

Abstract
The membrane-bound mucins, MUC17 (human) and Muc3 (mouse), are highly expressed on the apical surface of intestinal epithelia and are thought to be cytoprotective. The extracellular regions of these mucins contain EGF-like Cys-rich segments (CRD1 and CRD2) connected by an intervening linker domain (L). The purpose of this study was to determine the functional activity of human MUC17 membrane-bound mucin.
Methods
Endogenous MUC17 was inhibited in LS174T colon cells by stable transfection of a small hairpin RNA targeting MUC17 (LSsi cells). The effect of recombinant MUC17-CRD1-L-CRD2 protein on migration, apoptosis, and experimental colitis was determined.
Results
Reduced MUC17 expression in LSsi cells was associated with visibly reduced cell aggregation, reduced cell–cell adherence, and reduced cell migration, but no change in tumorigenicity. LSsi cells also demonstrated a 3.7-fold increase in apoptosis rates compared with control cells following treatment with etoposide. Exposure of colonic cell lines to exogenous recombinant MUC17-CRD1-L-CRD2 protein significantly increased cell migration and inhibited apoptosis. As a marker of biologic activity, MUC17-CRD1-L-CRD2 proteins stimulate ERK phosphorylation in colonic cell lines; and inhibition of ERK phosphorylation reduced the anti-apoptosis and migratory effect of MUC17-CRD1-L-CRD2. Finally, mice treated with MUC17-CRD1-L-CRD2 protein given per rectum demonstrated accelerated healing in acetic acid and dextran sodium sulfate induced colitis in vivo. These data indicate that both native MUC17 and the exogenous recombinant cysteine-rich domain of MUC17 play a role in diverse cellular mechanisms related to cell restitution, and suggest a potential role for MUC17-CRD1-L-CRD2 recombinant protein in the treatment of mucosal inflammatory diseases.
Keywords: Mucin, Cell migration, Inflammatory bowel disease, MUC17, Epidermal growth factor
1. Introduction
Mucin-type proteins, categorized as secretory or membrane bound (1), are located at the interface of epithelial surfaces and the environment. These proteins are thought to be integral to the epithelial defense of respiratory, digestive, ocular, and reproductive surfaces. The membrane-bound mucins are characterized by an extracellular region composed of a short amino terminal domain, followed by a large, heavily O-glycosylated tandem repeat domain which accounts in part for their protective function. This large (>4000 amino acids) structural domain is followed by a globular region just proximal to the membrane that is made up of two Cys-rich domains (CRD1 and CRD2), each with similarity to epidermal growth factor (EGF)-like motifs, separated by a linker (L) domain containing a SEA (sea-urchin sperm protein, enterokinase and agrin) module. C-terminal to the CRD1-L-CRD2 unit is a transmembrane segment followed by a small cytoplasmic domain. An overview of this structure of MUC17 is shown schematically in Fig. 1. The related membrane-bound mucin genes MUC3A/B, MUC12, and MUC17 are clustered on chromosome 7q22, and are highly expressed in intestinal tissues at the apical surface of enterocytes. The mouse Muc3 gene (Shekels et al., 1998) is most similar in sequence and chromosomal localization to the human MUC17 gene (Gum et al., 1997; Crawley et al., 1999; Williams et al., 1999).
Fig. 1.
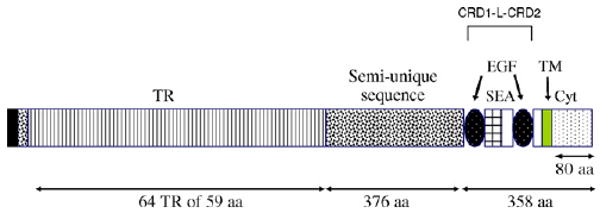
Schematic representation of MUC17 protein structures. The MUC17 amino acid sequence is comprised of a signal peptide, a large tandemly repeated central domain (TR), two EGF-like domains, a SEA domain, a transmembrane domain (TM), and an 80 amino acid cytoplasmic tail (Cyt). The recombinant proteins used are derived from the two Cys-rich domains (CRD1 and CRD2), separated by a linker (L) domain which includes the SEA domain. On this figure CRD1 corresponds to the first EGF-like domain, the linker region corresponds to the next domain that includes the SEA module, and CRD2 corresponds to the second EGF-like domain.
We have previously shown that GST-tagged recombinant mouse Muc3-CRD1-L-CRD2 (also termed Muc3 EGF1, 2) inhibits cellular apoptosis and accelerates cell migration over surfaces in vitro; and also promotes healing in mouse models of ulcerative colitis (Ho et al., 2006). The Muc3-GST-CRD1-L-CRD2 protein does not directly activate EGF receptors, but the specific mechanisms responsible for the actions of the mucin-derived recombinant proteins have not been determined to date. These findings constitute the rationale for studying the function of endogenous membrane-bound MUC17 mucin and the testing of a human recombinant mucin protein containing a bivalent display of Cys-rich EGF-like CRD domains for potential therapeutic use.
In the present work, we sought to determine: (1) if endogenous human MUC17 demonstrated biologic activity related to cell restitution in vitro; (2) if exogenous administration of recombinant human MUC17-CRD1-L-CRD2 proteins stimulated cell restitution processes in vitro; and (3) determine if recombinant human MUC17-CRD1-L-CRD2 enhances healing of experimental colitis in vivo. The findings from these studies begin to describe a CRD1-L-CRD2 unit from human MUC17 that is a candidate for further development as a potential mucosal restitution agent for the treatment of inflammatory bowel disease and other mucosal disorders.
2. Materials and methods
2.1. Recombinant proteins
Two MUC17 recombinant proteins were synthesized with either a GST or poly-His tag, as described previously (Ho et al., 2010). The first is labeled human MUC17-CRD1-Linker (L)-CRD2-GST (R-1 through S-260) containing a N-terminal GST tag, with the sequence: (glutathione-S-transferase)-R T T T C F G D G C Q N T A S R C K N G G T W D G L K C Q C P N L Y Y G E L C E E V V S S I D I G P P E T I S A Q M E L T V T V T S V K F T E E L K N H S S Q E F Q E F K Q T F T E Q M N I V Y S G I P E Y V G V N I T K L R L G S V V V E H D V L L R T K Y T P E Y K T V L D N A T E V V K E K I T K V T T Q Q I M I N D I C S D M M C F N T T G T Q V Q N I T V T Q Y D P E E D C R K M A K E Y G D Y F V V E Y R D Q K P Y C I S P C E P G F S V S K N C N L G K C Q M S L S G P Q C L C V T T E T H W Y S G E T C N Q G T Q K S. The extracellular, globular region of MUC17-CRD1-L-CRD2 coding region was amplified from human intestinal cDNA, cloned into the pGEX-2TK vector (Amersham, Piscataway, NJ), sequenced, and introduced into E. coli strain BL21 (Invitrogen, Carlsbad, CA). GST-fusion proteins were then expressed in E. coli by induction with 0.5 mM isopropylthio-beta-d-galactoside (IPTG; Fisher, Pittsburgh, PA) and purified by affinity chromatography using glutathione agarose (Sigma Chemical Co., St. Louis, MO), as described (Ho et al., 2010).
