

Abstract
Thioredoxin-interacting protein (Txnip) has important functions in regulating cellular metabolism including glucose utilization; the expression of the Txnip gene is sensitive to the availability of glucose and other fuels. Here, we show that Txnip expression is down-regulated at the transcriptional level by diverse inhibitors of mitochondrial oxidative phosphorylation (OXPHOS). The effect of these OXPHOS inhibitors is mediated by earlier identified carbohydrate-response elements (ChoREs) on the Txnip promoter and the ChoRE-associated transcription factors Max-like protein X (MLX) and MondoA (or carbohydrate-response element-binding protein (ChREBP)) involved in glucose-induced Txnip expression, suggesting that inhibited oxidative phosphorylation compromises glucose-induced effects on Txnip expression. We also show that the OXPHOS inhibitors repress the Txnip transcription most likely by inducing the glycolytic rate, and increased glycolytic flux decreases the levels of glycolytic intermediates important for the function of MLX and MondoA (or ChREBP). Our findings suggest that the Txnip expression is tightly correlated with glycolytic flux, which is regulated by oxidative phosphorylation status. The identified link between the Txnip expression and glycolytic activity implies a mechanism by which the cellular glucose uptake/homeostasis is regulated in response to various metabolic cues, oxidative phosphorylation status, and other physiological signals, and this may facilitate our efforts toward understanding metabolism in normal or cancer cells.
Keywords: Gene Expression, Glucose, Glycolysis, Metabolic Regulation, Respiration, Transcription Factors, MondoA-MLX, Txnip, Warburg Effect, Oxidative Phosphorylation
Introduction
Thioredoxin-interacting-protein (Txnip)3 plays important roles in diverse physiological or pathological processes (reviewed in Ref 1). In many cancer cell lines or cancer tissues, the expression of the Txnip gene is down-regulated; because Txnip can inhibit cell proliferation and promote apoptosis, it is thought that low Txnip expression may contribute to cancer development (2–8). Recently, Txnip was identified as a negative regulator for cellular glucose uptake (9–11); Txnip knock-out mice exhibit abnormalities in glucose and lipid metabolism (4, 12–14). These observations are in line with critical function(s) of Txnip in metabolic control.
The expression of the Txnip gene is sensitive to many nutritional factors. Txnip was originally identified as a vitamin D up-regulated protein (VDUP1 (15)), and the Txnip expression was later shown to be induced by glucose, which is mediated by carbohydrate-response elements (ChoREs (16–18)) and associated transcription factors Max-like protein X (MLX) and Mondo (MondoA or ChoRE-binding protein (ChREBP, which is also dubbed as MondoB) (18–20)). Moreover, the Txnip expression is modulated by glutamine, potentially through glutaminolysis, fatty acids, and an array of adenosine-containing molecules (21–23). The capability of the Txnip gene to sense diverse nutrients and to regulate their utilization implies a mechanism for cells to integrate different signaling pathways evoked by diverse nutritional factors.
In the current study, we wished to investigate whether the Txnip expression can be influenced by the cellular energy status. Here, we show that the Txnip expression is dramatically down-regulated when oxidative phosphorylation (OXPHOS) is inhibited. The effect of OXPHOS inhibitors is mediated by enhanced glycolytic flux and reduced occupancy of Mondo-MLX on the Txnip promoter, which leads to compromised Txnip gene transcription. Our results suggest that functional mitochondria capable of active OXPHOS are critical for Txnip expression.
EXPERIMENTAL PROCEDURES
Cell Culture
HeLa or HEK293 cells were maintained with 5% CO2 at 37 °C in complete Dulbecco's modified Eagle's medium (D5546, Sigma) that was supplemented with l-glutamine (Invitrogen), antibiotics (Invitrogen), and 10% fetal bovine serum (HyClone). To deplete glucose, cells were cultured overnight in glucose-free Dulbecco's modified Eagle's medium (Sigma) that was supplemented with serum, antibiotics, and l-glutamine; 2 mm sodium pyruvate (Invitrogen) was added as a carbon source.
DNA Constructs
Txnip promoter-driven luciferase reporters have been described in Ref 18. GFP-MondoA and FLAG-MLXβ coding sequences were inserted into pCI-neo vector for expression in mammalian cells. The GFP-Txnip expression plasmid was constructed by inserting the GFP-Txnip coding sequence into a lentiviral vector (pCDH-mCherry, kindly provided by Dr. Parth Patwari (11)); lentivirus produced was used to infect HeLa cells to obtain a cell line that stably expressed GFP-Txnip.
Promoter Assay
Cells at ∼80% confluence were transfected with firefly luciferase reporter plasmids (driven by wild-type or ChoRE-mutated Txnip promoters) and Renilla luciferase (driven by simian virus 40 (SV40) promoter) control plasmids using Lipofectamine 2000 and OPTI-MEM medium (both from Invitrogen) according to the manufacturer's instructions. Five hours later, the medium was replaced with complete Dulbecco's modified Eagle's medium. Cells were then treated with NaN3 (1 mm) or rotenone (1 μm) for 16 h or left untreated. Firefly or Renilla luciferase activity was measured using the Dual-Luciferase reporter assay system (Promega) on a TD-20/20 luminometer (Promega).
Western Blotting
HeLa or HEK293 cells were lysed using lysis buffer (25 mm Tris-HCl (pH 7.5), 100 mm NaCl, 2.5 mm EDTA and EGTA, 20 mm NaF, 1 mm Na3VO4, 20 mm Sodium β-glycerophosphate, 10 mm sodium pyrophosphate, 0.5% Triton X-100, and the protease inhibitor mixture (Roche Diagnostics) and 0.1% β-mercaptoethanol). Whole cell lysate proteins were then resolved by SDS-PAGE, and proteins were transferred onto a nitrocellulose membrane. Antibodies used were Txnip JY2 (K0205; MBL International), α-tubulin (236-10501; Invitrogen), ChREBP (NB400-135; Novus Biologicals), AMPKα (2532; Cell Signaling Technology), GFP (632381; Clontech), FLAG (F3165; Sigma-Aldrich), and in-house raised rabbit anti-GAPDH antibodies.
