

Abstract
Objective
Thrombin mediates the life-threatening cerebral edema that occurs following intracerebral hemorrhage. Therefore, we examined the mechanisms of thrombin-induced injury to the blood-brain barrier (BBB) and subsequent mechanisms of BBB repair.
Methods
Intracerebroventricular (i.c.v.) injection of thrombin (20 U) was used to model intraventricular hemorrhage in adult rats.
Results
Thrombin reduced brain microvascular endothelial cell (BMVEC) and peri-vascular astrocyte immunoreactivity –indicating either cell injury or death; and, functionally disrupted the BBB as measured by increased water content and extravasation of sodium fluorescein and Evans blue dyes 24h later. Administration of non-specific src family kinase inhibitor PP2 immediately following thrombin injections blocked brain edema and BBB disruption. At 7 to 14 days after thrombin injections newborn endothelial cells and astrocytes were observed around cerebral vessels at the time when BBB permeability and cerebral water content resolved. Delayed administration of PP2 on days 2 through 6 following thrombin injections prevented resolution of the edema and abnormal BBB permeability.
Interpretation
Thrombin, via its PAR receptors, is postulated to activate src kinase phosphorylation of molecules that acutely injure the BBB and produce edema. Thus, acute administration of src antagonists blocks edema. In contrast, src blockade for 2-6 days following thrombin injections is postulated to prevent resolution of edema and abnormal BBB permeability in part because src kinase proto-oncogene members stimulate proliferation of newborn BMVECs and peri-vascular astrocytes in the “neurovascular niche” that repair the damaged BBB. Thus, src kinases not only mediate acute BBB injury but also mediate chronic BBB repair after thrombin-induced injury.
Introduction
Intracerebral hemorrhage (ICH) activates thrombin 1, 2. Thrombin is the molecule that mediates the development of acute cerebral edema following ICH since acute edema can be prevented by thrombin inhibitors 1. Following thrombin injections into caudate-putamen of adult rat brain, edema increases within several hours, peaks around the first to third day, and then declines gradually over several weeks 3, 4. The cerebral edema changes in parallel with changes in BBB permeability 4. However, the mechanisms that lead to thrombin-induced BBB disruption are unknown, as are the mechanisms responsible for the subsequent repair of the BBB. These mechanisms are the subject of this study.
The BBB is a specialized system of brain microvascular endothelial cells (BMVEC), astrocytes, basement membrane, pericytes and neurons 5. BMVEC are the thin layer of cells that line the interior surface of blood vessels, forming an interface between circulating blood and the brain. Complex tight junctions between adjacent endothelial cells form a physical barrier, forcing most molecular traffic to take a transcellular/transporter route across the BBB, rather than moving paracellularly through the junctions, as in most endothelial cells 5-8. Astrocytes are an important component of the BBB, enveloping >99% of BMVEC 8. BMVEC and astrocytes influence each other's development, structure and function 5, 6, 9.
It is not known, however, how ICH leads to BBB dysfunction and brain edema. In this study we postulated that thrombin-induced BMVEC and astrocyte injuries would lead to disruption of the BBB and brain edema, and that the proliferation (birth) of BMVEC and astrocytes would correlate with restoration of the BBB and resolution of edema following ICH. To begin to address these hypotheses, we examined BMVEC and peri-vascular astrocytes during the BBB disruption and cerebral edema formation following i.c.v. thrombin injections to adult rats. The time course of BrdU incorporation into BMVEC and peri-vascular astrocytes was then examined during the period of resolution of brain edema and normalization of BBB permeability. To test whether the proliferating BMVEC and peri-vascular astrocytes played a causative role in repair of the BBB, the cell cycle and src kinase inhibitor, PP2, was given after thrombin injections. We predicted that PP2 should prolong the edema and prolong the abnormal BBB permeability. Thrombin injections into the cerebral ventricles were performed in this study because: (1) thrombin is the cause of acute edema following ICH 1; (2) ventricular injections of thrombin provide a simple model of intra-ventricular hemorrhage that occurs in humans 3, 4, 10, 11; and (3) intraventricular hemorrhage in humans is associated with particularly severe brain edema and high mortality rates 12.