2.1.1. His-tagged fusion proteins
The second recombinant protein was labeled human MUC17-CRD1-L-CRD2His8 (R-1 through K-259) with a 5-residue extension (shown in low case), followed by a C-terminal 8-His tag, with the sequence: M R T T T C F G D G C Q N T A S R C K N G G T W D G L K C Q C P N L Y Y G E L C E E V V S S I D I G P P E T I S A Q M E L T V T V T S V K F T E E L K N H S S Q E F Q E F K Q T F T E Q M N I V Y S G I P E Y V G V N I T K L R L G S V V V E H D V L L R T K Y T P E Y K T V L D N A T E V V K E K I T K V T T Q Q I M I N D I C S D M M C F N T T G T Q V Q N I T V T Q Y D P E E D C R K M A K E Y G D Y F V V E Y R D Q K P Y C I S P C E P G F S V S K N C N L G K C Q M S L S G P Q C L C V T T E T H W Y S G E T C N Q G T Q K slvyg H H H H H H H H. The MUC17-CRD1-L-CRD2 coding region was amplified as previously described (Ho et al., 2010). The PCR product was cloned into pCR2.1 and sequenced. The Muc17-CRD1-L-CRD2 coding sequence was subcloned into pET28a using NcoI and XhoI to create pET28 Muc17. This construct expresses MUC17-CRD1-L-CRD2 with His6 tags on both the N and C termini. This plasmid was transformed into BL21(DE3) bacteria. Freshly transformed BL21(DE3) isolates were inoculated and grown overnight in 5 ml of LB + 100 μg/ml kanamycin then induced with 1 mM IPTG. Induced cells were collected by centrifugation and lysed by sonication in lysis buffer (50 mM Tris (pH 8.0), 100 mM NaCl, 5 mM EDTA, 0.5% (v/v) Triton-X-100, 0.1% (v/v) 2-ME, 100 μM PMSF). The lysate was centrifuged and the insoluble fraction (inclusion bodies) was washed in lysis buffer. The resultant insoluble protein was resuspended in PS buffer (50 mM Na2HPO4, pH 7.6, containing 100 mM NaCl) by sonication and then dissolved with the addition of urea to 6 M (PSU buffer). The solubilized protein was next purified by binding to PSU equilibrated Ni-NTA resin and eluted with PSU + 0.5 M imidizole. The protein was re-folded by dialyzing in a 5kDa molecular weight cut-off membrane against 100 volumes of 50 mM Tris (pH 8.2) and 137 mM NaCl with three changes of buffer. The yield of proteins synthesized in E. coli ranged from 40 to 50 mg/l. The proteins were ∼95% pure based on SDS-PAGE analysis and N-terminal sequencing. In addition, recombinant proteins were endotoxin-purified using Detoxi-Gel Endotoxin Removal Gel (Pierce, Rockford, IL), which did not alter the activity of the proteins. We have previously shown that MUC17-CRD1-L-CRD2 recombinant protein is cleaved at the SEA domain within the linker region, and is reassociated when purified. The GST- and 8-His-tagged proteins have molecular weights of 45–50 and 30–35 kDa, respectively, on reducing gels, and have been shown to have similar activity (Ho et al., 2010).
2.2. Cell lines and cell culture
Several types of colonic cells lines were used for these experiments. LoVo cells are a human colon cancer cell line known to respond to Muc3CRD protein as described previously (Ho et al., 2006), and express ErbB1 and low level ErbB2 receptors (Magne et al., 2002; Nyati et al., 2004). Colon cell lines commonly used as models of “normal” colon cells were used, including the intestinal cell line IEC-6 (American Type Culture Collection (Manassas, VA)) and the Young Adult Mouse Colon (YAMC) cell line (Ho et al., 2006). YAMC are conditionally immortalized mouse colon cells grown in RPMI 1640 supplemented with 5% FCS + 50 U penicillin/ml and 0.05 μg streptomycin/ml (Invitrogen, Carlsbad, CA), as described previously (Kaiser and Polk, 1997; Frey et al., 2004). YAMC cells have previously been shown to respond to Muc3CRD protein and are known to express EGF-type receptors (Ho et al., 2006). Cells were grown in 24-well plates for cell migration and proliferation experiments or T-25 flasks for immunoblotting experiments. Lovo and IEC-6 cells were grown using DMEM supplemented with 10% fetal calf serum + 50 U penicillin/ml and 0.05 μg streptomycin/ml (Invitrogen, Carlsbad, CA). Cells were cultured at 37 °C, 5% CO2, 10% FCS until the desired confluence was reached. The monolayers were washed with PBS 24 h before experiments and switched to serum-free media for cell migration and immunoblotting experiments.
2.3. Antibodies
Anti-ERK 1 (C-16) and anti-phospho-ERK (E-4) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). The antibody against a MUC17 core protein (M17TR) was used as described previously (Moniaux et al., 2006). Antibody against β-actin purchased from Invitrogen (Carlsbad, CA).
2.4. Immunoblots
Cells were cultured on 6-well plates until confluency. Cells were then serum starved overnight prior to treatment with recombinant mucin proteins, EGF, or BSA. After designated time points total protein was extracted from cells with RIPA buffer (150 mM NaCl, 1.0% NP-40, 50 mM Tris base (pH 8.0)) with the addition of Phosphatase Inhibitor Cocktail Set V (sodium fluoride, sodium orthovanadate, sodium pyrophosphate, decarbohydrate, β-glycerophosphate) (Calbiochem, San Diego, CA). After quantification by DC Protein Assay (Bio-Rad, Hercules, CA) equal amounts of total protein were resolved by 10% SDS-PAGE and transferred to polyvinylidene difluoride membrane (Millipore, Billerica, MA). Membranes were blocked with 5% nonfat milk in Tris-buffered saline (TBS) (0.137 M NaCl, 2.68 mM KCl, 24.7 mM Tris base (pH 8.0)) for 1 h at room temperature. The immunoblots were then incubated overnight at 4 °C with primary antibodies in TBS plus 0.1% Tween-20 (TBST) and washed three times with TBST. The immunoblots were then incubated for 1 h at room temperature with secondary antibody and washed three times with TBST before being visualized by ECL (ThermoScientific, Rockford, IL).
2.5. Reverse transcriptase PCR
Cells were seeded onto T25 flasks, n = 3. When cells reached close to 85% confluency, RNA was extracted with TRI Reagent (Ambion, Austin, TX) and then treated with TurboDNase (Ambion, Austin, TX). RNA extract was reverse transcribed with M-MLV (Invitrogen, San Diego, CA) and then screened for MUC17 with PCR. Forward primer: GGG CCA GCA TAG CTT CGA. Reverse primer: GCT ACA GGA ATT GTG GGA GTT CA (Russo et al., 2006). PCR product was viewed by gel electrophoresis.
2.6. Wound healing assay
Cells were seeded onto 6-well plates, coated with poly-L-lysine, and cultured until confluent. When cells were 90% confluent, an area of cells was scraped with a razor, producing a sharp, clean scrape. Several scrapes were made per well. Cells surrounding the scrape were allowed to migrate over the scrape over 48 h in complete media. Cells were then fixed and stained with Dip Quick (Jorgensen Laboratories, Loveland, CO). For Lovo cells the number of cells migrated across the scrape were counted at 400×. For LS174T cells photomicrographs were made at 1600× and area of cell migration determined using NIH ImageJ program (http://rsbweb.nih.gov/ij/). For inhibition experiments, cells were pretreated with the ERK inhibitor, U0126 (Calbiochem, San Diego, CA), when indicated, for 1 h in fresh media followed by treatment with recombinant proteins.
2.7. Apoptosis assay
Apoptosis was induced by incubating cells with either interferon gamma 100 ng/ml for 24 h followed by removal of the interferon and the addition of anti-Fas antibody at 500 ng/ml for 48 h (R&D Systems, Minneapolis, MN) (Quirk et al., 1998), or 125 μM etoposide (Calbiochem, San Diego, CA). Lovo cells were previously shown to be sensitive to anti-Fas treatment (Ho et al., 2006), and LS174T cells were previously shown to be sensitive to etoposide (Kouniavsky et al., 2002). Cells were pretreated with or without recombinant proteins 1 h prior to addition of anti-Fas or etoposide. Cells were fixed in 4% paraformaldehyde in PBS pH 7.4 for 5 min, then washed twice in PBS. The cells were stained with the nuclear dye, Hoechst 33258 (Polysciences Inc., Warrington, PA), at a concentration of 5 μg/ml in PBS for 30 min, rinsed, cover-slipped with Slowfade Antifade (Molecular Probes, Eugene, OR), and then immediately imaged using an fluorescent microscope. Apoptotic nuclei were identified by morphology (Fan et al., 1996). The total number of normal and apoptotic nuclei were counted in three 400× lens fields per dish (representing >200 nuclei per dish). Three or more dishes were used for each experimental condition. In separate experiments LS174T cells were seeded at 1 × 106 cells/well in 6-well plates. After 24 h treatment with or without etoposide all cells, floating and attached, were collected and treated with Annexin V-FITC and propidium iodide. These were then submitted for flow cytometry analysis. In addition, LS174T cells were seeded at 5 × 104 cells/well in 8-well glass chamber slides. After 24 h incubation at 37 °C cells were treated with 125 μm etoposide for another 24 h. Cells were then fixed with 4% paraformaldehyde and stained with Hoescht dye. Cells were imaged at 400× at random fields with a fluorescent microscope. The number of apoptotic cells was compared to the total number of cells to find the percent of apoptotic cells.