RNA Interference
HeLa cells at 20% confluency were transfected with 100 nm small interfering RNAs (siRNAs) using Lipofectamine RNAiMAX and OPTI-MEM (Invitrogen). At 48 h, the medium was changed to complete Dulbecco's modified Eagle's medium, and at 72 h, cells were subjected to different treatments. The efficiency of the siRNA-mediated knockdown was verified by Western blotting. Nucleotide sequences of siRNAs used in this study were: random, 5′-CAGUGUCAUACGUACGACGdTdT-3′; AMPKα1, 5′-GAAUCCUGUGACAAGCACUdTdT-3′; AMPKα2, 5′-CGUCAUUGAUGAUGAGGCUdTdT-3′; GAPDH 1, 5′-CACAAGAGGAAGAGAGAGAdTdT-3′; GAPDH 2, 5′-UCAAGAAGGUGGUGAAGCAdTdT-3′.
RNA Extraction, Reverse Transcription, and Quantitative Real-time PCR
Following various treatments, HeLa cells were washed with cold phosphate-buffered saline and subjected to RNA extraction using an RNeasy mini kit (Qiagen); residual DNA was removed by on-column RNase-free DNase (Qiagen) treatment. RNA samples (0.5 μg) were reverse-transcribed to complementary DNA using SuperScript III reverse transcriptase along with random hexamers, dNTP, and RNaseOUT (all from Invitrogen). Complementary DNA was then diluted and used for quantification (with β-actin gene as a control) by real-time PCR, which was performed using KAPA SYBR FAST qPCR master mix (Kapa Biosystems) and the 7300 real-time PCR system (Applied Biosystems). PCR primers were synthesized by 1st BASE (Singapore), and their sequences can be found in Refs. 23 and 24.
Lactate Measurement
HeLa cells were washed with phosphate-buffered saline and incubated with fresh medium for 1 h, and then culture medium were collected before and after various treatments. For measuring lactate level, reactions were carried out in 0.2 m hydrazine sulfate, 0.4 m glycine, 2.5 mm EDTA, 2 mm NAD+ (final concentrations), varied amounts of medium, 1 unit of lactate dehydrogenase in a final volume of 100 μl. The conversion of NAD+ to NADH was monitored by a fluorometer (340 nm excitation and 460 nm emission).
Chromatin Immunoprecipitation (ChIP) Assays
Control cells or cells treated with OXPHOS inhibitors (1 h) were fixed with 1% formaldehyde for 10 min (room temperature), the fixation was stopped by adding excess glycine, and the cells were then harvested, washed with phosphate-buffered saline twice, and incubated in ice-cold MC buffer (25) for 30 min. Afterward, ChIP assays were carried out using the ChIP assay kit (Millipore) following the manufacturer's instructions, along with indicated antibodies. The recovered and enriched chromatin DNA fragments were quantified using quantitative real-time PCR assays with primers targeting the human or rat Txnip gene promoter. The information on primer sequences is available in Ref 18 and is also available upon request.
RESULTS
Inhibition of OXPHOS Down-regulates Txnip mRNA Levels
OXPHOS is an important metabolic pathway for the synthesis of ATP in the mitochondrial inner membrane, carried out by five protein complexes (complexes I–IV for electron transfer and complex V for ATP synthesis). When HeLa cells were treated with chemicals that can inhibit these complexes (rotenone for complex I, 2-thenoyltrifluoroacetone (TTFA) for complex II, antimycin A and myxothiazol for complex III, sodium azide (NaN3) and diethylenetriamine/NO (DETA/NO) for complex IV, and oligomycin A for complex V) in a time course, the Txnip mRNA levels were found to be swiftly down-regulated by inhibitors for complexes I, III, IV, and V but not by the inhibitor TTFA for complex II as determined by quantitative real-time PCR (Fig. 1). The lack of effect of TTFA is in line with a non-essential role of complex II for electron transfer. Carbonyl cyanide 3-chlorophenylhydrazone (CCCP), a known uncoupler that dissipates the H+ gradient across the mitochondrial inner membrane, was also found to repress the Txnip mRNA expression (Fig. 1). Conversely, the expression of the histone 2B (H2B) gene was found to be down-regulated by complex IV inhibitors NaN3 and DETA/NO but not other inhibitors (Fig. 1).
FIGURE 1.

OXPHOS inhibitors compromise Txnip mRNA expression. HeLa cells were treated with the indicated OXPHOS inhibitors for 0, 1, 2, and 4 h, and the Txnip mRNA expression levels were scored by quantitative real-time PCR. The dosages of the drugs used were within reported effective ranges, which are: rotenone, 1 μm; 2-thenoyltrifluoroacetone (TTFA), 2 μm; antimycin A, 1 μg/ml; myxothiazol, 1 μm; NaN3, 1 mm; DETA/NO, 1 mm; oligomycin A, 1 μg/ml; and carbonyl cyanide 3-chlorophenylhydrazone (CCCP), 1 μm. The same dosages were used throughout this study. H2B, histone 2B. Error bars indicate S.E.
The above results and the rapid onset of the repressed Txnip expression, largely in a gene-specific manner, indicate that the observed effects are unlikely due to a general toxicity. Thus, the Txnip expression is tightly correlated with the OXPHOS status, and a functional proton pumping electron transport chain, as well as ATP synthesis, is essential for maintaining normal Txnip mRNA levels. NO was earlier shown to be able to repress Txnip expression; however, this was without a revelation that it might be linked to a role in inhibiting OXPHOS (26). The effect of NaN3 and DETA/NO on Txnip mRNA expression was tested in other cell lines, U2OS, WI-39, Namalwa B, and Jurkat T cells included, and the Txnip expression was repressed dramatically in all tested cell lines (data not shown), suggesting that the action of OXPHOS inhibitors on the Txnip expression is most likely a general phenomenon.
Inhibition of OXPHOS Down-regulates Txnip Protein Levels
The Txnip protein level is under tight control under physiological conditions. We tested the Txnip protein stability in cultured cells. We first used a HeLa cell line that stably expressed a GFP-Txnip fusion protein under the control of a cytomegalovirus promoter (which was also used in Fig. 2C). When treated with cycloheximide, these cells exhibited rapid protein turnover of the pre-existing endogenous Txnip protein (t½ <1 h; Fig. 2A). HEK293 cells exhibited similar half-lives of the Txnip protein when protein synthesis was blocked; the Txnip protein accumulated in the presence of MG-132, a proteasome inhibitor (Fig. 2B). These results are in agreement with a recent report showing that the Txnip protein undergoes a rapid ubiquitin-mediated degradation (27). GFP-Txnip fusion protein also undergoes fast turnover similarly to endogenous Txnip protein (Fig. 2A). The unstable nature of the Txnip protein suggests that a steady-state Txnip level depends largely on Txnip mRNA translation; therefore, maintaining a proper Txnip mRNA level by transcription may be crucial for sustaining Txnip protein expression.