Materials and Methods
Injections of thrombin into cerebral ventricle
Male Sprague-Dawley rats (n=120 total), weighing 300-320 grams, were anesthetized with isoflurane (Minrad, New York, NY) and placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA). A heating blanket maintained body temperature at 37°C. Thrombin (from bovine plasma, Sigma, St. Louis, MO) was dissolved in 5μl saline (20 U/animal) and injected into the left cerebral ventricle (i.c.v. day 0) (coordinates: -0.9 AP, −1.4 ML, −4.6 DV, with respect to bregma) 13. The control group received 5μl saline injections (i.c.v.). The thrombin inhibitor hirudin (20U, Sigma, St. Louis, MO) was co-injected (i.c.v., day 0) with thrombin in some subjects. Rats that received i.c.v. thrombin without hirudin were divided into two groups. One group of rats received one intraperitoneal injection immediately (day 0) of the non-specific src family kinase inhibitor PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine, 1.0 mg/kg, Biomol International LP, Plymouth Meeting, PA). The second group of rats received a total of five i.p. injections of PP2, once per day, from days 2 to 6 (days 2-6).
After closure of the operative sites, rats were allowed to recover in an incubator maintained at 37°C, and then returned to their home cages with free access to food and water. All experimental procedures were performed in accordance with National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee, University of California at Davis. All reagents were purchased from Sigma (St. Louis, MO), unless otherwise stated.
BrdU administration and sample preparation
Please see details in Supplementary Text.
Immunohistochemistry and cell counting
Co-localization of RECA-1 or GFAP immunoreactive cells with BrdU immunoreactive nuclei was examined in the lacunosum moleculare layer (LMol) of the hippocampus as shown in Figure 1. Please see details in Supplementary Text.
Figure 1.

The Lacunosum moleculare layer (LMol) is located between CA1 pyramidal neurons and the molecular layer of the dentate gyrus (MoDG) in the hippocampus. The LMol is characterized by a series of large (10 - 50 μm diameter) blood vessels (marked with stars) that were perpendicular to the plane of these coronal sections in every animal in this study. Scale bar: 500 μm.
Blood-brain barrier (BBB) permeability
BBB permeability was measured using a recently developed sodium fluorescein (NF) / Evans blue (EB) technique 14. Please see details in Supplementary Text.
Brain water content
Please see details in Supplementary Text.
Results
The effects of thrombin on BMVEC
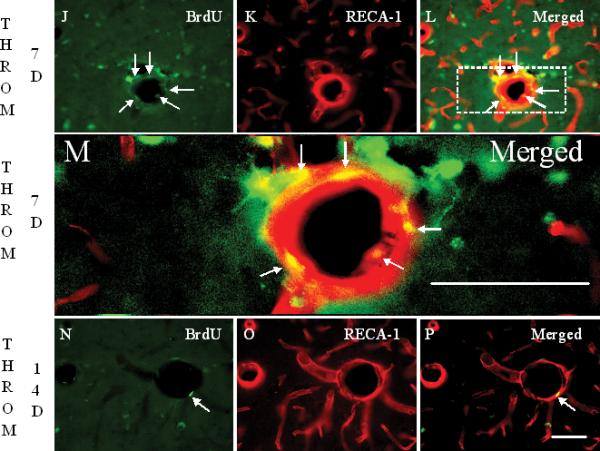
In sham operated rats, RECA-1+ cells delineated the tube-shaped brain capillaries (arrows in Figure 2C). BMVEC staining with RECA-1 markedly decreased (Figure 2E), and brain capillary shape changed (arrows in Figure 2F) at 1 day after thrombin injections. Acute PP2 administration, immediately after thrombin injection, blocked the thrombin-induced reductions in RECA-1 immunoreactivity (Figure 2H, I), and brain capillary shape changes (Figure 2I). At one and two weeks after thrombin injections, RECA-1 stained BMVEC were often co-labeled with BrdU (Figures 2J-Q). However, the numbers of BrdU+/GFAP+ cells at 14 days (arrows in Figure 2P) were less than those at 7 days (arrows in Figures 2L, 2M) after thrombin injections (Figure 2Q). Some brain capillaries regained their tube-shape at 7 days (Figures 2K, L, M), and more brain capillaries regained their tube-shape at 14 days (Figures 2O, P) after the thrombin injections.