2.8. Endogenous MUC17 inhibition
The human colonic cell line LS174T was chosen because of high baseline expression levels of MUC17 compared to other colonic cells lines, such as HT29, Caco2, LoVo, HCT15, SW480, WiDr, and Colo 205 (S. Batra et al., unpublished observations). The LS174T cells were stably transduced with a small hairpin RNA sequence, cloned and expressed in a retroviral expression vector pSUPER.Retro (OligoEngine, Inc., Seattle, WA) secreted from Phi-NX cells (293T cell-derived), targeting MUC17 expression, as described previously (Chaturvedi et al., 2007). The MUC17-specific sequence was determined using the search algorithm of Dharmacon's siDESIGN® Center (http://www.dharmacon.com/DesignCenter/DesignCenterPage.aspx, Dharmacon, Lafayette, CO) based on the previously reported sequence (Moniaux et al., 2006). Multiple controls were used to test the efficacy of the MUC17 inhibition. This included using multiple targeting sequences that resulted in knock down of MUC17 message and protein, quantification of knockdown of both MUC17 mRNA and protein, and the use of a nonsense scrambled small hairpin RNA sequence for an experimental control. Note that the MUC17 mRNA is too large to feasibly transfect to “rescue” the cellular changes caused by the interfering RNA. Two different sequences were used to investigate knock down of MUC17 message and protein. Finally, a 19 nucleotide target sequence 5′-ataccaacctcgactctta-3′ occurring 5445 downstream of the initiation codon was selected as the best candidate target. This resulted in a cell line with a suppressed expression of MUC17, labeled LSsi. A second small hairpin RNA with an unspecific scrambled sequence was used to create LSscram as an experimental control. Targeted clones were identified by antibiotic selection, 3 μg/ml puromycin (InvivoGen, San Diego, CA) for LSsi or G418 at 400 μg/ml (EMD Chemicals, Calbiochem, Gibbstown, NJ) for LSscram. Reverse transcriptase PCR and immunoblotting confirmed a decrease in MUC17 expression. Permanent cell lines were created by continuous antibiotic selection with either puromycin or G418.
LS174T cells were grown in MEM (Mediatech, Herndon, VA) supplemented with 10% FBS, HEPES, non-essential amino acids, L-glutamine, sodium pyruvate, sodium bicarbonate, and its corresponding antibiotic (Puromycin used at 3 μg/ml for Lssi, G418 (neomycin) use at 400 μg/ml for LSscram) maintained at 37 °C in 5% CO2 atmosphere. Antibiotics were removed at least 24 h prior to experiments.
2.9. Morphology and cell aggregation
Cells were examined for morphological differences under light microscope grown in normal tissue culture conditions. Cells were suspended in media at 2 × 105 cells/ml. 25 μl droplets, containing 5000 cells each, were placed onto the inner surface of 35 mm Petri dish lids. The lids were then inverted without disturbing the droplets and incubated for 30 h at 37 °C to allow cells to aggregate. The droplets were viewed under the microscope at 40× and 100× magnification.
2.10. Cell adhesion
Cells were seeded at 2 × 105 cells/well in 6-well plates in triplicates. After 5 h of incubation at 37 °C, unattached cells were separated from attached cells. The number of unattached cells was compared to the number of attached cells to find the percentage of cell adherence. Cell viability prior to seeding was checked with trypan blue.
2.11. Doubling time and growth curve
LS174T cells were seeded onto 6-well plates at 5 × 104 cells/well and allowed to proliferate over time. At each respective time point cells were trypsinized and counted by hemocytometer. Cell doubling time was found by the formula: Td = 0.693 × t/ln(Nt/N0), where t is the time, Nt is the number of cells at time t, and N0 is the number of cells at t0. Cell growth was checked over 192 h to determine doubling time and the overall cell growth curve.
2.12. Anchorage-independent growth in soft agar
Cells of different starting densities were suspended in warm agar (20% FBS, 0.3% agar, in growth medium), mimicking growth in soft tissue. Cells were allowed to form colonies over 14 days at 37 °C. Cells were imaged and number of colonies formed in agar was counted with the use of the NIH ImageJ program (http://rsbweb.nih.gov/ij/).
2.13. Subcutaneous tumor growth
1 × 106 cells were subcutaneously injected per site into 3-wk-old athymic Nu−/− female mice from Harlan Laboratories, as described previously (Kuan et al., 1987). Each mouse received injections at three separate sites. Mice received only either LSsi or LSscram cells. Mice were given ready access to food and water. Tumors were allowed to grow for 15 days. Tumor growth was monitored every other day with calipers. After 15 days the mice were sacrificed and the tumors were extracted and weighed. The tumor volume was found by: volume = (length × (width)2)/2.
2.14. Experimental colitis models
All experimental procedures were approved by the Institutional Animal Care and Use Committee. Acetic acid colitis: Female CD-1 mice (20–30 g, Harlan Sprague-Dawley, Indianapolis, IN) were fasted overnight and anesthetized with 3% isofluorane by inhalation. As described previously, colitis was induced by intrarectal administration of 0.1 ml of 5% acetic acid (Ho et al., 2006). The solutions were administered through a trocar needle approximately 3 cm proximal to the anus. Mice were subsequently treated 12 and 24 h later by intrarectal administration of 0.1 ml recombinant peptide in phosphate buffered saline or with 0.1 ml of control peptide in the same buffer at a similar concentration, using isofluorane anesthesia. Then all mice were harvested at 30 h after induction of colitis (6–12 h after last treatment enema), and the distal colons were removed and examined for gross ulceration and microscopic examination. This model has been described previously (Tomita et al., 1995; McCafferty et al., 1997). Dextran sodium sulfate (DSS) colitis: Acute colitis was induced in female CD-1 mice (20–30 g) by administration of 5% dextran sodium sulfate (molecular weight 40,000–50,000, USB, Cleveland, OH) in drinking water, as previously described (Okayasu et al., 1990; Cooper et al., 1993; Murthy et al., 1993; Ho et al., 2006). After 7 days the DSS was removed from the drinking water. Mice were treated 24 and 48 h after removal of DSS by intrarectal administration of 0.1 ml recombinant peptide in phosphate buffered saline or with 0.1 ml of control peptide in the same buffer, using isofluorane anesthesia. All mice were harvested at 72 h after removal of DSS and the colons examined histologically. Histologic mucosal injury score: Resected colons were fixed in 10% buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin. The severity of mucosal injury was graded similarly to that described previously (Okayasu et al., 1990; Murthy et al., 1993). The injury scale was graded from 0 to III, as follows: grade 0: normal; grade I: distortion and/or destruction of the bottom third of glands and focal inflammatory infiltrate; grade II: erosions/destruction of all glands or the bottom two-thirds of glands and inflammatory infiltrate with preserved surface epithelium; and grade III: loss of entire glands and surface epithelium. Specimens were examined without knowledge of the experimental group. The total number of low power (10×) fields exhibiting grade III colitis was determined for each specimen. An overall crypt damage score was also calculated by giving grades I, II, and III scores of 1, 2, and 3, respectively. Each low power field was graded, and the percentage of each specimen with each score was calculated and added to give the final crypt damage score (range 0–3.00), as described previously (Ho et al., 2006).
2.15. Statistical analysis
Mean ± SEM was calculated for variables in each experimental group and analyzed by ANOVA and Student's t-test (two-tailed) or Fishers exact test, as appropriate. A p-value of <0.05 was considered significant.
3. Results
3.1. Characterization of MUC17 knock down cell line LSsi
The LS174T colon cells exhibited high levels of endogenous MUC17 mRNA and therefore was chosen to create a stably transfected cell line with MUC17 siRNA in order to demonstrate whether endogenous MUC17 mucin may play a role in cellular behavior and processes related to cell restitution. Both Western blot and RT-PCR screening for MUC17 in LSsi and LSscram showed decreased MUC17 protein and mRNA expression in LSsi as compared to LSscram (Fig. 2). LSsi and LSscram cells also exhibited morphological differences in standard cell culture environment, with the LSsi cells appearing less aggregated and demonstrating a reduced ability to adhere to plastic. The hanging droplet environment forces cells to aggregate, as depicted in Fig. 3. Reduced MUC17 expression in LSsi cells was associated with visibly reduced cell aggregation. Reduced MUC17 expression also resulted in a reduced cell adherence after cell seeding (LSscram 96.25% vs. LSsi 91.67%, p < 0.002) (Fig. 3B). LSsi cells demonstrated a slightly increased doubling time and a slightly slower growth rate (Fig. 3C) compared with LSscram cells. However, when examining colony formation in soft agar and nude mouse tumorigenicity, no significant differences were seen between the two cell lines (Fig. 4).
Fig. 2.
