

Abstract
The mechanism of doxorubicin is compared with that of doxazolidine, a doxorubicin-formaldehyde conjugate. The IC50 for growth inhibition of 67 human cancer cell lines, but not cardiomyocytes, is 32-fold lower with doxazolidine than with doxorubicin. Growth inhibition by doxazolidine correlates better with growth inhibition by DNA crosslinking agents than with growth inhibition by doxorubicin. Doxorubicin induces G2/M arrest in HCT-116 colon cancer cells and HL-60 leukemia cells through a well-documented topoisomerase II-dependent mechanism. Doxazolidine fails to induce a G2/M arrest in HCT-116 cells, but induces apoptosis 4-fold better than doxorubicin. The IC50 for doxazolidine growth inhibition of HL-60/MX2 cells, a topoisomerase II-deficient derivative of HL-60 cells, is 1420-fold lower than the IC50 for doxorubicin, and doxazolidine induces apoptosis 15-fold better. Further, Doxazolidine has little effect in a topoisomerase II activity assay. These data indicate that doxorubicin and doxazolidine induce apoptosis via different mechanisms and doxazolidine cytotoxicity is topoisomerase II-independent.
Keywords: Doxorubicin, Doxazolidine, Topoisomerase II, Virtual Crosslink, HL-60/MX2
Introduction
Doxorubicin, an anthracycline antibiotic, was discovered as a Streptomyces metabolite in 1969.1 Since then, it has been developed as a chemotherapeutic for leukemia, lymphomas, sarcomas and solid breast, ovary and lung tumors and continues to be clinically useful.2 Doxorubicin also displays antiangiogenic properties when applied frequently in small doses.3 Doxorubicin is commonly used in combination therapy with other anticancer chemotherapeutics with different mechanisms of action. Doxorubicin is also commonly delivered in a pegylated liposome to enhance accumulation in tumors.4
The mechanism of doxorubicin cytotoxicity is not totally understood; however the following events likely play key roles: 1) induction of topoisomerase II-mediated DNA strand breaks and 2) induction of reactive oxygen species (ROS)-mediated lipid peroxidation.5–7 Despite the controversy about the mechanism of action, doxorubicin is commonly described as a topoisomerase II poison.8 Topoisomerases catalyze DNA unwinding for transcription and replication, which involves formation of an intermediate called the “cleavable complex.” Doxorubicin binds the topoisomerase II cleavable complex, resulting in double-strand DNA breaks.9 More recent studies have implicated formaldehyde in cancer cell growth inhibition by doxorubicin.10
Doxorubicin reacts with formaldehyde to form doxazolidine, a doxorubicin-formaldehyde conjugate that bears an oxazolidine ring at its amino sugar (Scheme 1). Further reaction yields the dimeric conjugate doxoform as shown in Scheme 1. On average, doxazolidine and doxoform inhibit the growth of several human cancer cell lines at 600-fold lower concentration than doxorubicin.11 Further, doxazolidine and doxoform inhibit human cancer cell line growth at 200-fold lower concentration than doxsaliform, a prodrug to an acyclic doxorubicin-formaldehyde conjugate (Scheme 2).11 Doxazolidine reacts with DNA at 5′-NGC-3′ sites in the minor groove to form a virtual crosslink of the DNA strands, where N is an unspecified base.11 A virtual crosslink involves intercalation of drug coupled with hydrogen bonding to one strand and covalent bonding to the other strand mediated by formaldehyde as shown in Scheme 1.12–19 Identical virtual crosslinks are formed from reaction of DNA with doxorubicin and formaldehyde, but more slowly.20 The high activity of doxazolidine and doxoform is proposed to result from direct virtual crosslinking of DNA with synchronous ring opening of the oxazolidine.11 The lower activity of doxsaliform, which releases an acyclic doxorubicin-formaldehyde conjugate, likely stems from loss of formaldehyde from the conjugate in competition with cyclization to doxazolidine as shown in Scheme 2.
Scheme 1.

Synthesis of doxazolidine and doxoform by reaction of doxorubicin with formaldehyde and virtual crosslinking of DNA at a 5′-NGC-3′ site by doxazolidine. N and N’ represent unspecified, complementary bases. The structure of the virtual crosslink is also shown with a three-dimensional model, with N equal to C and N’ equal to G, created using coordinates from Rutgers Protein Database (PDB code 1D33) for the Wang crystal structure 15 of the daunorubicin virtual crosslink of 5′-CGCGCG-3′ using Chem 3D and Pymol.
Scheme 2.

Formation and reaction of acyclic doxorubicin-formaldehyde conjugate from hydrolysis of doxsaliform.
Formation of doxazolidine from reaction of doxorubicin with formaldehyde, together with one cell-free and two cell-based observations led the Koch laboratory to propose that doxazolidine is an active metabolite of doxorubicin and that doxorubicin and doxazolidine share similar mechanisms of action. The cell-free observation was of iron-doxorubicin catalysis of formaldehyde formation using spermine as the carbon source and glutathione/dioxygen as a redox system.17,21 The second observation was a 3 mM elevation of formaldehyde concentration in doxorubicin- and daunorubicin-treated cancer cells.22,23 The third observation was that both doxorubicin and doxoform, induced apoptosis determined by both annexin V staining and DNA fragmentation measured by the TUNEL assay and gel electrophoresis.24 The difference in activity between doxazolidine and doxorubicin was explained by inefficiency in production of formaldehyde and subsequent formation of a doxorubicin-formaldehyde conjugate.24 The results presented here now suggest that doxazolidine induces cell death by a mechanism distinct from that of doxorubicin. The new data include a comparison of doxorubicin and doxazolidine with respect to 1) cell growth inhibition in cardiomyocytes and a wide variety of cancer cells, 2) cell cycle distribution and induction of apoptosis in topoisomerase II positive and deficient isogenic cell lines and 3) inhibition of topoisomerase II enzymatic activity.