FIGURE 2.

Decreased protein expression levels in cells treated by OXPHOS inhibitors. A, HeLa cells stably expressing a transgenic cytomegalovirus-driven GFP-Txnip fusion gene were treated with 40 μm cycloheximide (CHX) for the indicated times followed by immunoblot analysis of whole cell lysates. B, HEK293 cells were treated with 40 μm cycloheximide for the indicated times or with 20 μm proteasome inhibitor MG-132 for 12 h followed by analysis of endogenous Txnip expression. C and D, HeLa cells (C) and HEK293 cells (D) were treated with mitochondrial inhibitors for the indicated times followed by immunoblot analysis of whole cell lysates.
We also examined the Txnip protein level in cells treated with different OXPHOS inhibitors. In HeLa cells such as those used in Fig. 2A, NaN3, rotenone, or oligomycin A treatment dramatically decreased the expression level of the endogenous Txnip protein; however, the GFP-Txnip fusion protein was not affected (Fig. 2C). We reason that although the GFP-Txnip fusion protein is unstable (Fig. 2A), a high level of the fusion protein was sustained because the cytomegalovirus promoter, which was unlikely repressed by the OXPHOS inhibitors, was driving GFP-Txnip transcription constantly. On the other hand, the endogenous Txnip (mRNA and protein) expression was severely reduced given that the transcription from the endogenous Txnip promoter was repressed (see below). In HEK293 cells treated with antimycin A, rotenone, or NaN3, the Txnip protein level was also swiftly down-regulated (Fig. 2D); the kinetics of the expression reduction of the Txnip mRNA and protein in cells treated with OXPHOS inhibitors were similar (Figs. 1 and 2, C and D), hence suggesting that OXPHOS inhibitors mainly target Txnip transcription and that sustainable Txnip transcription is critical for maintaining steady-state Txnip protein expression.
ChoREs Mediate the Effect of OXPHOS Inhibitors on the Txnip Gene Transcription
To confirm that the Txnip transcription was repressed by OXPHOS inhibitors, we tested the effect of these inhibitors on the activity of a luciferase reporter driven by a Txnip promoter in HeLa cells. In the presence of NaN3 or rotenone, the activity of a wild-type (WT) Txnip promoter was down-regulated by ∼10-fold, as was that of a shorter truncated Txnip promoter (WT-Short), which contains two ChoREs and CCAAT boxes and is known to be sufficient to confer glucose-induced expression to the gene (18); however, NaN3 or rotenone only slightly down-regulated the activity of a Txnip promoter with mutations at two ChoRE sites (Mut-ChoRE) (∼2-fold; Fig. 3, A and B). The mild inhibitory effect of NaN3 or rotenone on ChoRE-mutated Txnip promoter might be due to residual activities of the mutated ChoREs or, more likely, was not Txnip promoter-specific. For instance, the OXPHOS inhibitor NO has been shown to repress the luciferase expression in a promoter-independent fashion (28). Thus, we conclude that OXPHOS inhibitors repress the Txnip expression at the transcriptional level, which is mediated largely by the ChoREs on the Txnip promoter.
FIGURE 3.

Repressed Txnip promoter activity in cells treated with NaN3 or rotenone. A, a diagram shows the luciferase reporter constructs used in B; Mut-ChoRE contains mutations at two ChoRE sites, and WT-Short is a hybrid promoter that contains a shorter Txnip promoter segment and the TATA box in front of the luciferase gene. WT, wild type. B, luciferase activities of the indicated reporter genes in HeLa cells treated with NaN3 or rotenone, expressed as the percentages of the corresponding activities in untreated cells. Error bars indicate S.E.
Reduced Txnip Promoter Occupancy by Mondo-MLX in the Presence of OXPHOS Inhibitors
Both ChoREs on the Txnip promoter are essential for the full promoter activity and are able to recruit glucose-responsive transcription factors Mondo-MLX (18). ChoREs are clearly important for mediating the OXPHOS inhibitor-repressed Txnip transcription (Fig. 3). We thus investigated whether these inhibitors repressed the expression of Mondo-MLX or compromised the recruitment of Mondo-MLX to the Txnip promoter as a basis for transcriptional repression.
As seen (Fig. 4A), although the Txnip protein level was significantly reduced in the rotenone- or NaN3-treated HeLa cells, which is in agreement with earlier data (Fig. 2, C and D), the protein levels of ectopically expressed GFP-MondoA or FLAG-MLXβ were not changed in the presence of NaN3 or rotenone; additionally, these inhibitors did not suppress the mRNA expression levels of endogenous MondoA or MLX (not shown). Hence, a reduced Mondo-MLX expression is unlikely the basis for Txnip promoter repression by OXPHOS inhibitors, prompting us to explore possible changes in Txnip promoter occupancy by MondoA-MLX. To this end, we subjected HeLa cells, untreated or treated with NaN3 for 1 h, to ChIP assays and found that the occupancy of MondoA and MLX was reduced in NaN3-treated cells as that of RNA polymerase II (Pol II); however, the recruitment of nuclear factor Y subunit A (NF-YA) to the CCAAT boxes, as well as the acetylation status of histone H3, on the promoter was not significantly changed (Fig. 4B). Rotenone exhibited similar effects (Fig. 4C). Thus, although an overall cellular MondoA-MLX expression is not reduced, their nuclear availability and recruitment to the Txnip promoter are significantly reduced by the OXPHOS inhibitors.
FIGURE 4.

The occupancy of Mondo-MLX on the Txnip promoter is reduced by NaN3 or rotenone. A, the protein level of ectopic GFP-MondoA and FLAG-MLX or endogenous ChREBP was not affected by NaN3 or rotenone treatment (1.5 h) in HeLa or INS-1 cells, whereas Txnip expression was repressed. B and C, ChIP assays in HeLa cells, respectively, treated (1 h) with NaN3 and rotenone, using antibodies corresponding to the indicated proteins. NF-YA, nuclear factor Y, subunit A; Pol II, RNA polymerase II. D, ChIP assay in pancreatic INS-1 cells treated with NaN3 (1 h) using antibodies corresponding to the indicated proteins. The occupancy was scored by quantitative real-time PCR, expressed as percentages of input total chromatin DNA. Error bars in B and D indicate S.E.