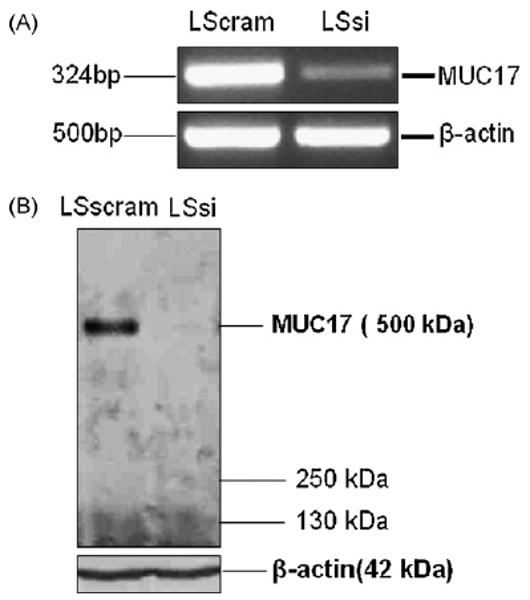
Expression analysis of MUC17. (A) RT-PCR for MUC17 mRNA expression in LSscram and LSsi cells. RT-PCR performed with B-actin primers were used to indicate mRNA in all lanes. (B) Western blot of cell lysates from LSscram and LSsi using anti-MUC17 antibody. Protein lysates from were resolved on 2% agarose gel containing SDS and transferred passively onto PVDF membrane. A unique band was revealed with a molecular weight consistent with the 500 kDa expected for MUC17. Loading control indicated by immunoreactivity using anti-actin antibody. Samples were tested in duplicate on two different occasions with similar results.
Fig. 3.
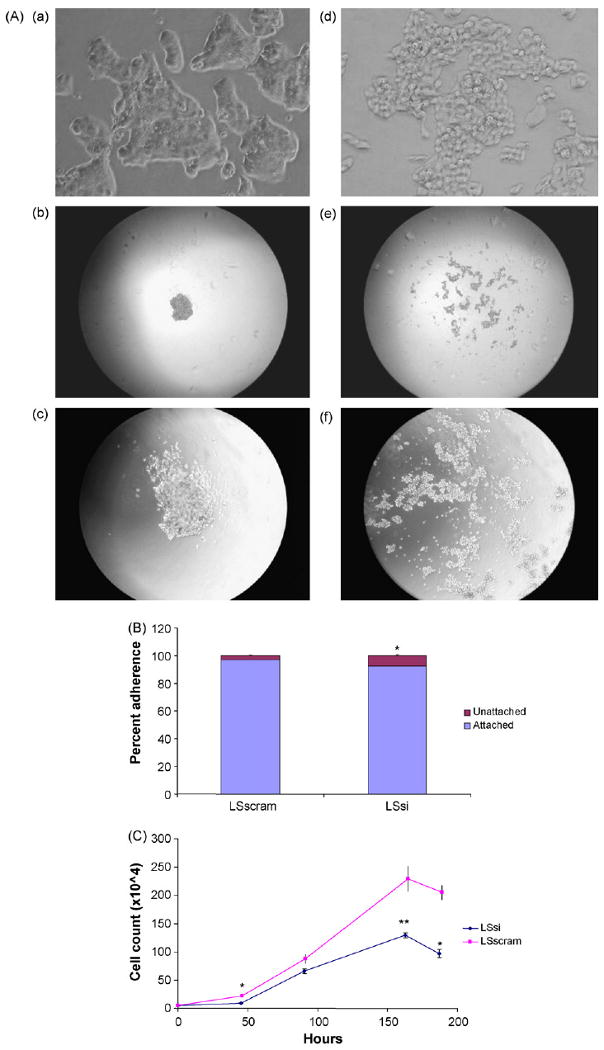
LS174T cell morphology, adhesion, and cell growth. (A) Cell morphology in standard cell culture environment: 1600× magnification images of LSscram (a) and LSsi (d). Cell aggregates in a droplet environment: LS scram at 40× (b) and 100× (c) magnification and LSsi at 40× (e) and 100× (f) magnification. (B) Cell Adhesion assay. LS174T cells were seeded at 2 × 105 cells/well into 6-well plates, in triplicate and performed twice. After 5 h of incubation at the medium was aspirated and unattached cells were counted and expressed as a percentage of total cells seeded, *p < 0.002 vs. LSscram. (C) Cell growth assay. LSsi and LSscram cells seeded in triplicate onto 6-well plates at 5 × 104 cells/well and counted at 48 and 96 h. Mean ± SEM, *p < 0.002, **p < 0.01 vs. LSscram.
Fig. 4.
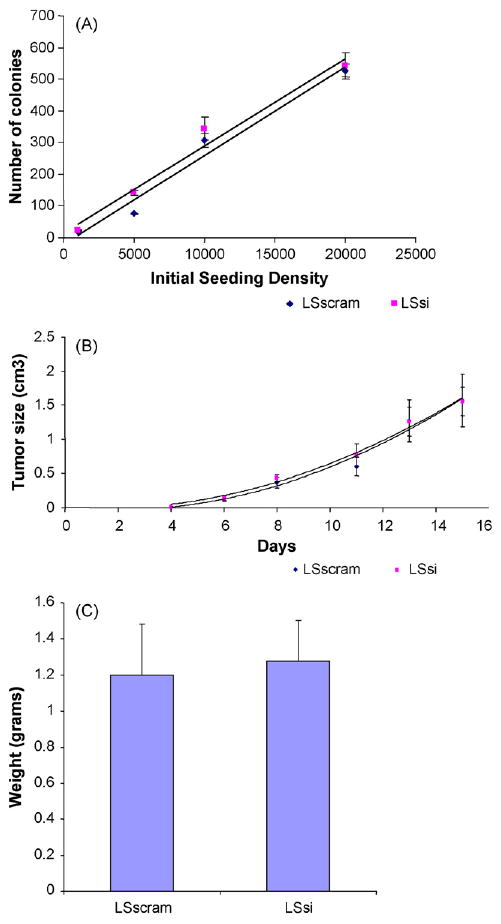
(A) Colony formation in soft agar with different initial seeding densities. Cells were seeded at indicated densities, allowed to incubate for 14 days at 37 °C, then the plates were imaged and number of colonies counted with the use of ImageJ. n = 4 per initial seeding density. Mean ± SEM. (B) Tumor size in nude mice over time after initial injection of 1 × 106 LSsi cells per site (n = 9) and 1 × 106 LSscram cells per site (n = 9). Mean ± SEM. (C) Final tumor weights after 15 days in LS scram (n = 9) and LSsi (n = 9), Mean ± SEM.
3.2. Inhibition of endogenous MUC17 impairs anti-apoptosis and cell migration response
Exogenous recombinant MUC17 and Muc3 cysteine-rich domain protein has been shown to stimulate cell migration in colon cells, and also inhibits apoptosis in response to toxins (Ho et al., 2006, 2010). Similarly, reduction of MUC17 expression in LSsi cells was associated with 67 ± 5% less migration than control LSscram cells over 48 h (p < 0.0001) in a wound healing assay (Fig. 5A and B). Furthermore, LSsi cells demonstrated increased apoptosis rates (6.53 ± 1.44%) compared with LSscram cells (1.75 ± 0.33%) following treatment with etoposide (p < 0.002) (Fig. 5C).
Fig. 5.
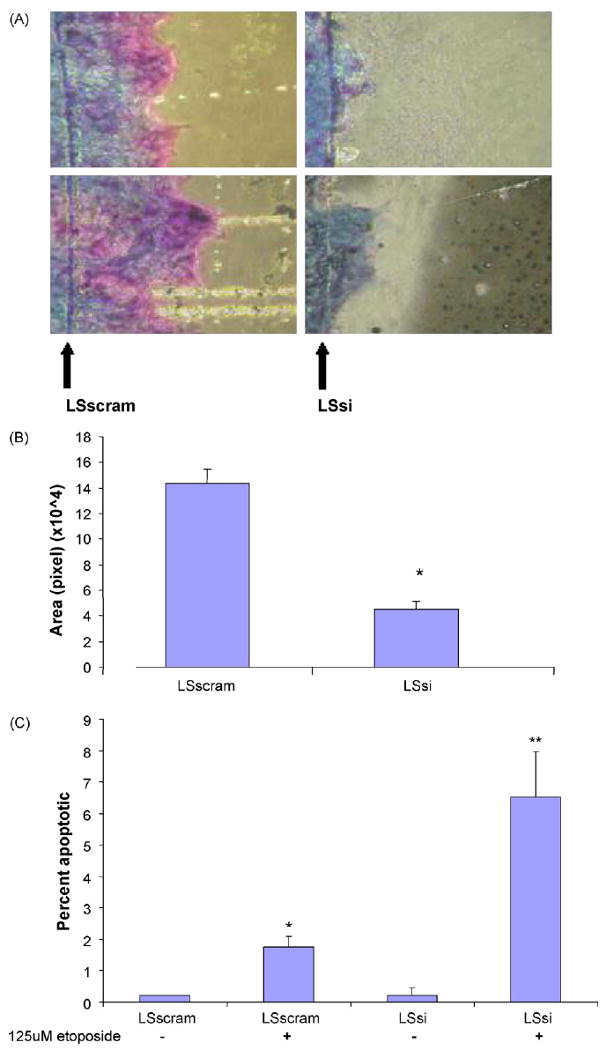
Cell migration and apoptosis. (A) The number of cells migrating across a razor scrape (arrow) was measured after 48 h at 37 °C; two images of LSscram cells and two images of LSsi cells (1600×). (B) Surface area of migration over wound over 48 h measured by computer imaging. (Mean ± SEM); *p < 0.00001. (C) Percent of apoptotic cells determined by Hoescht dye staining with and without 24 h treatment with etoposide. LSsi and LSscram cells were imaged at 400× at random fields. N = 4 per sample. Mean ± SEM. *p < 0.002 vs. LSscram no treatment, p < 0.03 vs. LSsi + etoposide; **p < 0.008 vs. LSsi no treatment.