Materials and Methods
Cell Lines and Chemicals
Human pancreatic carcinoma cell lines MiaPaCa-2 and BxPC3 were from David Ross (University of Colorado Health Sciences Center). Human prostate carcinoma cell lines were from Andrew Kraft (University of Colorado Health Sciences Center). Human non-small cell lung carcinoma cell line H2122 and human small cell lung carcinoma cell line SHP-77 were from Daniel Chan (University of Colorado Health Sciences Center). Human melanoma cell line A375 was from Natalie Ahn (University of Colorado). Human breast carcinoma cell line MDA-MB-43525 was from Renata Pasqualini (M.D. Anderson Cancer Center). Human breast carcinoma cell line MCF-7/ADR was from William Wells (Michigan State University). Human leukemic cell line HL-60 was from Kira Glover (University of Colorado Health Sciences Center). Human colon cancer cell line HCT-116 was from Bert Vogelstein (John Hopkins University). The mitoxantrone resistant and topoisomerase II deficient variant HL-60/MX2,26 human breast carcinoma cell line MCF-7, human liver carcinoma cell lines Hep G2 and SK-HEP-1 and rat cardiomyocyte cell line H9c2(2-1) were obtained from American Type Culture Collection (Rockville, MD). The MDA-MB-435 cell line was maintained in DMEM medium supplemented with 10% FBS, penicillin (100 units/mL), streptomycin (0.1 mg/mL), L-glutamine (2 mM), sodium pyruvate (1 mM), nonessential amino acids and vitamins. HCT-116 cell line was maintained in McCoy’s medium supplemented with 10% FBS, penicillin (100 units/mL) and streptomycin (0.1 mg/mL). SHP-77, PC-3, DU-145, BxPC3, MCF-7, MCF-7/ADR, HL-60 and HL-60/MX2 cell lines were maintained in RPMI 1640 medium supplemented with 10% FBS, L-glutamine (2 mM), HEPES buffer (10 mM), penicillin (100 units/mL) and streptomycin (0.1 mg/mL). MiaPaCa-2, A375, Hep G2 and SK-HEP-1 cell lines were maintained in DMEM supplemented with 10% FBS, penicillin (100 units/mL) and streptomycin (0.1mg/mL). Doxorubicin hydrochloride was from PolyMed Therapeutics (Houston, TX). Doxazolidine was synthesized as previously described.11
Growth Inhibition Assay
Growth inhibition was determined as previously described27 with minor modifications. All cell lines, except HL-60 and HL-60/MX2, were treated with drug for 3 h, then allowed to grow until control wells reached ~80% confluence (3–5 days). Cells were quantified by measuring crystal violet staining or cellular metabolism of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Promega, Madison, WI). The MTT and crystal violet assays are described in the literature.20,28–30 HL-60 and HL-60/MX2 cells were treated with drug for 48 h, fixed with 50 μL of 80% trichloroacetic acid for 1 h at 4 °C and quantified by crystal violet staining. For every experiment, each condition was done in hexuplicate; experiments were done at least twice.
Cell Cycle Distribution and Apoptosis Analyses
For both analyses, cells were treated with drug in appropriate media described in Cell Lines and Chemicals. HCT-116 cells were treated for 3 h and allowed to grow for an additional 21 or 45 h prior to analysis. HL-60 and HL-60/MX2 cells were treated for 24 or 48 h prior to analysis. For cell cycle distribution, adherent cells were trypsinized and harvested by centrifugation. Suspension cells were centrifuged. All cells were washed twice in cold PBS and resuspended in cold 2 mM EDTA in PBS. Cells were fixed in 50% ethanol, washed in cold 2 mM EDTA in PBS, treated with RNase A, and stained with propidium iodide at a final concentration of 50 μg/mL. For apoptotic index assays, adherent cells were trypsinized and harvested by centrifugation. Suspension cells were centrifuged. All cells were washed with cold PBS, resuspended in annexin binding buffer (140 mM NaCl, 10 mM HEPES pH 7.4 and 2.5 mM CaCl2) and stained with propidium iodide and annexin V- fluorescein (Molecular Probes, Eugene, OR) following the manufacturer’s instructions. Flow cytometric analyses were performed using a Beckton-Dickinson FACScan instrument.
Topoisomerase II Decatenation Assay
The assay was performed according to the protocol of TopoGen, Inc. (Port Orange, Florida). The total reaction volume was held at 20 μL. Assay buffer (120 mM KCl, 50 mM Tris-HCl, 10 mM MgCl2, 0.5 mM dithiothreitol, 0.5 mM ATP and 30 μg/mL BSA) containing 143 ng of catenated kinetoplast DNA(KDNA) and one unit of human recombinant topoisomerase II in the presence or absence of drug was incubated for 30 min at 37 °C. The reaction was stopped upon addition of 5 μL of stop buffer containing loading dye (5% glycerol, 1% sarkosyl and 0.025% bromphenol blue), then analyzed by agarose gel electrophoresis.
Results
Growth Inhibition of Cells by Doxorubicin and Doxazolidine
Doxorubicin and doxazolidine were evaluated for their growth inhibitory effects on thirteen human cancer cell lines and rat cardiomyocyte cell line H9c2(2-1). Cells were treated for 3 h and control wells allowed to grow to ~80% confluence (3–5 days). The results are summarized in Table I. On average, doxazolidine is over 100-fold more cytotoxic than doxorubicin, but equitoxic to cardiomyocytes. Preferential cancer cell cytotoxicity is an important result because the dose-limiting chronic side effect of doxorubicin therapy is cardiotoxicity.7 Doxazolidine was also evaluated by the National Cancer Institute Developmental Therapeutics Program (NCI-DTP) in their human cancer cell line screen. One cell line from each cell type is shown in Table II. The Developmental Therapeutics Program treats for 48 h with no additional growth period. On average, doxazolidine was over 80-fold more toxic than doxorubicin. Pooling of the National Cancer Institute’s data with our data resulted in doxazolidine being over 32-fold more toxic than doxorubicin to 67 human cancer cell lines, on average. Interestingly, using the National Cancer Institute’s compare feature on their website (http://dtp.nci.nih.gov/), we found that doxazolidine growth inhibition data correlate modestly (0.68) with doxorubicin growth inhibition data, but correlate best with data for the DNA crosslinking agents shown in Chart 1. In contrast, doxorubicin growth inhibition data correlate best with that of other anthracyclines and intercalating agents, also shown in Chart 1. These correlations suggest doxazolidine inhibits cancer cell growth more similarly to DNA crosslinking agents than to doxorubicin, namely by virtually crosslinking DNA. These data from NCI are reported here because IC50 data for doxoform/doxazolidine are currently proprietary to our group.