Figure 2.
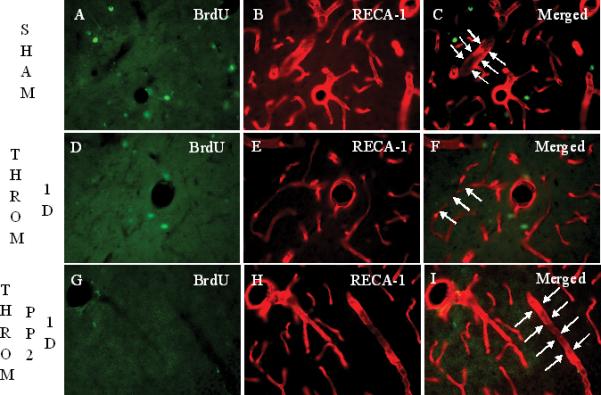

Intracerebroventricular (i.c.v.) injection of thrombin (20 U/animal, i.c.v.) causes reductions in brain microvascular endothelial cell (BMVEC) immunoreactivity after 1 day, and subsequent BMVEC proliferation around the rat brain vessels in lacunosum moleculare layer (LMol) of the hippocampus after 7 days and 14 days. Panels A-C show rats following sham operations labeled for BrdU, bromodeoxyuridine (A), RECA-1, rat endothelial antigen-1 (B) and the overlay or Merged image (C). RECA-1+ cells demonstrate the tube-shape of brain capillaries (arrows in panel C). Panels D to F show BrdU (D), RECA-1 (E) and the Merged image (F) at 1 day after thrombin injections. Compared with the sham group, RECA-1+ cells tend to lose their tube-shape at 1 day following thrombin injections (arrows in panel F) and there were no BrdU+ cells co-labeled with RECA-1 at one day (panel F). Panels G-I show the staining for BrdU (G), RECA-1 (H) and the Merged image (I) 1 day after thrombin plus PP2 injections. PP2 administration at day 0, immediately after thrombin injection, blocks the thrombin-induced loss of tube-shape of RECA-1+ cells. (Legend for first portion of Figure 2, Panels A-I).
Panels J-L show the staining of BrdU (J), RECA-1 (K) and the Merged image (L) 7 days after thrombin injection. Compared to 1 day, BrdU+ cells are increased 7 days after thrombin injection. Some of these BrdU+ cells are co-labeled with RECA-1 (arrows in panel L and M). A few brain capillaries regained their tube-shape, though not completely. Panel M shows a higher power image of Panel L (area within dashed lines). RECA-1 stained BMVEC are red. The BrdU+/RECA-1+ double-labeled new born BMVEC nuclei are yellow. Panels N to P show the staining for BrdU (M), RECA-1 (N) and the Merged image (P) 14 days after thrombin injection. Compared to 7 days, BrdU+ cells are decreased, but some BrdU+ cells (N) remain co-labeled with RECA-1 (arrow in panel P), and more and more brain capillaries regained the tube-shape 14 days after the thrombin injection. Scale bars: A-P, 50 μm. (Legend for second portion of Figure 2, Panels J-P).
Panel Q shows the density of BrdU+ RECA-1+ double-labeled cells counted in LMol layer of hippocampus in each experimental group. Each column and vertical bar represents the mean ± standard error of the mean. ** p<0.01 vs. Cont (one-way ANOVA followed by Tukey's post hoc test). Cont = control. PP2 = non-specific src family kinase inhibitor. Throm = thrombin. RECA-1 = rat endothelial cell antigen-1. BrdU = bromodeoxyuridine, a marker of cell proliferation. (Legend for third portion of Figure 2, Panel Q).