3.3. Characterization of recombinant MUC17-CRD proteins
We have previously reported that a GST-tagged recombinant mouse Muc3 cysteine-rich domain protein demonstrated anti-apoptotic and pro-migratory activity in colonic cell lines (Ho et al., 2006); and that human recombinant MUC17-CRD1-L-CRD2 protein synthesized with either a GST tag or a smaller poly-His tag inhibited anti-Fas-induced apoptosis and enhanced migration of colonic cells across plastic (Ho et al., 2010). The recombinant proteins used in this study were tested for biologic activity related to anti-apoptosis and migration, and were verified to inhibit Fas-induced apoptosis in a dose-dependent manner (Fig. 6A). Recombinant MUC17-CRD1-L-CRD2 at a dose of 10 μg/ml was also shown to enhance migration of Lovo colonic cells across plastic to a similar degree as 10 ng/ml purified EGF (Fig. 6B).
Fig. 6.
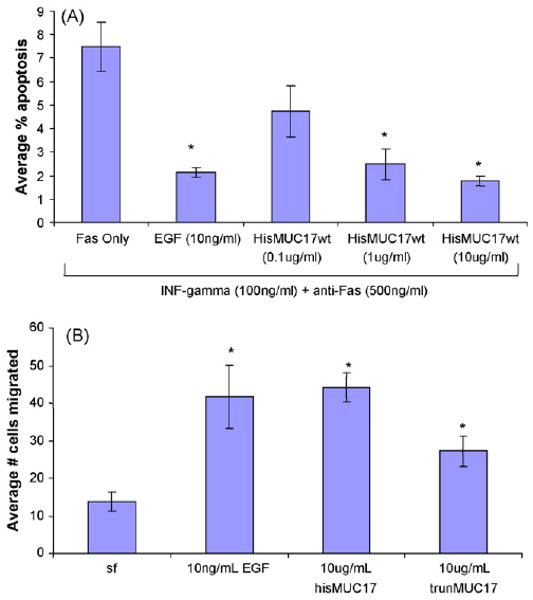
(A) Apoptosis was induced in Lovo colon cells using anti-Fas. His-tagged MUC17-CRD1-L-CRD2 (HisMUC17wt) inhibits Fas-induced apoptosis in a dose-dependent fashion. N = 4 per sample, *p < 0.001 vs. Fas Only treatment. (B) His-tagged MUC17-CRD1-L-CRD2 (hisMUC17wt) as well as His-tagged MUC17-L-CRD2 without CRD1, indicated as trunMUC17, stimulated cell migration in Lovo colon cells, N = 6 per sample, *p < 0.01 vs. serum free (sf). EGF at 10 ng/ml was a positive control for anti-apoptosis and cell migration.
3.4. ERK phosphorylation as a marker of recombinant MUC17-CRD1-L-CRD2 activity
Since prior studies have indicated that exogenous agents may stimulate cell migratory and anti-apoptotic pathways by ERK phosphorylation (Frey et al., 2004), we sought to determine if exposure to exogenous MUC17-CRD1-L-CRD2 would give measurable ERK activation. The differentiated colon cell lines IEC6 and YAMC were tested following treatment with His-MUC17-CRD1-L-CRD2 derived from E. coli. As seen in Fig. 7A, MUC17 treatment of IEC6 cells induced ERK phosphorylation above baseline by 30 min, which was blocked with pretreatment with the ERK inhibitor U0176. This was tested in an additional colon cell line, YAMC, which has been shown to respond to EGF exposure by stimulation of ERK phosphorylation (Frey et al., 2004). As shown in Fig. 7B, MUC17-CRD1-L-CRD2 activates ERK phosphorylation beginning at 5 min with maximal stimulation at 20 min, followed by a decrease in ERK phosphorylation by 60 to 120 min. This is in contrast to more rapid ERK phosphorylation induced by EGF 100 ng/ml. Note that EGF-stimulated tyrosine phosphorylation has also been shown to dissipate in colon cell lines after 60 min of exposure (Polk, 1995).
Fig. 7.
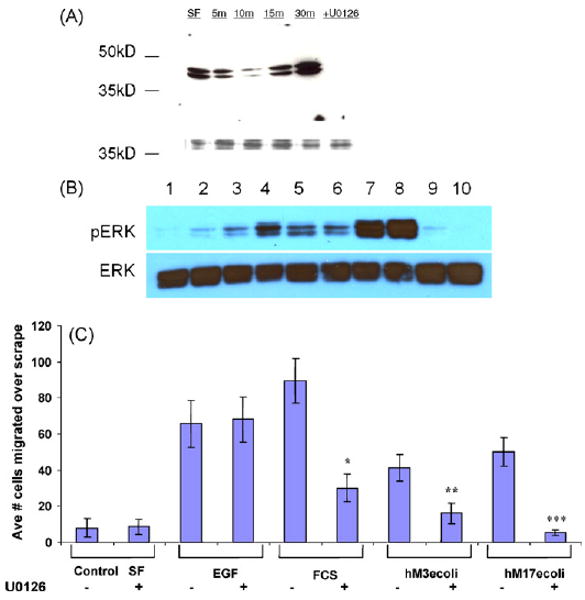
Activation of ERK cell signaling pathways. (A) IEC6 cells were treated with control serum-free medium (SF) or E. coli MUC17-CRD1-L-CRD2His8 (10 μg/ml) for 5–30 min and lysates were blotted with anti-phosphoERK1. Cells pretreated with ERK inhibitor U0126 before 15 min exposure to the MUC17 protein resulted in no phosphoERK1. The lower molecular weight bands indicate coumassie-stained protein demonstrating equal loading of gel. (B) YAMC cells were treated with control serum-free medium (SF. Lane 1); E. coli MUC17-CRD1-L-CRD2His8 (10 μg/ml) for 5, 10, 20, 60 or 120 min (lanes 2–6); EGF (100 ng/ml) for 5 and 10 min (lanes 7 and 8); and BSA (10 μg/ml) for 5 and 10 min; and lysates were blotted with anti-phosphoERK1. (C) Cell migration of Lovo cells treated with EGF (20 ng/ml), 10% fetal calf serum (FCS), Muc3-CRD1-L-CRD2 (hM3ecoli; 10 μg/ml), or MUC17-CRD1-L-CRD2 (hM17ecoli; 10 μg/ml); with or without pretreatment with 10 μM ERK inhibitor U0126 (u). *p < 0.01 vs. SF; **p < 0.01 vs. SF and p < 0.04 vs. hM3ecoli+u; ***p < 0.005 vs. SF and p < 0.02 vs. hM17ecoli+u. ERK inhibition has no effect on EGF-induced cell migration, partially blocks migration induced by 10% fetal calf serum (FCS), and completely blocks migration induced by both Muc3 and MUC17-CRD proteins.
Due to the established effect of mucin CRD proteins in the Lovo human cell line (Ho et al., 2006 and Fig. 6), we tested whether inhibition of ERK pathways altered the effect on cell migration induced by His-MUC17-CRD1-L-CRD2 from E. coli. As depicted in Fig. 7C, ERK inhibition by U0126 had no effect on EGF-induced cell migration, partially blocked migration induced by 10% fetal calf serum (FCS), and completely blocked migration induced by both His-tagged Muc3-CRD1-L-CRD2 and MUC17-CRD1-L-CRD2 proteins. These data suggest that ERK phosphorylation can be used as a measure of mucin CRD protein activity and that cell migration is induced at least in part via ERK-related cell signaling pathways.