Table I.
IC50 data for growth inhibition of cancer cells by doxorubicin and doxazolidine. Cells were treated with drug for 3 h and growth was measured when controls reached 80% confluence.
| log IC50 (M) | |||
|---|---|---|---|
| Cell Line | Cancer Type | Doxorubicin | Doxazolidine |
| SHP-77 | S. Cell Lung | > −6.0 | −8.7 ± 0.07 |
| A375 | Melanoma | −6.7 ± 0.04 | −9.0 ± 0.02 |
| PC-3 | Prostate | −6.5 ± 0.14 | −8.0 ± 0.15 |
| DU-145 | Prostate | −6.6 ± 0.0744 | −8.5 ± 0.0744 |
| LNCaP | Prostate | −7.6 ± 0.0644 | −8.9± 0.0644 |
| MCF-7 | Breast | −6.543 | −8.5 ± 0.0343 |
| MCF-7/ADR | Breast | −5.2 ± 0.143 | −9.020 |
| MDA-MB-435 | Breast | −6.8 ± 0.745 | −8.0 ± 0.02 |
| Hep G2 | Liver | −6.7 ± 0.0943 | −8.0 ± 0.0843 |
| SK-HEP-1 | Liver | −7.0 ± 0.143 | −8.4 ± 0.143 |
| MiaPaCa-2 | Pancreas | −6.5 ± 0.09 | −8.5 ± 0.08 |
| BxPC-3 | Pancreas | −6.5 ± 0.14 | −8.0 ± 0.15 |
| HeLaS3 | Cervix | −7.424 | −8.524 |
| Mean | > −6.6 ± 0.14 | −8.5 ± 0.15a | |
| H9c2(2-1) | Heart | −7.5 ± 0.243 | −7.5 ± 0.243 |
p<0.000005 in comparison with doxorubicin.
Table II.
IC50 data for growth inhibition of cancer cells by doxorubicin and doxazolidine from the Developmental Therapeutics Program at the National Cancer Institute. Cells were treated with drug for 48 h.
| log IC50 (M) | |||
|---|---|---|---|
| Cell Line | Cancer Type | Doxorubicina | Doxazolidineb |
| HL-60(TB) | Leukemia | −7.23 | −9.48 |
| NCI-H460 | NSCLC | −8.22 | < −9.68 |
| HCT-116 | Colon | −7.15 | −8.25 |
| SF-268 | CNS | −7.04 | −8.58 |
| UACC-257 | Melanoma | −6.68 | < −8.82 |
| IGROV1 | Ovarian | −6.96 | < −8.86 |
| RXF 393 | Renal | −6.75 | < −9.01 |
| DU-145 | Prostate | −6.96 | −8.50 |
| MCF-7/ADR | Breast | −4.88 | < −7.73 |
| Mean | −6.87 | < −8.77c | |
Average of 1837 determinations with log maximum concentration (M) = −4.6.
Average of 2 determinations (RXF 393 data are from 1 determination) with log maximum concentration (M) = −6.0.
p<0.00001 in comparison with doxorubicin.
Chart 1.

Compare groups using growth inhibition data in the National Cancer Institute-Developmental Therapeutics Program database. The correlation number is given with the structure, and the NSC number for each compound is shown in parentheses. A generic name is also given when available. Further information about each compound can be found at the data search section of the NCI-DTP web site, http://dtp.nci.nih.gov, using the NSC number.
Effect of Doxorubicin and Doxazolidine on HCT-116 Colon Cancer Cells
Doxorubicin and doxazolidine were further evaluated with HCT-116 cells because the effect of doxorubicin on cell cycle distribution in these cells is well-documented.31 Cell cycle distribution was determined by flow cytometric analysis of propidium iodide-stained cells 24 h after a 3 h, 400 nM treatment. Doxorubicin induced a prominent G2/M arrest (Figure 1A). G2/M arrest is a well-documented effect of topoisomerase II inhibitors, such as doxorubicin.32 G2/M arrest is generally thought to provide additional time for repair of DNA lesions, increasing not only genomic integrity, but also cellular survival.32 Doxazolidine failed to arrest HCT-116 cells in G2/M (Figure 1A), but induced apoptosis instead, as evident by the increase in the sub G1 fraction compared to control cells. These data suggest that doxazolidine’s mechanism of cytotoxicity is topoisomerase-II independent. Apoptosis was also measured by staining with annexin V, which detects increased phosphatidyl-serine exposure in the outer leaflet of the cell membrane, an early event in apoptosis. Both doxorubicin and doxazolidine induced apoptosis in HCT-116 cells (Figure 1B); however, doxazolidine at 400 nM was much more effective, 59% versus 15% annexin V positive cells, respectively. Further, the percent of apoptotic cells from doxazolidine treatment was dose-dependent as shown in Figure 1C rising from 20% at 50 nM to 60% at 200 nM. Previous experiments, that included annexin V staining and DNA fragmentation detection by both the TUNEL assay and gel electrophoresis, confirm that both doxorubicin and doxazolidine induce apoptosis.24
Figure 1.