Unlike other cell types, which employ MondoA as a transcription factor partner for MLX (18, 19), pancreatic cells appear to employ ChREBP as a dominant partner (over MondoA) for MLX on regulating Txnip expression (18, 20). In rat insulinoma INS-1 cells, the Txnip expression was also significantly repressed by NaN3 or rotenone, and the ChREBP protein level was not affected by these inhibitors (Fig. 4A). We performed ChIP assays in INS-1 cells and found that the recruitment of ChREBP-MLX-RNA polymerase II to the Txnip promoter was significantly reduced in cells treated with NaN3 (Fig. 4D). Taken together, results in Fig. 4 suggest that both the MondoA-MLX and the ChREBP-MLX transcription factors are sensitive to the OXPHOS inhibitors, which trigger reduced occupancy of these factors on the Txnip promoter, leading to a reduced RNA polymerase II recruitment, thus repressing Txnip transcription.
OXPHOS Inhibitors Negatively Regulate Glucose-induced Txnip Expression
ChoREs and associated transcription factors Mondo-MLX mediate the glucose-induced Txnip expression (17–20); we reasoned that OXPHOS inhibitors might repress Txnip expression by interrupting a glucose-induced signaling pathway. Indeed, when HeLa cells were cultured in glucose-free medium, the measurable (albeit low) Txnip mRNA level was not affected by administering NaN3, rotenone, or oligomycin A to cells, whereas the glucose-induced Txnip expression was drastically compromised by these OXPHOS inhibitors (Fig. 5A). A glucose analog, 2-deoxyglucose (2DG), could dramatically boost Txnip expression, which coincided with enhanced nuclear MondoA-MLX accumulation (19, 23). Here, we found that the stimulatory effect of 2DG on Txnip expression was totally abolished by co-treating HeLa cells with OXPHOS inhibitors (Fig. 5B). The observation that the repression of Txnip expression is glucose-dependent (Fig. 5A) and is mediated by ChoREs and associated transcription factors (Figs. 3 and 4) suggests that a glucose-induced signaling pathway critical for Txnip expression is negatively regulated by OXPHOS inhibitors. The fact that occupancy of the Txnip promoter by Mondo-MLX is up-regulated by glucose (18–20) and down-regulated by OXPHOS inhibitors (Fig. 4) is a strong indication that the positive (glucose) and adverse (OXPHOS inhibitors) effects are mediated by a common signaling molecule(s) that normally triggers the activation of transcription factors Mondo-MLX.
FIGURE 5.
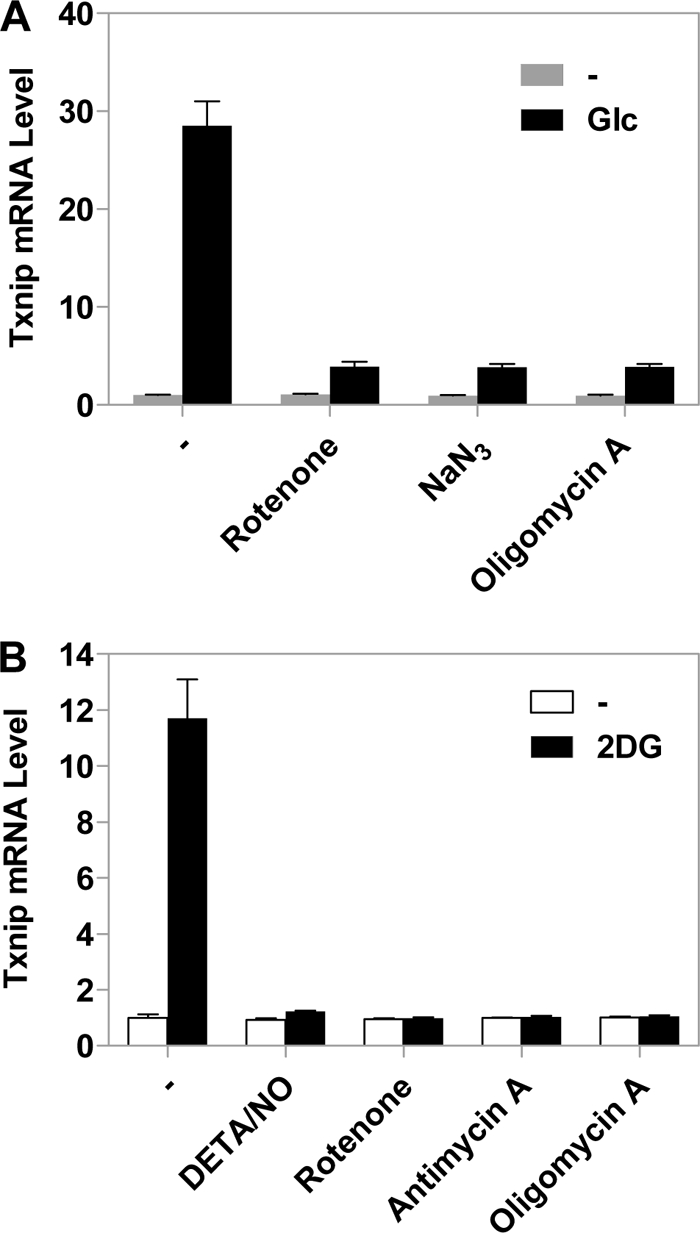
OXPHOS inhibitors repress glucose-induced Txnip expression. HeLa cells were first incubated in glucose-free medium overnight and were left untreated or treated with 10 mm glucose for 2 h (A) or 10 mm 2DG for 1 h (B) with or without OXPHOS inhibitors. Txnip mRNA levels were scored by real-time PCR. Error bars indicate S.E.
The Effects of OXPHOS Inhibitors Are Not Mediated by AMPK
The various mitochondrial OXPHOS inhibitors have different consequences for OXPHOS. Blockage of complexes I, III, and IV inhibits electron transport and the formation of the transmembrane electrochemical potential (ΔμH+) as well as ATP synthesis. In contrast, ATP synthase inhibition with oligomycin inhibits electron transport and ATP synthesis but increases ΔμH+, whereas the uncoupler carbonyl cyanide 3-chlorophenylhydrazone increases electron transport but abolishes ΔμH+ and ATP synthesis. Given that the mitochondrial inhibitors regulate Txnip expression in the same manner, it can be concluded that Txnip regulation is not mediated through changes in the inner membrane electrochemical gradient and electron transport, which are known to control cellular redox state and the production of cellular reactive oxygen species. However, all OXPHOS inhibitors used in this study are able to inhibit ATP synthesis. Hence, certain signaling pathway(s) evoked by low ATP levels could be responsible for the Txnip expression repressed by OXPHOS inhibitors, for which a candidate is 5′-AMP-activated protein kinase (AMPK) that is activated by a high intracellular AMP/ATP ratio, a possible outcome when OXPHOS is inhibited (29–31).