3.5. Exogenous MUC17-CRD1-L-CRD2 accelerates healing in experimental models of colitis
Colitis was induced in mice using rectal infusion of acetic acid or by administration of DSS in drinking water. Treatment of mice with GST-tagged MUC17-CRD1-L-CRD2 at 100 μg/dose per rectum at 12 and 24 h after acetic acid infusion resulted in a trend for reduced overall crypt damage score and a significant improvement in the amount of severe or grade III ulceration (Fig. 8A and B). A second experiment using smaller sized mice showed a significant survival benefit of mice treated with the MUC17 protein given at 200 μg/dose (Fig. 8C). Due to the deaths in the control animals in this experiment comparison of histology was not performed. Similarly, mice treated with hisMUC17-CRD1-L-CRD2 (100 μg/dose) at 12, 24, and 36 h following induction of colitis with DSS exhibited significantly improved crypt damage scores and length of severe grade III ulceration compared with animals treated with control buffer alone (Fig. 9).
Fig. 8.
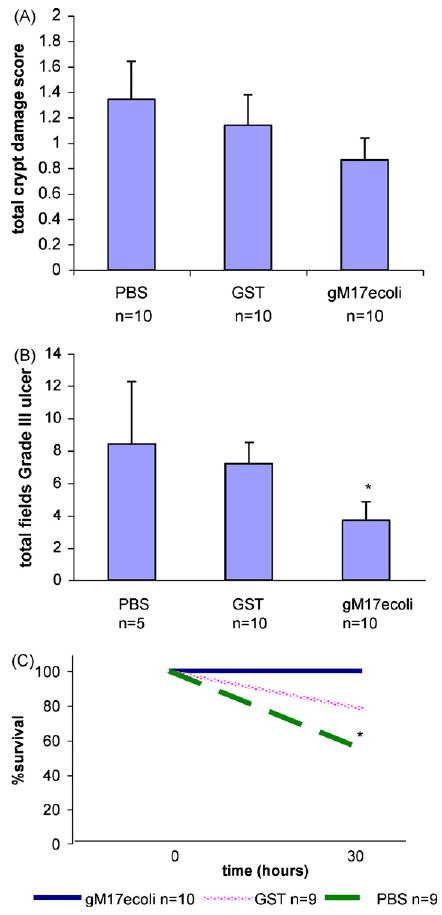
Colitis induced by acetic acid. Mice received 5% acetic acid per rectum at time 0, then treated with PBS, GST control protein (100 μg/dose), or GST-tagged MUC17-CRD1-L-CRD2 (gM17ecoli); 100 μg/dose per rectum at time 12 and 24 h. Mice were euthanized at 30 h for histologic examination of the distal colon for (A) crypt damage score and (B) length of total (grade III) ulceration. *p < 0.056 vs. GST. (C) Survival experiment: mice were given 5% acetic acid per rectum at time 0, then treated with PBS buffer, GST control protein (100 μg/dose), or GST-tagged MUC-17 (gM17ecoli) protein at 200 μg/dose at 12 h and 24 h (note mice treated with 100 μg/dose showed similar survival in previous experiment). Mice were then euthanized at 30 h. In this experiment, 100% of mice treated with the MUC17 protein survived, compared to 78% treated with GST, and 55% treated with PBS (*p = 0.032).
Fig. 9.
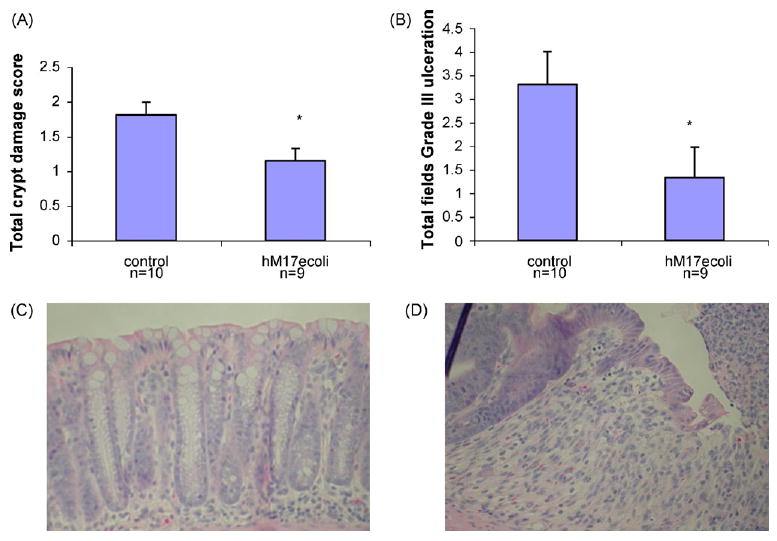
Colitis induced by dextran sodium sulfate. Mice received 5% DSS for 5 days and then received control buffer or hisMUC17-CRD1-L-CRD2 (hM17ecoli) (100 μg/dose) at 12, 24, and 36 h. Mice were harvested at 48 h and distal colons examined. (A) Crypt damage score. *p < 0.02 vs. control. (B) Total length of grade III ulceration. *p < 0.056 vs. control. (C) Representative histology of undamaged distal colon crypts in mouse treated with hisMUC17-CRD1-L-CRD2. (D) Representative histology of early grade III ulceration in distal colon of control mouse treated with buffer only.
4. Discussion
Membrane-bound mucins such as MUC17 are highly expressed on the surface of intestinal cells, but to date little is known about their function. We demonstrate that inhibition of expression of endogenous MUC17 mucin in a high-MUC17 expressing cell line resulted in an altered morphology, reduced cellular aggregation, increased susceptibility to apoptosis, and reduced spontaneous cell migration, indicating the potential importance of endogenous membrane-bound mucins in cellular processes related to cell–cell interactions and cell restitution. Recombinant protein corresponding to the extracellular cysteine-rich EGF-like domains of MUC17 stimulate ERK phosphorylation and result in enhanced cell migration and inhibition of apoptosis in response to toxins, indicating that these domains of membrane-bound mucins may be the active site for eliciting these processes.
Following intestinal injury the ability of cells to migrate and close a wound allows for restitution of the epithelial barrier more rapidly than by enhanced proliferation. Increased cell migration has been shown to occur in response to experimental intestinal injury and peptic ulcer disease (Feil et al., 1987; Argenzio, 1999), and inflammatory diseases of the bowel (Hermiston and Gordon, 1995; Karayiannakis et al., 1998). In addition, apoptosis is a determinant of the amount of epithelial damage that occurs in inflammatory conditions (Borges et al., 2005; Greten et al., 2004). Increased cell migration and anti-apoptosis are often associated and share some common pathways (Ridley et al., 2003). Thus, agents that enhance intestinal cell migration and reduce apoptosis are potentially therapeutic for inflammatory bowel disease. A recombinant mouse GST-Muc3 cysteine-rich domain (CRD) protein with two cysteine-rich EGF-like domains was previously shown to inhibit apoptosis and stimulate cell migration in colonic epithelial cells, and was able to enhance healing of experimental mouse models of colitis (Ho et al., 2006). We have now extended these studies to demonstrate that the human ortholog of the mouse Muc3 cysteine-rich protein, MUC17-CRD1-L-CRD2, has similar biologic activity in vitro, and have demonstrated that rectally administered MUC17-CRD1-L-CRD2 is also effective in accelerating histologic healing in mouse models of colitis. Taken together, these results indicate that full length human MUC17 mucin-derived CRD protein produced in E. coli expression system is a feasible and promising candidate for further development as a therapeutic for inflammatory bowel disease targeting epithelial restitution.