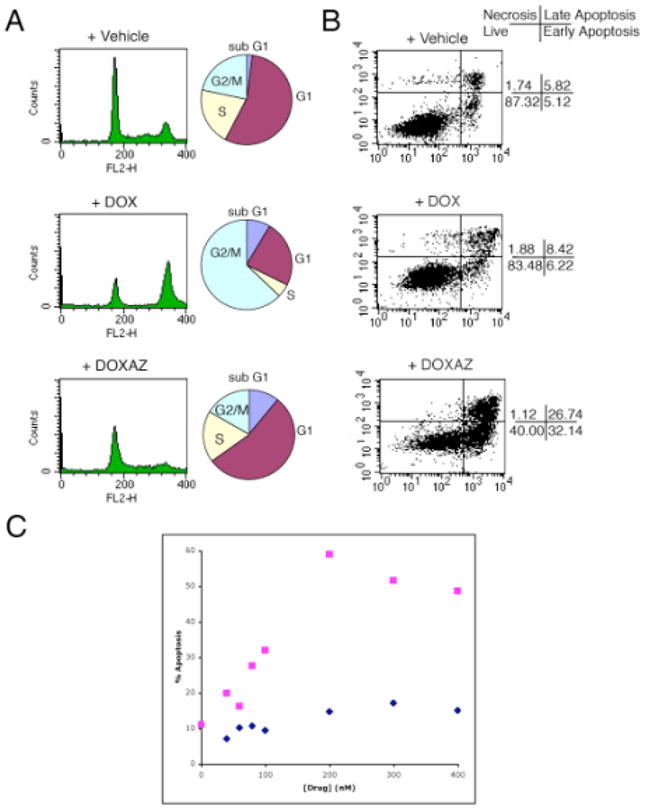
Cell cycle distribution and apoptotic index analyses of HCT-116 cells exposed to doxorubicin or doxazolidine. HCT-116 cells were treated with 400 nM doxorubicin (DOX), doxazolidine (DOXAZ) or vehicle for 3 h and analyzed for (A) cell cycle distribution at 24 post-treatment and (B) apoptosis at 48 h post-treatment. (C) HCT-116 cells were treated with various concentrations of doxorubicin ( ) and doxazolidine (
) and doxazolidine ( ) for 3 h and analyzed for apoptosis at 48 h post-treatment by annexin V staining.
) for 3 h and analyzed for apoptosis at 48 h post-treatment by annexin V staining.
Doxazolidine Induces Apoptosis by a Topoisomerase II-independent Mechanism
To further investigate the role of topoisomerase II in doxorubicin and doxazolidine cytotoxicity, we utilized the human leukemia HL-60 cell line and its topoisomerase II deficient HL-60/MX2 subline. HL-60/MX2 cells exhibit decreased topoisomerase IIα expression and sub-detectable levels of topoisomerase IIβ.26 The cells were evaluated for growth inhibition and induction of apoptosis by doxorubicin and doxazolidine (Figure 2). Our standard 3 h growth inhibition treatment protocol was not amenable for suspension cells, such as HL-60 and HL-60/MX2; so, the cells were treated for 48 h and assayed using a method similar to that employed by NCI (See Materials and Methods). For this reason, the growth inhibition data are not included in Table I or II, but shown in Figure 2. Again, doxazolidine is much more effective at inducing apoptosis than doxorubicin. In HL-60 cells, doxorubicin induced a 29% annexin V positive population and doxazolidine induced a 77% annexin V positive population. In HL-60/MX2 cells, doxorubicin induced a 5% annexin V positive population and doxazolidine induced a 75% annexin V positive population. Growth inhibition studies revealed that HL-60/MX2 cells were 8-fold resistant to doxorubicin relative to the parental cell line HL-60 (Figure 2). However, HL-60/MX2 cells are only 1.5 fold resistant to doxazolidine relative to the parental line. Both methods indicate that HL-60/MX2 cells are resistant to doxorubicin, but not to doxazolidine. Next, the cell cycle distributions of both cell lines were evaluated at 24 h post treatment (Figure 3). HL-60 cells treated with doxorubicin arrested in G2/M as has been reported previously.33 Doxorubicin-induced G2/M arrest seems to be dependent on topoisomerase II activity, as it is not observed in HL-60/MX2 cells. In contrast, doxazolidine induced robust apoptosis with no signs of G2/M arrest in both cell lines, indicating that this drug acts through topoisomerase II-independent mechanisms. Overall, these data are in agreement with the data obtained with the HCT-116 cell line and further support the conclusion that doxorubicin induces apoptosis via a topisomerase II-dependent mechanism, but doxazolidine induces apoptosis via a topoisomerase II-independent-mechanism.
Figure 2.
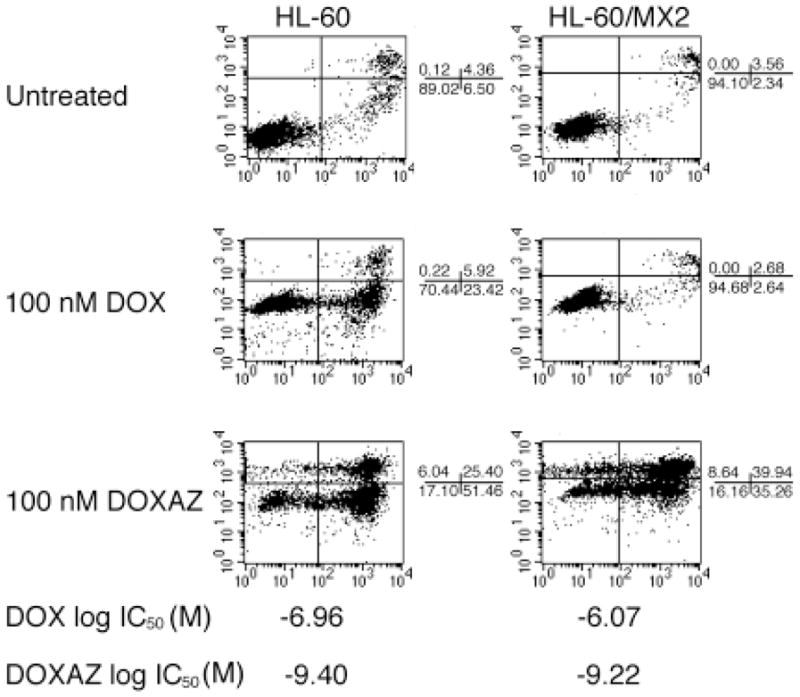
Apoptotic index and growth inhibition analyses of HL-60 and HL-60/MX2 cells. HL-60 and HL-60/MX2 cells were treated with 100 nM doxorubicin (DOX), doxazolidine (DOXAZ) or vehicle for 24 h and analyzed for apoptosis or for 48 h and analyzed for growth inhibition. HL-60 is a human leukemic cell line and HL-60/MX2 is a topoisomerase-II deficient subline of HL-60.