To test this possibility, we treated HeLa cells with compound C, an AMPK inhibitor, before adding NaN3 or rotenone to the cultured cells. As shown in Fig. 6A, compound C alone did not exhibit a significant effect on the Txnip expression, which remained dramatically down-regulated by NaN3 or rotenone regardless of compound C. In addition, we have down-regulated the expression of AMPKα, the catalytic subunit of AMPK, using siRNAs targeting AMPKα1 and/or AMPKα2 in HeLa cells, and we found that the Txnip mRNA level was still significantly down-regulated by rotenone in the presence of AMPKα siRNAs (Fig. 6B). Thus, we believe that the repressed Txnip expression by OXPHOS inhibitors is unlikely mediated through activating AMPK.
FIGURE 6.
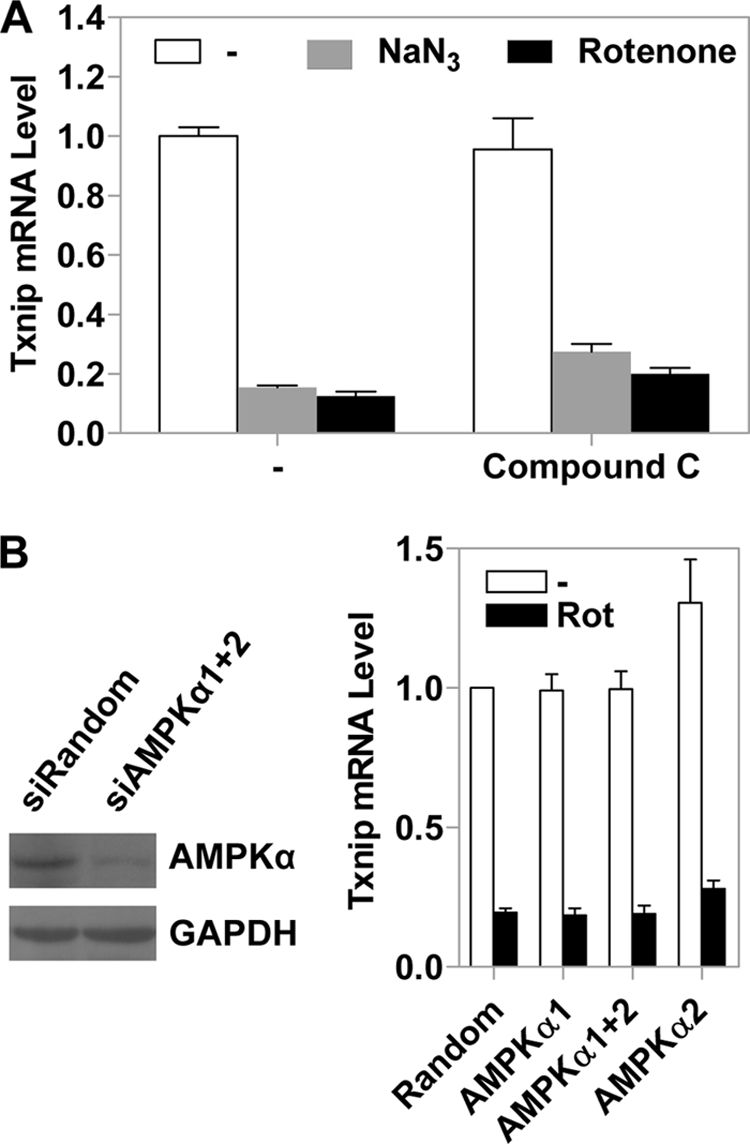
AMPK does not affect the Txnip expression patterns. A, control HeLa cells or cells preincubated (20 min) with 10 μm compound C were treated with NaN3 or rotenone (1 h) or left untreated, and the Txnip mRNA levels were scored by real-time PCR. B, the AMPKα protein level in HeLa cells transfected with a mixture of siRNAs targeting AMPKα1 and AMPKα2 was significantly reduced, as indicated by immunoblot analysis of whole cell lysates (left; please note that anti-AMPKα antibodies detect both isoforms). The effect of rotenone (Rot) on Txnip mRNA expression was not reversed by AMPKα siRNAs either singly or in combination (right). Error bars indicate S.E.
OXPHOS Inhibitors Induce Glycolytic Rate to Repress Txnip Expression
Given the above results (Fig. 6), we explored other potential mechanism(s) by which OXPHOS inhibitors could repress Txnip expression. The glycolytic rate is known to be elevated when OXPHOS is inhibited to maintain cellular ATP levels (30); indeed, the production of lactate in cells treated with different OXPHOS inhibitors for 1, 2, or 4 h was significantly increased (Fig. 7A), which was in parallel with the repressed Txnip expression upon the addition of OXPHOS inhibitors (Figs. 1 and 2). Consistent with an earlier observation (24), ATP levels were not significantly reduced in a tested time frame from 0 to 4 h, although a longer treatment (overnight) did exhibit significantly reduced ATP levels (data not shown). Hence, HeLa cells can respond to compromised OXPHOS by boosting intrinsic glycolysis, which along with pre-existing ATP sustains overall ATP levels at least in a short term (<4 h).
FIGURE 7.

Txnip transcription can sense glycolytic rate. A, HeLa cells were left untreated or treated with indicated OXPHOS inhibitors for 1, 2, or 4 h, and lactate levels in the medium were determined. B, HeLa cells maintained in glucose-containing medium were treated with the indicated glycolytic intermediates for 2 h (G6P, 25 mm; GADP, 5 mm; 3-phosphoglycerate (3PG) 5 mm) with or without rotenone. C, HeLa cells, preincubated with 0.5 mm iodoacetate (IodoAc), 0.5 mm sodium acetate (NaAC), or 10 mm oxamate for 20 min, were incubated in the absence or presence of NaN3 or rotenone for 2 h. D, the GAPDH protein level in HeLa cells transfected with two siRNAs targeting GAPDH was significantly reduced, as indicated by immunoblot analysis of whole cell lysates (left). The effect of rotenone on Txnip expression was reversed by GAPDH-specific siRNAs (right). In B–D, Txnip mRNA levels were scored by real-time PCR. Error bars in B–D indicate S.E.