The mechanisms whereby membrane-bound mucins and their cysteine-rich domains influence cell migration and anti-apoptosis are largely unknown. We hypothesized that, similar to other growth factors, recombinant MUC17-CRD1-L-CRD2 may act by stimulating the raf/MEK/ERK pathway, which has been shown to function in anti-apoptotic and pro-migratory pathways in response to growth factors in multiple cell types (Dieckgraefe et al., 1997; El-Assal and Besner, 2005; Tan et al., 2008; Satoh et al., 2009). We showed that recombinant MUC17-CRD1-L-CRD2 induced phosphorylation of ERK in two colonic epithelial cell lines, and that pretreatment with an ERK phosphorylation inhibitor blocked migration induced by Muc3CRD1-L-CRD2 and by MUC17-CRD1-L-CRD2 recombinant proteins. Unlike the EGF growth factor property of inducing cell proliferation, the Muc3- and MUC17-CRD1-L-CRD2 proteins (EGF-like domains) minimally stimulate cell proliferation (Ho et al., 2006, 2010). In addition, EGF-stimulated cell migration was less susceptible to ERK inhibition compared with the mucin recombinant CRD proteins, suggesting that EGF and mucin CRD protein pathways may be distinct. There are numerous other parallel pathways related to apoptosis and cell migration that could also be involved, including phospholipase C-gamma, phosphatidylinositol (PI) 3-kinase/Akt, Src kinase, and p38 MAPK (Polk, 1998; Polk and Tong, 1999; Frey et al., 2004). The finding of recombinant mucin CRD protein activation of ERK is similar to the trefoil TFF2, which stimulates cell migration and activates ERK in bronchial epithelial cells (Graness et al., 2002), but is in contrast to the activity of intestinal trefoil factor, or TFF3, which has been shown to down-regulate ERK phosphorylation despite inducing cell migration in intestinal cells (Kanai et al., 1998). The purpose of the present experiments was not to fully test these possibilities, but rather show that at least one potential pathway related to processes of cellular restitution is stimulated by the mucin recombinant proteins, which may be used as an intermediate marker of activity for testing recombinant mucin proteins. The entire range of cell signaling pathways that could be induced by the mucin proteins is currently under investigation.
Therapeutic strategies aimed at improving the ability of the mucosal lining to restore itself have been shown to have potential and may augment current treatments that are designed primarily to inhibit the immune system. In this study we have shown that recombinant human MUC17-CRD1-L-CRD2 given per rectum is associated with decreased histologic colitis from both acetic acid-induced and dextran sodium sulfite-induced colitis. Because of the effects on anti-apoptosis and cell migration established for this recombinant protein, we believe that the beneficial effect is due to accelerated restitution of the epithelial barrier in these models, however further studies are required to explore the potential effects of these proteins in vivo. Targeting epithelial restitution has shown promising results with the use of topical growth factors such as epidermal growth factor (EGF), recombinant trefoil proteins, cell membrane components such as phospholipids, and intestinal energy sources such as short chain fatty acids, indicating that approaches to enhance the mucosal barrier are feasible (Taupin et al., 2000; Sinha et al., 2003; Hoffmann, 2004; Gibson and Muir, 2005). Agents that enhance cell restitution may also be expected to increase the risk of neoplastic transformation in the colon, however agents such as MUC17-CRD1-L-CRD2 which do not stimulate cell proliferation as much as a EGF may be preferred because of a potential lesser risk of neoplasia. Further animal and clinical trials will be necessary to delineate these risks.
Other mucin proteins have been implicated as playing a protective role in experimental colitis. A mouse model with the deletion of the secretory Muc2 mucin demonstrated enhanced susceptibility to colitis induced by dextran sodium sulfate (Van der Sluis et al., 2006). A mouse model with deletion of the membrane-bound Muc1 mucin demonstrated no alterations in intestinal function under normal germ free conditions, but was found to have increased susceptibility to invasion and damage by the pathogenic bacteria, Campylobacter jejuni (McAuley et al., 2007). Lastly, altering the glycosylation of mucin proteins, such as is found in mouse models deleting core 3-type and core 2-type protein O-glycosyltransferases, has been shown to confer increased susceptibility to colitis from dextran sodium sulfate (An et al., 2007; Stone et al., 2009). Further gene deletion studies of more intestine specific membrane-bound mucins, such as the mouse Muc3, are needed to establish the specific role of membrane-bound mucins in colonic mucosal protection. In humans, the membrane-bound mucin locus on Chromosome 7 may be included in the large area that has been demonstrated to be linked to susceptibility to inflammatory bowel disease in large population linkage studies (Satsangi et al., 1996), but no specific data linking mucin defects to colitis has been reported in humans to date. The relevance of various intestinal epithelial barrier functions to inflammatory bowel disease susceptibility has been reviewed (McGuckin et al., 2008).
In conclusion, the data presented in this paper demonstrate that endogenous MUC17 plays an important role in cell restitution processes, such as aggregation, apoptosis, and cell migration. The data also demonstrate the efficacy of synthetic proteins derived from cysteine-rich regions of the human MUC17 surface membrane mucin to promote restitution of wounded colonic cells and accelerate healing in mouse models of colitis. Use of recombinant mucin-related proteins for mucosal restitution may potentially augment current therapies for inflammatory bowel disease and other mucosal diseases.
Acknowledgments
This study was supported by a VA Merit Review grant (SBH), NIH STTR grant G1 R43 DK072629-01 (SBH, RLH), NIH center grant (DK080506) (SBH), NIH CA78590 (SKB), and the Research Service of the Department of Veterans Affairs.
References
- An G, Wei B, Xia B, McDaniel JM, Ju T, Cummings RD, et al. Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J Exp Med. 2007;204(6):1417–29. doi: 10.1084/jem.20061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argenzio RA. Comparative pathophysiology of nonglandular ulcer disease: a review of experimental studies. Equine Vet J Suppl. 1999;29:19–23. doi: 10.1111/j.2042-3306.1999.tb05163.x. [DOI] [PubMed] [Google Scholar]
- Borges HL, Bird J, Wasson K, Cardiff RD, Varki N, Eckmann L, et al. Tumor promotion by caspase-resistant retinoblastoma protein. Proc Natl Acad Sci USA. 2005;102(43):15587–92. doi: 10.1073/pnas.0503925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi P, Singh AP, Moniaux N, Senapati S, Chakraborty S, Meza JL, et al. MUC4 mucin potentiates pancreatic tumor cell proliferation, survival, and invasive properties and interferes with its interaction to extracellular matrix proteins. Mol Cancer Res. 2007;5(4):309–20. doi: 10.1158/1541-7786.MCR-06-0353. [DOI] [PubMed] [Google Scholar]
- Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69(2):238–49. [PubMed] [Google Scholar]
- Crawley SC, Gum JR, Jr, Hicks JW, Pratt WS, Aubert JP, Swallow DM, et al. Genomic organization and structure of the 3′ region of human MUC3: alternative splicing predicts membrane-bound and soluble forms of the mucin. Biochem Biophys Res Commun. 1999;263(3):728–36. doi: 10.1006/bbrc.1999.1466. [DOI] [PubMed] [Google Scholar]
- Dieckgraefe BK, Weems DM, Santoro SA, Alpers DH. ERK and p38 MAP kinase pathways are mediators of intestinal epithelial wound-induced signal transduction. Biochem Biophys Res Commun. 1997;233(2):389–94. doi: 10.1006/bbrc.1997.6469. [DOI] [PubMed] [Google Scholar]
- El-Assal ON, Besner GE. HB-EGF enhances restitution after intestinal ischemia/reperfusion via PI3K/Akt and MEK/ERK1/2 activation. Gastroenterology. 2005;129(2):609–25. doi: 10.1016/j.gastro.2005.05.054. [DOI] [PubMed] [Google Scholar]
- Fan G, Ma X, Kren BT, Steer CJ. The retinoblastoma gene product inhibits TGF-beta1 induced apoptosis in primary rat hepatocytes and human HuH-7 hepatoma cells. Oncogene. 1996;12(9):1909–19. [PubMed] [Google Scholar]
- Feil W, Wenzl E, Vattay P, Starlinger M, Sogukoglu T, Schiessel R. Repair of rabbit duodenal mucosa after acid injury in vivo and in vitro. Gastroenterology. 1987;92(6):1973–86. doi: 10.1016/0016-5085(87)90632-9. [DOI] [PubMed] [Google Scholar]