Figure 3.
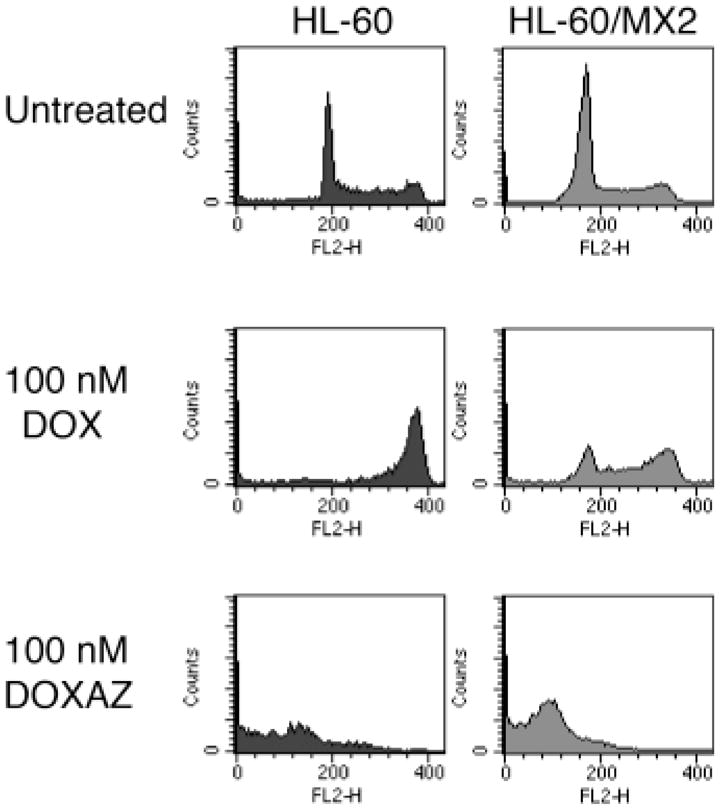
Cell cycle distribution analyses of HL-60 and HL-60/MX2 cells as a function of exposure to doxorubicin or doxazolidine. HL-60 and HL-60/MX2 cells were treated with 100 nM doxorubicin (DOX), doxazolidine (DOXAZ) or vehicle for 24 h and analyzed for cell cycle distribution. Cells lines are defined in the legend to Figure 2.
Antitumor activity of doxorubicin and other anthracyclines mainly results from inhibition of mammalian topoisomerase II.34,35 To investigate the inhibitory effects of doxorubicin and doxazolidine on the enzymatic activity of topoisomerase II, a decatenation assay was utilized. The decatenation assay36 is specific for both isoforms of topoisomerase II because it relies on the conversion of catenated DNA to its decatenated form, which requires double-strand breakage and subsequent strand rotation and ligation uniquely done by topoisomerase II. This decatenation can be detected by agarose gel electrophoresis. The results of topoisomerase II inhibition by doxorubicin and doxazolidine are shown in Figure 4. Doxorubicin inhibits topoisomerase II activity at both 500 and 50 nM. Doxazolidine only slightly inhibits topoisomerase II activity at both 500 and 50 nM. The slight inhibition of topoisomerase II activity can be attributed to the presence of doxorubicin from doxazolidine hydrolysis during the treatment period.11
Figure 4.
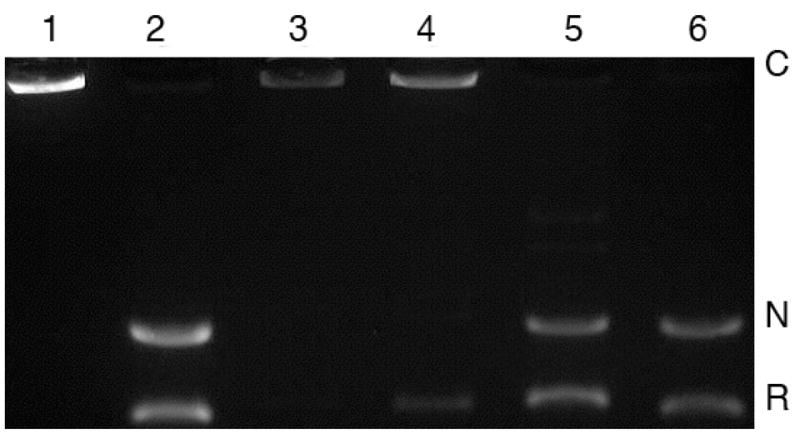
Effects of doxorubicin and doxazolidine on decatenation of KDNA (kinetoplast DNA) by topoisomerase II. Lane 1, KDNA without topoisomerase II (catenated form); lane 2, KDNA with topoisomerase II (decatenated form); lane 3, doxorubicin, 500 nM; lane 4, doxorubicin, 50 nM; lane 5, doxazolidine, 500 nM; lane 6, doxazolidine, 50 nM. C: catenated DNA; N: nicked, decatenated DNA; R: relaxed, decatenated DNA.
Discussion
Phillips and coworkers discovered that doxorubicin-DNA adducts induce a topoisomerase II-independent form of cell death.33 They formed doxorubicin-DNA adducts in cells by pre-incubation with a formaldehyde-releasing prodrug.33 Since this discovery and concomitant reevaluation of the National Cancer Institute’s growth inhibition data, namely the correlations between doxorubicin, doxazolidine and other DNA crosslinking agents, we reconsidered our view of doxazolidine as an active metabolite of doxorubicin and investigated the existence of different mechanisms for each anthracycline.