An elevated glycolysis rate may facilitate the depletion of certain intermediate molecule(s), which normally transmits the glucose signal to induce Txnip expression in which a critical step is the activation of Mondo-MLX. We reasoned that when the glycolytic rate is increased by OXPHOS inhibitors, the levels of this/these glycolytic intermediate(s) may drop, which in turn is sensed by Mondo-MLX, thus repressing Txnip expression.
Glucose-6-phosphate (G6P) is recognized as a glycolytic metabolite that transmits the glucose signaling to Txnip expression. This action can be mimicked by 2DG-6-phosphate, which is derived from 2DG upon its phosphorylation in cells, but 2DG-6-phosphate is not further metabolized; thus, it accumulates and likely functions as a G6P analog to boost Txnip expression (19). If the Txnip transcriptional repression by OXPHOS inhibitors was mediated solely by depletion of G6P, 2DG in principle should be able to rescue the Txnip transcription. Our observation, however, was on the contrary; the stimulatory effect of 2DG on Txnip expression was completely repressed in the presence of OXPHOS inhibitors (Fig. 5B). We reasoned that the effect of OXPHOS inhibitors might be mediated by certain other downstream glycolytic intermediate(s) involved in activating Txnip expression, which may function in addition to, or in conjunction with, G6P.
To test this hypothesis, we treated HeLa cells with other available glycolytic metabolites, which included G6P, glyceraldehyde-3-phosphate (GADP), and 3-phosphoglycerate (3PG), and found that GADP stimulated the Txnip expression as potently as G6P, whereas glyceraldehyde-3-phosphate was without an effect (Fig. 7B). Interestingly, in the presence of G6P (but not GADP), the Txnip expression was no longer repressed by rotenone (Fig. 7B), suggesting that G6P can sustain a proper level of itself, and possibly, that of certain downstream intermediates such as GADP, which likely are critical for Mondo-MLX function in a combinatorial fashion. To explain the observation that in contrast to G6P, GADP did not block the repression by OXPHOS inhibitors (Fig. 7B), we reason that GADP may result in an overall increase in the cellular GADP level as well as that of some upstream glycolytic intermediates such as G6P, thus boosting Txnip expression in the absence of OXPHOS inhibitors. In the presence of OXPHOS inhibitors, however, glycolysis is enhanced to compensate for reduced mitochondrial ATP production (also see below and see “Discussion”). Given that the two ATP generation steps during glycolysis are downstream of GADP, enhanced glycolysis would result in GADP being metabolized in an ATP-generating direction rather than the reverse. We also tested several farther downstream glucose-derived glycolytic metabolites, such as pyruvate or lactate, which exhibited no effect on Txnip expression (data now shown).
G6P and GADP are glycolytic metabolites upstream of the enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH); blocking the GAPDH enzyme function might potentially lead to accumulation of these metabolites, which in turn feeds back to the Txnip expression. Indeed, HeLa cells incubated with iodoacetate (IodoAc), an inhibitor of the GAPDH enzyme, which in principle causes a decreased glycolytic rate, exhibited slightly up-regulated Txnip expression; importantly, the Txnip expression was no longer repressed by OXPHOS inhibitor NaN3 or rotenone (Fig. 7C). On the other hand, sodium acetate as a control or oxamate (an inhibitor of lactate dehydrogenase) failed to stimulate Txnip expression and to overcome the repressed Txnip expression by NaN3 or rotenone (Fig. 7C). Lactate dehydrogenase is at the end of glycolysis; blocking the function of this enzyme may not be able to lead to significant accumulation of G6P, GADP, and other intermediate metabolites much farther upstream of glycolysis (Fig. 8).
FIGURE 8.

A model linking Txnip expression, glucose transport, glycolysis, OXPHOS, and other physiological cues. OXPHOS inhibitors (this work), glutamine (22), and insulin signaling (9) have all been shown to repress Txnip expression; these factors are also known to increase the glycolytic rate. The augmented glycolytic flux may dynamically deplete glycolytic intermediate metabolites normally involved in transmitting glucose signaling to Mondo-MLX activation, which in turn represses Txnip expression. Our data suggest that the metabolites upstream of the GAPDH enzyme, G6P and GADP in particular but others are not ruled out, are candidates that link Txnip expression to glycolytic flux. How Mondo-MLX transcription factors are activated, the nuclear translocation included, and whether the above mentioned relevant metabolites are directly involved in this activation process are a challenging problem to be addressed in the future. Txnip has an inhibitory role on glucose transport; hence, glucose uptake in cells with repressed Txnip expression will be induced, a common theme in many cancerous cells that are addictive to glucose. Glut, glucose transporters; 3PG, 3-phosphoglycerate.
We also knocked down, in HeLa cells, the GAPDH protein expression with two different siRNAs; in these (partially) GAPDH-deficient cells, the repressive effect of rotenone on Txnip expression was significantly reversed (Fig. 7D), reminiscent of the iodoacetate effects (Fig, 7C). This again suggests that slowing down glycolysis at the GAPDH step leads to accumulation of upstream metabolites such as G6P and GADP that are critical for Txnip expression. Taken together, the above results support the notion that OXPHOS inhibitors repress Txnip expression via depleting glycolytic metabolites as a result of increased glycolytic flux (of which early intermediate metabolites such as G6P and GADP are the most likely candidates), that allowing accumulation of G6P, GADP, and possibly other glycolytic intermediates in between can significantly boost Txnip expression, and that these metabolites can directly or indirectly activate the transcription factors Mondo-MLX, thus enhancing Txnip expression (Fig. 8).
DISCUSSION
We show that Txnip expression is tightly linked to the status of mitochondrial OXPHOS (Figs. 1 and 2). In the presence of OXPHOS inhibitors, the Txnip gene transcription is dramatically down-regulated, and the inhibitory effects of OXPHOS inhibitors on Txnip expression were mediated by ChoREs and the associated transcription factor complex Mondo-MLX (Figs. 3 and 4). This suggests that a glucose-dependent signaling pathway that is responsible for induction of Txnip expression is specifically repressed by the OXPHOS inhibitors. Indeed, without glucose, the basal (measurable) Txnip expression was not repressed by OXPHOS inhibitors (Fig. 5).
