1968) J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 28.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 29.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 30.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 31.Vriend G. (1990) J. Mol. Graph. 8, 52–56 [DOI] [PubMed] [Google Scholar]
- 32.DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 33.Becke A. D. (1993) J. Chem. Phys. 98, 5648–5652 [Google Scholar]
- 34.Lee C., Yang W., Parr R. G. (1988) Phys. Rev. B Condens. Matter 37, 785–789 [DOI] [PubMed] [Google Scholar]
- 35.Hehre W. J., Ditchfield R., Pople J. A. (1972) J. Chem. Phys. 56, 2257–2261 [Google Scholar]
- 36.Clark M., Cramer R. D., 3rd, Van Opdenboch N. (1989) J. Comput. Chem. 10, 982–1012 [Google Scholar]
- 37.Gasteiger J., Marsili M. (1980) Tetrahedron 36, 3219–3228 [Google Scholar]
- 38.Rarey M., Wefing S., Lengauer T. (1996) J. Comput. Aided Mol. Des. 10, 41–54 [DOI] [PubMed] [Google Scholar]
- 39.Jain A. N. (2007) J. Comput. Aided Mol. Des. 21, 281–306 [DOI] [PubMed] [Google Scholar]
- 40.Dietmann S., Holm L. (2001) Nat. Struct. Biol. 8, 953–957 [DOI] [PubMed] [Google Scholar]
- 41.Wierenga R. K. (2001) FEBS Lett. 492, 193–198 [DOI] [PubMed] [Google Scholar]
- 42.Lim L. W., Mathews F. S., Steenkamp D. J. (1988) J. Biol. Chem. 263, 3075–3078 [PubMed] [Google Scholar]
- 43.Trickey P., Basran J., Lian L. Y., Chen Z., Barton J. D., Sutcliffe M. J., Scrutton N. S., Mathews F. S. (2000) Biochemistry 39, 7678–7688 [DOI] [PubMed] [Google Scholar]
- 44.Tsutsumi M., Fujieda N., Tsujimura S., Shirai O., Kano K. (2008) Biosci. Biotechnol. Biochem. 72, 786–796 [DOI] [PubMed] [Google Scholar]
- 45.Pace C. P., Stankovich M. T. (1991) Arch. Biochem. Biophys. 287, 97–104 [DOI] [PubMed] [Google Scholar]
- 46.Dixon D. A., Lindner D. L., Branchaud B., Lipscomb W. N. (1979) Biochemistry 18, 5770–5775 [DOI] [PubMed] [Google Scholar]
- 47.Zheng Y. J., Ornstein R. L. (1996) J. Am. Chem. Soc. 118, 9402–9408 [Google Scholar]
- 48.Basran J., Jang M. H., Sutcliffe M. J., Hille R., Scrutton N. S. (1999) J. Biol. Chem. 274, 13155–13161 [DOI] [PubMed] [Google Scholar]
- 49.Basran J., Sutcliffe M. J., Scrutton N. S. (2001) J. Biol. Chem. 276, 42887–42892 [DOI] [PubMed] [Google Scholar]
- 50.Mewies M., Basran J., Packman L. C., Hille R., Scrutton N. S. (1997) Biochemistry 36, 7162–7168 [DOI] [PubMed] [Google Scholar]
- 51.Leys D., Basran J., Talfournier F., Sutcliffe M. J., Scrutton N. S. (2003) Nat. Struct. Biol. 10, 219–225 [DOI] [PubMed] [Google Scholar]
- 52.Basran J., Chohan K. K., Sutcliffe M. J., Scrutton N. S. (2000) Biochemistry 39, 9188–9200 [DOI] [PubMed] [Google Scholar]
- 53.Burgess S. G., Messiha H. L., Katona G., Rigby S. E., Leys D., Scrutton N. S. (2008) Biochemistry 47, 5168–5181 [DOI] [PubMed] [Google Scholar]
- 54.Jones M., Talfournier F., Bobrov A., Grossmann J. G., Vekshin N., Sutcliffe M. J., Scrutton N. S. (2002) J. Biol. Chem. 277, 8457–8465 [DOI] [PubMed] [Google Scholar]
- 55.Bellamy H. D., Lim L. W., Mathews F. S., Dunham W. R. (1989) J. Biol. Chem. 264, 11887–11892 [PubMed] [Google Scholar]
- 56.Raine A. R., Yang C. C., Packman L. C., White S. A., Mathews F. S., Scrutton N. S. (1995) Protein Sci. 4, 2625–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Horton J. R., Sawada K., Nishibori M., Zhang X., Cheng X. (2001) Structure 9, 837–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paesen G. C., Adams P. L., Harlos K., Nuttall P. A., Stuart D. I. (1999) Mol. Cell 3, 661–671 [DOI] [PubMed] [Google Scholar]
- 59.Mans B. J., Ribeiro J. M., Andersen J. F. (2008) J. Biol. Chem. 283, 18721–18733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mans B. J., Calvo E., Ribeiro J. M., Andersen J. F. (2007) J. Biol. Chem. 282, 36626–36633 [DOI] [PubMed] [Google Scholar]
- 61.Fukui H., Fujimoto K., Mizuguchi H., Sakamoto K., Horio Y., Takai S., Yamada K., Ito S. (1994) Biochem. Biophys. Res. Commun. 201, 894–901 [DOI] [PubMed] [Google Scholar]
- 62.Gantz I., Schäffer M., DelValle J., Logsdon C., Campbell V., Uhler M., Yamada T. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 429–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lovenberg T. W., Roland B. L., Wilson S. J., Jiang X., Pyati J., Huvar A., Jackson M. R., Erlander M. G. (1999) Mol. Pharmacol. 55, 1101–1107 [PubMed] [Google Scholar]
- 64.Nakamura T., Itadani H., Hidaka Y., Ohta M., Tanaka K. (2000) Biochem. Biophys. Res. Commun. 279, 615–620 [DOI] [PubMed] [Google Scholar]
- 65.Oda T., Matsumoto S. (2001) Nippon Yakurigaku Zasshi 118, 36–42 [DOI] [PubMed] [Google Scholar]
- 66.Jongejan A., Leurs R. (2005) Arch. Pharm. 338, 248–259 [DOI] [PubMed] [Google Scholar]
- 67.Kelley M. T., Bürckstümmer T., Wenzel-Seifert K., Dove S., Buschauer A., Seifert R. (2001) Mol. Pharmacol. 60, 1210–1225 [DOI] [PubMed] [Google Scholar]
- 68.Yao B. B., Hutchins C. W., Carr T. L., Cassar S., Masters J. N., Bennani Y. L., Esbenshade T. A., Hancock A. A. (2003) Neuropharmacology 44, 773–786 [DOI] [PubMed] [Google Scholar]
- 69.Shin N., Coates E., Murgolo N. J., Morse K. L., Bayne M., Strader C. D., Monsma F. J., Jr. (2002) Mol. Pharmacol. 62, 38–47 [DOI] [PubMed] [Google Scholar]
- 70.Jongejan A., Bruysters M., Ballesteros J. A., Haaksma E., Bakker R. A., Pardo L., Leurs R. (2005) Nat. Chem. Biol. 1, 98–103 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.