Doxazolidine Induces Apoptosis Via a Topoisomerase II-independent Mechanism
Both HCT-116 and HL-60 cells displayed different cell cycle responses when treated with doxazolidine compared to doxorubicin (Figures 1 and 3). Doxorubicin caused a pronounced G2/M arrest, which is topoisomerase II-dependent.32 Doxorubicin induces topoisomerase-II mediated DNA strand breaks, and G2/M arrest provides additional time for cells to increase genomic integrity and improve cellular survival rates.32 Doxazolidine-treated cells failed to arrest in G2/M, but still underwent dose-dependent apoptosis, as indicated by cell cycle distribution and annexin V staining analyses (Figures 1 and 3), suggesting a topoisomerase II-independent mechanism of action. Our own studies using HL-60/MX2 cells confirmed that the doxorubicin-induced G2/M arrest is at least partly topoisomerase II-dependent (Figure 3). Growth inhibition data indicate that HL-60/MX2 cells are resistant to doxorubicin but highly susceptible to doxazolidine, further establishing that doxazolidine induces apoptosis via a topoisomerase II-independent mechanism. Finally, doxorubicin inhibited topoisomerase II activity better than doxazolidine as measured with a topoisomerase II decatenation assay (Figure 4). A high level of decatenation was observed even at 500 nM doxazolidine and very little decatenation was observed at 500 nM doxorubicin. The small amount of inhibition by doxazolidine can be attributed to doxorubicin resulting from hydrolysis of doxazolidine to doxorubicin during the assay.11
Two Anthracyclines: Different Mechanisms
The discovery that doxorubicin and doxazolidine induce apoptosis by different mechanisms of action is surprising for three reasons. First and foremost, the structures of both drugs differ by only one carbon atom (Scheme 1). We are unaware of other natural products that increase potency and change mechanism of action upon addition of a single carbon atom to their molecular structure. However, other alkylating anthracyclines have been reported and shown to be more potent than doxorubicin.37–41 An example is the bioactivation product of nemorubicin.39,42 Nemorubicin, a non-alkylating anthracycline, is less than 3-fold more toxic than doxorubicin, but its bioactivation product, 1 (PNU-15968239), an alkylating anthracycline, is over three orders of magnitude more toxic than doxorubicin (Scheme 3).39 Second, increased potency of doxazolidine occurs in spite of a half-life with respect to hydrolysis to doxorubicin and formaldehyde of only 3 min.11 The rapid hydrolysis may lead one to believe that treatment with formaldehyde or co-treatment with doxorubicin and formaldehyde could produce the same effect as doxazolidine. However, cells treated with 400 nM formaldehyde displayed an identical cell cycle distribution as untreated cells (data not shown). In addition, doxorubicin and formaldehyde co-treatment (400 nM each) resulted in an identical cell cycle distribution as doxorubicin-treatment (data not shown). Third, doxazolidine does not induce an S phase accumulation in HL-60 or HL-60/MX2 cells. Phillips and coworkers observed an S phase accumulation in doxorubicin treated HL-60 and HL-60/MX2 cells pretreated with a formaldehyde releasing prodrug.33 This combination treatment is thought to cause doxorubicin-DNA adducts as confirmed by [14C]doxorubicin experiments, but not topoisomerase II-mediated DNA strand breaks as confirmed by the comet assay.33 Doxazolidine induces apoptosis much better than the formaldehyde releasing prodrug and doxorubicin combination, suggesting that doxazolidine is better at doxorubicin-DNA virtual crosslink formation. This is consistent with anticipated kinetics because doxazolidine, as a single compound, is prepared to virtually crosslink DNA.11 In contrast, the formaldehyde releasing prodrug and doxorubicin combination first requires the doxorubicin and the formaldehyde released from the prodrug to find each other and then the resulting conjugate to crosslink DNA. The increased tumor cell growth inhibition activity may also result from doxazolidine being a poorer substrate for ABC transporters. This is evident from an earlier comparison of growth inhibition of MCF-7 and resistant MCF-7/Adr cells by doxorubicin and doxazolidine. MCF-7/Adr cells are 50-fold resistant to doxorubicin in large part because of over-expression of the P-170 glycoprotein efflux pump but are equally sensitive to doxazolidine.11 Doxazolidine may be a poorer substrate for P-170 glycoprotein because it is not a cation at physiological pH and may be less available to P-170 glycoprotein because it rapidly crosslinks DNA.
Scheme 3.

Structure of nemorubicin and its oxidative activation to the DNA alkylating derivative 1.
Doxazolidine Therapeutic Applications
Because doxazolidine is highly potent and hydrolytically unstable, it should be delivered locally or targeted to tumor cells or neovasculature in a stable, inactive form. Doxazolidine is stable and inactive when derivatized as an N-carbamate. Further conjugation of the carbamate to enzyme substrates allows for specific activation by both endogenous and exogenous enzymes.43 An example of a hydrolytically stable, nontoxic doxazolidine conjugate targeted to cancer cells that overexpress a carboxylesterase enzyme was recently reported. 43 Other enzymes currently being evaluated for activation of doxazolidine prodrugs at the site of tumors are plasmin and carboxypeptidase G2. Plasmin is of particular interest because of its role in metastatic tumor cell invasion of new tissue and subsequent tumor angiogenesis.
Acknowledgments
We thank the Developmental Therapeutics Program of the National Cancer Institute for testing doxoform/doxazolidine in their human tumor cell line screen. B.T.K. and T.H.K. were supported by NIH-NCI Grant # R01CA-92107. J.M.E. was supported by DOD-CDMRP Grant # CM50054, NIH-NCI Grant # 1R01CA117907-01, Basil O’Connor-March of Dimes Grant #5-FY05-1217 and SPORE in Lung Cancer. M.B.M. was supported by the Undergraduate Research Opportunity Program at CU-Boulder.