The repression of Txnip expression by OXPHOS inhibitors is most likely mediated by an increased rate of glycolysis because glycolytic intermediates could also induce Txnip expression, and the effect of OXPHOS inhibitors on Txnip expression was reversed when cellular glycolysis was inhibited by a GAPDH inhibitor or siRNAs targeting GAPDH (Fig. 7). We envision that an increased glycolytic rate would deplete certain glycolytic intermediates including G6P and GADP, which are critical for Mondo-MLX function and Txnip transcription (Fig. 8). It remains unclear how the mitochondrial OXPHOS status feeds back to glycolysis; however, AMPK, a known mediator of feedback from OXPHOS inhibition, is unlikely to be involved in transmitting inhibitory signals to the Txnip gene (Fig. 6). Thus, although AMPK is able to sense an ATP shortage in the (long term) presence of OXPHOS inhibitors (29), mammalian cells may use other swift mechanisms to couple OXPHOS status to glycolysis. Consistent with a non-essential role of AMPK (Fig. 6), the cellular ATP level was not significantly changed in cells treated with different OXPHOS inhibitors in a 0–4-h time course (data not shown), whereas within the same time frame, inhibited Txnip expression (Figs. 1 and 2), increased glycolysis (Fig. 7A), as well as decreased Mondo-MLX recruitment to the Txnip promoter (Fig. 4, B–D) were all manifested.
Our results suggest that the transcription of the Txnip gene is regulated in response to the rate of glycolysis. Given that Txnip negatively regulates glucose uptake (9–11), this connection is of physiological relevance for cellular glucose utilization. When cells require more energy or building blocks for synthesis of macromolecules, the glycolytic rate will increase, accompanied by a decline of certain glycolytic metabolites, which is sensed by the Txnip transcriptional machinery to repress Txnip expression. In turn, the reduced Txnip level allows cells to take up more glucose (Fig. 8). On the other hand, under conditions of lower metabolic activity, the glycolytic rate is expected to be decreased. In this scenario, the Txnip expression would be induced, which feeds back to inhibit the glucose influx (Fig. 8). Thus, a tight link between Txnip expression and glycolytic rate enables cells to efficiently regulate the cellular glucose homeostasis.
The linkage between the glycolytic rate and Txnip transcription may also explain several other phenomena related to Txnip expression. For instance, Txnip expression is down-regulated by insulin (9, 32), and it has been proposed that insulin may regulate Txnip transcription via an AKT- and a FOXO-dependent signaling pathway (33). There is a FOXO binding site on the Txnip promoter; however, it is not involved in glucose-induced Txnip expression (18, 23). Thus, instead of a direct control of Txnip transcription by FOXO1, insulin might modulate Txnip transcription by increasing the glycolytic rate, which in turn reduces the activity of Mondo-MLX (Fig. 8). To support this notion, a positive role of insulin on certain glycolytic enzymes is well documented (34). The Txnip expression is also significantly repressed by glutamine (22), which had been shown to increase glycolysis (35). Hence, the insulin signaling and glutamine might regulate Txnip expression through a similar pathway evoked by OXPHOS inhibitors, which increase the glycolytic rate and decrease some critical glycolytic intermediates; the decreased intracellular level of these metabolites, singly or in combination, may in turn repress Txnip expression by decreasing the occupancy of Mondo-MLX on the Txnip promoter (Fig. 8).
Most cancer cells are addicted to glucose; glucose in these cells is metabolized at a much faster rate through glycolysis. The majority of pyruvate produced by glycolysis, instead of entering the more efficient mitochondrial tricarboxylic acid cycle for energy (ATP) production, is fermented into lactate even when oxygen is present and mitochondria are potentially functional in OXPHOS and tricarboxylic acid cycle (Fig. 8). This phenomenon is called aerobic glycolysis or the Warburg effect, and the underlying mechanisms remain not fully understood (reviewed in Ref. 36). The expression of Txnip in many cancer cell lines and tissues is dramatically down-regulated (2) and, based on our results in the current study, the underlying mechanism is most likely attributed to increased glycolysis and reduced OXPHOS in cancer cells. Low Txnip expression would greatly favor cellular glucose uptake, thus contributing to massive/addictive glucose utilization in cancer cells. We propose that Txnip acts as a mediator to integrate cellular metabolic activity and energetic needs with cellular glucose supply; this may have important implications for regulation of glucose homeostasis in normal cells and the development of the Warburg effect in cancer cells (Fig. 8).
This work was supported by grants from the Agency for Science, Technology and Research (A*STAR), Singapore.
- Txnip
- thioredoxin interacting protein
- 2DG
- 2-deoxy-glucose
- AMPK
- 5′-adenosine monophosphate-activated protein kinase
- ChIP
- chromatin immunoprecipitation
- ChoRE
- carbohydrate-response element
- ChREBP
- carbohydrate-response element-binding protein
- FOXO
- forkhead box O
- DETA/NO
- diethylenetriamine/NO
- MLX
- Max-like protein X
- G6P
- glucose-6-phosphate
- GADP
- glyceraldehyde-3-phosphate
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- OXPHOS
- oxidative phosphorylation
- siRNA
- small interference RNA
- GFP
- green fluorescent protein.