The effects of thrombin on astrocytes
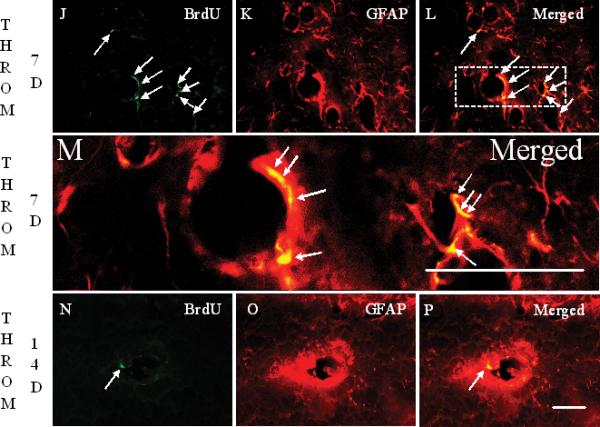
In sham operated rats, GFAP+ cells enveloped brain vessels (arrows in Figure 3B). Astrocyte staining with GFAP markedly decreased around brain vessels 24 h after thrombin injections (Figure 3E). PP2 administration at day 0, immediately after thrombin injection, blocked the thrombin-induced reductions in GFAP immunoreactivity (Figure 3H, I). GFAP stained astrocytes, that often were co-labeled with BrdU, increased around blood vessels at 7 days (Figures 3J-M, Q) and at 14 days (Figures 3N-P, Q) after thrombin injections. However, the numbers of BrdU+/GFAP+ cells at 14 days (arrows in Figure 3P) were less than those at 7 days (arrows in Figures 3L, M) after thrombin injections (Figure 3Q).
Figure 3.
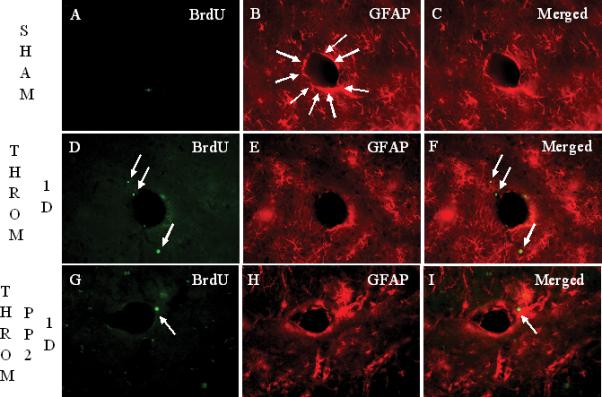

Intracerebroventricular injection of thrombin (20 U/animal, i.c.v.) causes reductions in astrocyte GFAP immunoreactivity after 1 day, and subsequent astrocyte proliferation around the rat brain vessels in lacunosum moleculare layer (LMol) of the hippocampus after 7 days and 14 days. Panels A-C show rats with sham operation labeled for BrdU (A), GFAP (B), and Merged image (C). GFAP+ cells envelop most all of the brain vessel (arrows in panel B). Panels D-F show BrdU (D), GFAP (E) and the Merged image (F) at 1 day after thrombin injection. Compared with the sham group, there is decreased GFAP immunoreactivity around brain vessels. There are a few BrdU+/GFAP- cells located close to the vessel (arrows in panel F). G-I show the staining for BrdU (G), RECA-1 (H) and the Merged image (I) 1 day after thrombin plus PP2 injections. PP2 administration at day 0, immediately after thrombin injection, blocks the thrombin-induced reductions in GFAP immunoreactivity. (Legend for first portion of Figure 3, Panels A-I).