Non-standard abbreviations
- DOX
doxorubicin
- DOXAZ
doxazolidine
- KDNA
kinetoplast DNA
- N,N’
unspecified DNA base pair
- NCI-DTP
National Cancer Institute Developmental Therapeutics Program
- ROS
reactive oxygen species
References
- 1.Di Marco A, Gaetani M, Scarpinato B. Adriamycin (NSC-123,127): a new antibiotic with antiumor activity. Cancer Chemother Rep (Part I) 1969;53:33–37. [PubMed] [Google Scholar]
- 2.Young RC, Ozols RF, Myers CE. Medical Progress: The anthracycline antineoplastic drugs. New Engl J Med. 1981;305:139–153. doi: 10.1056/NEJM198107163050305. [DOI] [PubMed] [Google Scholar]
- 3.Bocci G, Nicolaou K, Kerbel R. Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res. 2002;62:6938–6943. [PubMed] [Google Scholar]
- 4.Gabizon A, Shmeeda H, Zalipsky S. Pros and Cons of the Liposome Platform in Cancer Drug Targeting. J Liposome Res. 2006;16:175–183. doi: 10.1080/08982100600848769. [DOI] [PubMed] [Google Scholar]
- 5.Rajski SR, Williams RM. DNA interstrand cross-linking agents as antitumor drugs. Chem Rev. 1998;98:2723–2796. doi: 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y. Anthracycline Antibiotics: New Analogues, Methods of Delivery, and Mechanisms of Action. American Chemical Society; Washington, DC: 1995. DNA topoisomerases and their inhibition by anthracyclines; pp. 183–203. [Google Scholar]
- 7.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 8.DeVita VT, Hellman S, Rosenberg SA. Cancer: Principles and Practice of Oncology. 6. Lippincott, Williams and Wilkins; Philadelphia: 2001. [Google Scholar]
- 9.Liu LF, Rowe TC, Yang L, Tewey KM, Chen GC. Cleavage of DNA by mammalian DNA topoisomerase II. J Biol Chem. 1983;258:15365–15370. [PubMed] [Google Scholar]
- 10.Cutts SM, Nudelman A, Rephaeli A, Phillips DR. The power and potential of doxorubicin-DNA adducts. IUBMB Life. 2005;5:73–81. doi: 10.1080/15216540500079093. [DOI] [PubMed] [Google Scholar]
- 11.Post GC, Barthel BL, Burkhart DJ, Hagadorn JR, Koch TH. Doxazolidine, a proposed active metabolite of doxorubicin that cross-links DNA. J Med Chem. 2005;48:7648–7657. doi: 10.1021/jm050678v. [DOI] [PubMed] [Google Scholar]
- 12.Cullinane C, van Rosmalen A, Phillips DR. Does adriamycin induce interstrand cross-links in DNA? Biochemistry. 1994;33:4632–4638. doi: 10.1021/bi00181a025. [DOI] [PubMed] [Google Scholar]
- 13.Cullinane C, Cutts MS, van Rosmalen A, Phillips DR. Formation of Adriamycin-DNA adducts in vitro. Nucleic Acids Res. 1994;22:2296–2303. doi: 10.1093/nar/22.12.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skladanowski A, Konopa J. Interstrand DNA crosslinking induced by anthracyclines in tumour cells. Biochem Pharmacol. 1994;47:2269–2278. doi: 10.1016/0006-2952(94)90265-8. [DOI] [PubMed] [Google Scholar]
- 15.Wang AHJ, Gao YG, Liaw YC, Li YK. Formaldehyde cross-links daunorubicin and DNA efficiently: HPLC and X-ray diffraction studies. Biochemistry. 1991;30:3812–3815. doi: 10.1021/bi00230a002. [DOI] [PubMed] [Google Scholar]
- 16.Leng F, Savkur R, Fokt I, Przewloka T, Priebe W, et al. Base specific and regiospecific chemical cross-linking of daunorubicin to DNA. J Am Chem Soc. 1996;118:4731–4738. [Google Scholar]
- 17.Taatjes DJ, Gaudiano G, Resing K, Koch TH. A redox pathway leading to the alkylation of DNA by the anthracycline, anti-tumor drugs, adriamycin and daunomycin. J Med Chem. 1997;40:1276–1286. doi: 10.1021/jm960835d. [DOI] [PubMed] [Google Scholar]
- 18.Zeman SM, Phillips DR, Crothers DM. Characterization of covalent Adriamycin-DNA adducts. Proc Natl Acad Sci USA. 1998;95:11561–11565. doi: 10.1073/pnas.95.20.11561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Podell ER, Harrington DJ, Taatjes DJ, Koch TH. Crystal structure of epidoxorubicin-formaldehyde virtual crosslink of DNA and evidence for its formation in human breast-cancer cells. Acta Cryst. 1999;D55:1516–1523. doi: 10.1107/s0907444999008161. [DOI] [PubMed] [Google Scholar]
- 20.Fenick DJ, Taatjes DJ, Koch TH. Doxoform and Daunoform: Anthracycline-formaldehyde conjugates toxic to resistant tumor cells. J Med Chem. 1997;40:2452–2461. doi: 10.1021/jm970237e. [DOI] [PubMed] [Google Scholar]
- 21.Taatjes DJ, Gaudiano G, Koch TH. Production of formaldehyde and DNA-adriamycin or -daunomycin adducts, initiated through redox chemistry of DTT/iron, xanthine oxidase/NADH/iron, or glutathione/iron. Chem Res Toxicol. 1997;10:953–961. doi: 10.1021/tx970064w. [DOI] [PubMed] [Google Scholar]
- 22.Kato S, Burke PJ, Fenick DJ, Taatjes DJ, Bierbaum VM, et al. Mass spectrometric measurement of formaldehyde generated in breast cancer cells upon treatment with anthracycline antitumor drugs. Chem Res Toxicol. 2000;13:509–516. doi: 10.1021/tx000008m. [DOI] [PubMed] [Google Scholar]
- 23.Kato S, Burke PJ, Koch TH, Bierbaum VM. Formaldehyde in human cancer cells: detection by preconcentration-chemical ionization mass spectrometry. Anal Chem. 2001;73:2992–2997. doi: 10.1021/ac001498q. [DOI] [PubMed] [Google Scholar]