REFERENCES
- 1.Kim S. Y., Suh H. W., Chung J. W., Yoon S. R., Choi I. (2007) Cell Mol. Immunol. 4, 345–351 [PubMed] [Google Scholar]
- 2.Butler L. M., Zhou X., Xu W. S., Scher H. I., Rifkind R. A., Marks P. A., Richon V. M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 11700–11705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen C. L., Lin C. F., Chang W. T., Huang W. C., Teng C. F., Lin Y. S. (2008) Blood 111, 4365–4374 [DOI] [PubMed] [Google Scholar]
- 4.Chen J., Saxena G., Mungrue I. N., Lusis A. J., Shalev A. (2008) Diabetes 57, 938–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han S. H., Jeon J. H., Ju H. R., Jung U., Kim K. Y., Yoo H. S., Lee Y. H., Song K. S., Hwang H. M., Na Y. S., Yang Y., Lee K. N., Choi I. (2003) Oncogene 22, 4035–4046 [DOI] [PubMed] [Google Scholar]
- 6.Jeon J. H., Lee K. N., Hwang C. Y., Kwon K. S., You K. H., Choi I. (2005) Cancer Res. 65, 4485–4489 [DOI] [PubMed] [Google Scholar]
- 7.Sheth S. S., Bodnar J. S., Ghazalpour A., Thipphavong C. K., Tsutsumi S., Tward A. D., Demant P., Kodama T., Aburatani H., Lusis A. J. (2006) Oncogene 25, 3528–3536 [DOI] [PubMed] [Google Scholar]
- 8.Wang Z., Rong Y. P., Malone M. H., Davis M. C., Zhong F., Distelhorst C. W. (2006) Oncogene 25, 1903–1913 [DOI] [PubMed] [Google Scholar]
- 9.Parikh H., Carlsson E., Chutkow W. A., Johansson L. E., Storgaard H., Poulsen P., Saxena R., Ladd C., Schulze P. C., Mazzini M. J., Jensen C. B., Krook A., Björnholm M., Tornqvist H., Zierath J. R., Ridderstråle M., Altshuler D., Lee R. T., Vaag A., Groop L. C., Mootha V. K. (2007) PLoS Med. 4, e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshioka J., Imahashi K., Gabel S. A., Chutkow W. A., Burds A. A., Gannon J., Schulze P. C., MacGillivray C., London R. E., Murphy E., Lee R. T. (2007) Circ. Res. 101, 1328–1338 [DOI] [PubMed] [Google Scholar]
- 11.Patwari P., Chutkow W. A., Cummings K., Verstraeten V. L., Lammerding J., Schreiter E. R., Lee R. T. (2009) J. Biol. Chem. 284, 24996–25003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donnelly K. L., Margosian M. R., Sheth S. S., Lusis A. J., Parks E. J. (2004) J Nutr. 134, 1475–1480 [DOI] [PubMed] [Google Scholar]
- 13.Sheth S. S., Castellani L. W., Chari S., Wagg C., Thipphavong C. K., Bodnar J. S., Tontonoz P., Attie A. D., Lopaschuk G. D., Lusis A. J. (2005) J. Lipid Res. 46, 123–134 [DOI] [PubMed] [Google Scholar]
- 14.Oka S., Liu W., Masutani H., Hirata H., Shinkai Y., Yamada S., Yoshida T., Nakamura H., Yodoi J. (2006) FASEB J. 20, 121–123 [DOI] [PubMed] [Google Scholar]
- 15.Chen K. S., DeLuca H. F. (1994) Biochim. Biophys. Acta 1219, 26–32 [DOI] [PubMed] [Google Scholar]
- 16.Schulze P. C., Yoshioka J., Takahashi T., He Z., King G. L., Lee R. T. (2004) J. Biol. Chem. 279, 30369–30374 [DOI] [PubMed] [Google Scholar]
- 17.Minn A. H., Hafele C., Shalev A. (2005) Endocrinology 146, 2397–2405 [DOI] [PubMed] [Google Scholar]
- 18.Yu F. X., Luo Y. (2009) PLoS One 4, e8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoltzman C. A., Peterson C. W., Breen K. T., Muoio D. M., Billin A. N., Ayer D. E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 6912–6917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cha-Molstad H., Saxena G., Chen J., Shalev A. (2009) J. Biol. Chem. 284, 16898–16905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J., Fontes G., Saxena G., Poitout V., Shalev A. (2010) Diabetes 59, 440–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaadige M. R., Looper R. E., Kamalanaadhan S., Ayer D. E. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 14878–14883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu F. X., Goh S. R., Dai R. P., Luo Y. (2009) Mol. Endocrinol. 23, 932–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai R. P., Yu F. X., Goh S. R., Chng H. W., Tan Y. L., Fu J. L., Zheng L., Luo Y. (2008) J. Biol. Chem. 283, 26894–26901 [DOI] [PubMed] [Google Scholar]
- 25.Aparicio O., Geisberg J. V., Sekinger E., Yang A., Moqtaderi Z., Struhl K. (2005) in Current Protocols in Molecular Biology (Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K. eds) unit 21.3, John Wiley & Sons, Inc. Hoboken, NJ: [DOI] [PubMed] [Google Scholar]
- 26.Schulze P. C., Liu H., Choe E., Yoshioka J., Shalev A., Bloch K. D., Lee R. T. (2006) Arterioscler. Thromb. Vasc. Biol. 26, 2666–2672 [DOI] [PubMed] [Google Scholar]
- 27.Zhang P., Wang C., Gao K., Wang D., Mao J., An J., Xu C., Wu D., Yu H., Liu J. O., Yu L. (2010) J. Biol. Chem. 285, 8869–8879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan X., Roy E., Zhu L., Murphy T. C., Kozlowski M., Nanes M. S., Rubin J. (2003) J. Biol. Chem. 278, 10232–10238 [DOI] [PubMed] [Google Scholar]
- 29.Marsin A. S., Bertrand L., Rider M. H., Deprez J., Beauloye C., Vincent M. F., Van den Berghe G., Carling D., Hue L. (2000) Curr. Biol. 10, 1247–1255 [DOI] [PubMed] [Google Scholar]
- 30.Marsin A. S., Bouzin C., Bertrand L., Hue L. (2002) J. Biol. Chem. 277, 30778–30783 [DOI] [PubMed] [Google Scholar]
- 31.Suter M., Riek U., Tuerk R., Schlattner U., Wallimann T., Neumann D. (2006) J. Biol. Chem. 281, 32207–32216 [DOI] [PubMed] [Google Scholar]
- 32.Shaked M., Ketzinel-Gilad M., Ariav Y., Cerasi E., Kaiser N., Leibowitz G. (2009) Diabetologia 52, 636–644 [DOI] [PubMed] [Google Scholar]
- 33.Papadia S., Soriano F. X., Léveillé F., Martel M. A., Dakin K. A., Hansen H. H., Kaindl A., Sifringer M., Fowler J., Stefovska V., McKenzie G., Craigon M., Corriveau R., Ghazal P., Horsburgh K., Yankner B. A., Wyllie D. J., Ikonomidou C., Hardingham G. E. (2008) Nat. Neurosci. 11, 476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertrand L., Alessi D. R., Deprez J., Deak M., Viaene E., Rider M. H., Hue L. (1999) J. Biol. Chem. 274, 30927–30933 [DOI] [PubMed] [Google Scholar]
- 35.Lanks K. W. (1986) J. Cell Physiol. 126, 319–321 [DOI] [PubMed] [Google Scholar]
- 36.Vander Heiden M. G., Cantley L. C., Thompson C. B. (2009) Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]