- Frey MR, Golovin A, Polk DB. Epidermal growth factor-stimulated intestinal epithelial cell migration requires Src family kinase-dependent p38 MAPK signaling. J Biol Chem. 2004;279(43):44513–21. doi: 10.1074/jbc.M406253200. [DOI] [PubMed] [Google Scholar]
- Gibson PR, Muir JG. Reinforcing the mucus: a new therapeutic approach for ulcerative colitis? Gut. 2005;54(7):900–3. doi: 10.1136/gut.2004.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graness A, Chwieralski CE, Reinhold D, Thim L, Hoffmann W. Protein kinase C and ERK activation are required for TFF-peptide-stimulated bronchial epithelial cell migration and tumor necrosis factor-alpha-induced interleukin-6 (IL-6) and IL-8 secretion. J Biol Chem. 2002;277(21):18440–6. doi: 10.1074/jbc.M200468200. [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Gum JR, Jr, Ho JJ, Pratt WS, Hicks JW, Hill AS, Vinall LE, et al. MUC3 human intestinal mucin, analysis of gene structure, the carboxyl terminus, and a novel upstream repetitive region. J Biol Chem. 1997;272(42):26678–86. doi: 10.1074/jbc.272.42.26678. [DOI] [PubMed] [Google Scholar]
- Hermiston ML, Gordon JI. In vivo analysis of cadherin function in the mouse intestinal epithelium: essential roles in adhesion, maintenance of differentiation, and regulation of programmed cell death. J Cell Biol. 1995;129:489–506. doi: 10.1083/jcb.129.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SB, Dvorak LA, Moor RE, Jacobson AC, Frey MR, Corredor J, et al. Cysteine-rich domains of muc3 intestinal mucin promote cell migration, inhibit apoptosis, and accelerate wound healing. Gastroenterology. 2006;131(5):1501–17. doi: 10.1053/j.gastro.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Ho SB, Luu Y, Baskerville C, Shekels LL, Batra SK, Evans DB, et al. Activity of recombinant cysteine-rich domain proteins derived from the membrane-bound MUC17/Muc3 family mucins. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbagen.2010.03.010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann W. Trefoil factor family (TFF) peptides: regulators of mucosal regeneration and repair, and more. Peptides. 2004;25(5):727–30. doi: 10.1016/j.peptides.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Kaiser GC, Polk DB. Tumor necrosis factor alpha regulates proliferation in a mouse intestinal cell line. Gastroenterology. 1997;112(4):1231–40. doi: 10.1016/s0016-5085(97)70135-5. [DOI] [PubMed] [Google Scholar]
- Kanai M, Mullen C, Podolsky DK. Intestinal trefoil factor induces inactivation of extracellular signal-regulated protein kinase in intestinal epithelial cells. Proc Natl Acad Sci USA. 1998;95(1):178–82. doi: 10.1073/pnas.95.1.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayiannakis AJ, Syrigos KN, Efstathiou J, Valizadeh A, Noda M, Playford RJ, et al. Expression of catenins and E-cadherin during epithelial restitution in inflammatory bowel disease. J Pathol. 1998;185(4):413–8. doi: 10.1002/(SICI)1096-9896(199808)185:4<413::AID-PATH125>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Kouniavsky G, Khaikin M, Zvibel I, Zippel D, Brill S, Halpern Z, et al. Stromal extracellular matrix reduces chemotherapy-induced apoptosis in colon cancer cell lines. Clin Exp Metastasis. 2002;19(1):55–60. doi: 10.1023/a:1013880326925. [DOI] [PubMed] [Google Scholar]
- Kuan SF, Byrd JC, Basbaum CB, Kim YS. Characterization of quantitative mucin variants from a human colon cancer cell line. Cancer Res. 1987;47:5715–24. [PubMed] [Google Scholar]
- Magne N, Fischel JL, Dubreuil A, Formento P, Poupon MF, Laurent-Puig P, et al. Influence of epidermal growth factor receptor (EGFR), p53 and intrinsic MAP kinase pathway status of tumour cells on the antiproliferative effect of ZD1839 (“Iressa”) Br J Cancer. 2002;86(9):1518–23. doi: 10.1038/sj.bjc.6600299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley JL, Linden SK, Png CW, King RM, Pennington HL, Gendler SJ, et al. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. J Clin Invest. 2007;117(8):2313–24. doi: 10.1172/JCI26705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty DM, Mudgett JS, Swain MG, Kubes P. Inducible nitric oxide synthase plays a critical role in resolving intestinal inflammation. Gastroenterology. 1997;112(3):1022–7. doi: 10.1053/gast.1997.v112.pm9041266. [DOI] [PubMed] [Google Scholar]
- McGuckin MA, Eri R, Simms LA, Florin TH, Radford-Smith G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis. 2008 doi: 10.1002/ibd.20539. [DOI] [PubMed] [Google Scholar]
- Moniaux N, Junker WM, Singh AP, Jones AM, Batra SK. Characterization of human mucin MUC17, complete coding sequence and organization. J Biol Chem. 2006;281(33):23676–85. doi: 10.1074/jbc.M600302200. [DOI] [PubMed] [Google Scholar]
- Murthy SN, Cooper HS, Shim H, Shah RS, Ibrahim SA, Sedergran DJ. Treatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporin. Dig Dis Sci. 1993;38(9):1722–34. doi: 10.1007/BF01303184. [DOI] [PubMed] [Google Scholar]
- Nyati MK, Maheshwari D, Hanasoge S, Sreekumar A, Rynkiewicz SD, Chinnaiyan AM, et al. Radiosensitization by pan ErbB inhibitor CI-1033 in vitro and in vivo. Clin Cancer Res. 2004;10(2):691–700. doi: 10.1158/1078-0432.ccr-1041-03. [DOI] [PubMed] [Google Scholar]
- Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98(3):694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- Polk DB. Shc is a substrate of the rat intestinal epidermal growth factor receptor tyrosine kinase. Gastroenterology. 1995;109(6):1845–51. doi: 10.1016/0016-5085(95)90751-3. [DOI] [PubMed] [Google Scholar]
- Polk DB. Epidermal growth factor receptor-stimulated intestinal epithelial cell migration requires phospholipase C activity. Gastroenterology. 1998;114(3):493–502. doi: 10.1016/s0016-5085(98)70532-3. [DOI] [PubMed] [Google Scholar]
- Polk DB, Tong W. Epidermal and hepatocyte growth factors stimulate chemotaxis in an intestinal epithelial cell line. Am J Physiol. 1999;277(6 Pt 1):C1149–59. doi: 10.1152/ajpcell.1999.277.6.C1149. [DOI] [PubMed] [Google Scholar]
- Quirk SM, Porter DA, Huber SC, Cowan RG. Potentiation of fas-mediated apoptosis of murine granulosa cells by interferon-gamma, tumor necrosis factor-alfa and cycloheximide. Endocrinology. 1998;139:4860–9. doi: 10.1210/endo.139.12.6353. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Russo CL, Spurr-Michaud S, Tisdale A, Pudney J, Anderson D, Gipson IK. Mucin gene expression in human male urogenital tract epithelia. Hum Reprod. 2006;21(11):2783–93. doi: 10.1093/humrep/del164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Saitoh D, Takeuchi A, Ojima K, Kouzu K, Kawakami S, et al. ERK2 dependent signaling contributes to wound healing after a partial-thickness burn. Biochem Biophys Res Commun. 2009;381(1):118–22. doi: 10.1016/j.bbrc.2009.02.039. [DOI] [PubMed] [Google Scholar]
- Satsangi J, Parkes M, Louis E, Hashimoto L, Kato N, Welsh K, et al. Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet. 1996;14(2):199–202. doi: 10.1038/ng1096-199. [DOI] [PubMed] [Google Scholar]
- Shekels LL, Hunninghake DA, Tisdale AS, Gipson IK, Kieliszewsji M, Kozak CA, et al. Cloning and characterization of mouse intestinal Muc3 mucin: 3′ sequence contains epidermal-growth-factor-like domains. Biochem J. 1998;330:1301–8. doi: 10.1042/bj3301301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha A, Nightingale J, West KP, Berlanga-Acosta J, Playford RJ. Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N Engl J Med. 2003;349(4):350–7. doi: 10.1056/NEJMoa013136. [DOI] [PubMed] [Google Scholar]
- Stone EL, Ismail MN, Lee SH, Luu Y, Ramirez K, Haslam SM, et al. Glycosyltransferase function in core 2-type protein O glycosylation. Mol Cell Biol. 2009;29(13):3770–82. doi: 10.1128/MCB.00204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CT, Chu CY, Lu YC, Chang CC, Lin BR, Wu HH, et al. CXCL12/CXCR4 promotes laryngeal and hypopharyngeal squamous cell carcinoma metastasis through MMP-13-dependent invasion via the ERK1/2/AP-1 pathway. Carcinogenesis. 2008;29(8):1519–27. doi: 10.1093/carcin/bgn108. [DOI] [PubMed] [Google Scholar]
- Taupin DR, Kinoshita K, Podolsky DK. Intestinal trefoil factor confers colonic epithelial resistance to apoptosis. Proc Natl Acad Sci USA. 2000;97:799–804. doi: 10.1073/pnas.97.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita M, Itoh H, Ishikawa N, Higa A, Ide H, Murakumo Y, et al. Molecular cloning of mouse intestinal trefoil factor and its expression during goblet cell changes. Biochem J. 1995;311:293–7. doi: 10.1042/bj3110293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131(1):117–29. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Williams SJ, Munster DJ, Quin RJ, Gotley DC, McGuckin MA. The MUC3 gene encodes a transmembrane mucin and is alternatively spliced. Biochem Biophys Res Commun. 1999;261:83–9. doi: 10.1006/bbrc.1999.1001. [DOI] [PubMed] [Google Scholar]