- 24.Burke P, Koch T. Doxorubicin-formaldehyde conjugate, Doxoform: Induction of apoptosis relative to doxorubicin. Anticancer Res. 2001;21:2753–2760. [PubMed] [Google Scholar]
- 25.Calileau R, Olive M, Cruciger QV. Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro. 1978;14:911–915. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- 26.Harker W, Dlade D, Parr R, Feldhoff P, Sullivan D, et al. Alterations in the topoisomerase II alpha gene, messenger RNA, and subcellular protein distribution as well as reduced expression of the DNA topoisomerase II beta enzyme in a mitoxantrone-resistant HL-60 human lekemia cell line. Cancer Res. 1995;55:1707–1716. [PubMed] [Google Scholar]
- 27.Burkhart DJ, Kalet BT, Koch TH. Doxorubicin-formaldehyde conjugates targeting alpha-v beta-3 integrin. Mol Cancer Therap. 2004;3:1593–1604. [PubMed] [Google Scholar]
- 28.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 29.Gilles RJ, Didier N, Denton M. Determination of cell number in monolayer cultures. Anal Biochem. 1986;159:109–113. doi: 10.1016/0003-2697(86)90314-3. [DOI] [PubMed] [Google Scholar]
- 30.Reile H, Birnböck F, Bernhardt G, Spruss T, Schönenberger H. Computerized determination of growth kinetic curves and doubling times from cells in microcultures. Anal Biochem. 1990;187:262–267. doi: 10.1016/0003-2697(90)90454-h. [DOI] [PubMed] [Google Scholar]
- 31.Bunz F, Hwang P, Torrance C, Waldman T, Zhang Y, et al. Disruption of p53 in human cancer cells alters the repsonses to therapeutic agents. J Clin Invest. 1999;104:263–269. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larsen AE, Skladanowski AE. A From DNA Damage to G2 Arrest: the many roles of topoisomerase II. Prog Cell Cycle Res. 2003;5:295–300. [PubMed] [Google Scholar]
- 33.Swift LP, Rephaeli A, Nudelman A, Phillips DR, Cutts SM. Doxorubicin-DNA Adducts Induce a Non-Topoisomerase II-Mediated Form of Cell Death. Cancer Res. 2006;66:4863–4871. doi: 10.1158/0008-5472.CAN-05-3410. [DOI] [PubMed] [Google Scholar]
- 34.Liu LF. DNA topoisomerase poisons as antitumor drugs. Ann Rev Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- 35.Asche C. Antitumor Quinones. Mini-Rev Med Chem. 2005;5:449–467. doi: 10.2174/1389557053765556. [DOI] [PubMed] [Google Scholar]
- 36.Rhee H, Park HJ, Lee SK, Lee C, Choo HP. Synthesis, cytotoxicity, and DNA topoisomerase II inhibitory activity of benzoquinolinediones. Bioorg Med Chem. 2007;15:1651–1658. doi: 10.1016/j.bmc.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 37.Acton EM, Tong GL, Taylor DL, Filippi JA, Wolgemuth RL. New cyanomorpholinyl byproduct of doxorubicin reductive alkylation. J Med Chem. 1986;29:1225–1230. doi: 10.1021/jm00157a019. [DOI] [PubMed] [Google Scholar]
- 38.Priebe W, Przewloka T, Fokt I, Ling Y-H, Perez-Soler R. Methods and Compositions for the Manufacture of Highly Potent Anthracycline-based. Antitumor Agents; US: 2002. [Google Scholar]
- 39.Quintieri L, Geroni C, Fantin M, Battaglia R, Rosato A, et al. Formation and Antitumor Activity of PNU-159682, A Major Metabolite of Nemorubicin in Human Liver Microsomes. Clin Cancer Res. 2005;11:1608–1617. doi: 10.1158/1078-0432.CCR-04-1845. [DOI] [PubMed] [Google Scholar]
- 40.Uchida T, Imoto M, Takahashi Y, Odagawa A, Sawa T, et al. New potent anthracyclines, barminomycins I and II. J Antibiot. 1988;41:404–408. doi: 10.7164/antibiotics.41.404. [DOI] [PubMed] [Google Scholar]
- 41.Nagy A, Armatis P, Schally AV. High yield conversion of doxorubicin to 2-pyrrolinodoxorubicin, an analog 500–1000 times more potent: Structure-activity relationship of daunosamine-modified derivatives of doxorubicin. Proc Natl Acad Sci USA. 1996;93:2464–2469. doi: 10.1073/pnas.93.6.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cherif A, Farquhar D. N-(5,5-Diacetoxypent-1-yl)doxorubicin: A New Intensely Potent Doxorubicin Analogue. J Med Chem. 1992;35:3208–3214. doi: 10.1021/jm00095a017. [DOI] [PubMed] [Google Scholar]
- 43.Burkhart D, Barthel BL, Post GC, Kalet BT, Nafie JW, et al. Design, Synthesis, and Preliminary Evaluation of Doxazolidine Carbamates as Prodrugs Activated by Carboxylesterases. J Med Chem. 2006;49:7002–2012. doi: 10.1021/jm060597e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taatjes DJ, Koch TH. Growth inhibition, nuclear uptake, and retention of anthracycline-formaldehyde conjugates in prostate cancer cells relative to clinical anthracyclines. Anticancer Res. 1999;19:1201–1208. [PubMed] [Google Scholar]
- 45.Burke P, Kalet B, Koch T. Antiestrogen binding site (AEBS) and estrogen receptor (ER) mediate uptake and distribution of 4-hydroxytamoxifen-targeted doxorubicin-formaldehyde conjugate in breast cancer cells. J Med Chem. 2004;47:6509–6518. doi: 10.1021/jm049496b. [DOI] [PubMed] [Google Scholar]