J-L show the staining for BrdU (J), GFAP (K) and the Merged image (L) 7 days after thrombin injection. Compared to 1 day, BrdU+ cells are increased 7 days after thrombin injection (J, arrows). Many of these BrdU+ cells are co-labeled with GFAP (arrows in panel L). Panel M shows a higher power image of Panel L (area within dashed lines). GFAP stained astrocytes are red. The BrdU+ GFAP+ double-labeled new born astrocytic nuclei are yellow (arrows, Panel M). Panels N-P show the staining for BrdU (N), GFAP (O) and the Merged image (P) 14 days after thrombin injection. Compared to 7 days, BrdU+ cells are decreased 14 days after thrombin injection. Some BrdU+ cells (N, arrow) remain co-labeled with GFAP (arrow in panel P) 14 days after the thrombin injection. Scale bars: A-P, 50 μm. (Legend for second portion of Figure 3, panels J-P).
Panel Q shows density of BrdU+ GFAP+ double-labeled cells counted in the LMol of hippocampus in different conditions. Each column and vertical bar represents the mean ± standard error of the mean. ** p<0.01 vs. Cont (one-way ANOVA followed by Tukey's post hoc test). Abbreviations are the same as Figure 2. GFAP = astrocyte marker. Cont=control. Throm = thrombin. PP2 = non-specific src family kinase inhibitor. (Legend for third portion of Figure 3, Panel Q).
The effects of thrombin on brain NF/EB extravasation
One day after thrombin injections, the extravasation of NF (sodium flourescein) into brains of thrombin-treated rats was markedly greater than that detected in controls [Figure 4A; 611.1 ± 15.8 ng/ml (Throm/1day), vs. 242.5 ± 11.6 ng/ml (Cont); ** p<0.01]. Similarly, at 1 day after thrombin injections the EB extravasation was greater than that in controls [Figure 4B; 2588.6 ± 188.5 ng/ml (Throm/1day) vs. 967.1 ± 65.9 ng/ml (Cont); ** p<0.01].
Figure 4.

Brain sodium fluorescein (NF, panel A) and Evans blue (EB, panel B) extravasation increased 1 day after thrombin (Throm) injections (20U, i.c.v.), and decreased at 7 and 14 days. The thrombin inhibitor hirudin (Hir, 20U) blocked thrombin-induced NF/EB extravasation at 1 day after co-injection into the cerebral ventricles. PP2 (src family kinase inhibitor) administered with thrombin (day 0) blocked the NF/EB extravasation at 1 day after thrombin injection, whereas delayed PP2 administration (days 2-6) postponed alleviation of NF/EB extravasation at 7 days post-thrombin injection. Each column and vertical bar represents the mean ± standard error of the mean. ** p<0.01 vs. Cont; #p<0.05, ##p<0.01 vs. Throm/1day, ‡‡ p<0.01 vs. Throm/7days (one-way ANOVA followed by Tukey's post hoc test).
When co-injected with thrombin, hirudin blocked both thrombin-induced NF extravasation [Figure 4A; 384.2 ± 26.0 ng/ml (Throm/Hir/1day) vs. 611.1 ± 15.8 ng/ml (Throm/1day); ## p<0.01] and EB extravasation [Figure 4B; 1498.1 ± 143.3 ng/ml (Throm/Hir/1day) vs. 2588.6 ± 188.5 ng/ml (Throm/1day); # p<0.05] at 1 day after the injections (Figure 4).
PP2 given immediately after thrombin injections also blocked both thrombin-induced NF extravastion [Figure 4A; 242.4 ± 6.6 ng/ml (Throm/PP2/1day ) vs. 611.1 ± 15.8 ng/ml (Throm/1day); ## p<0.01] and EB extravasation [Figure 4B; 955.2 ± 90.8 ng/ml (Throm/PP2/1day) vs. 2588.6 ± 188.5 ng/ml (Throm/1day); ## p<0.01] at 1 day after thrombin injections (Figure 4).
In the absence of hirudin or PP2, NF in the brain samples decreased from 611.1 ± 15.8 ng/ml (Throm/1day) to 353.9 ± 23.8 ng/ml after 7 days (##p<0.05 for Throm/7days) and to 253.1 ± 15.7 ng/ml after 14 days (##p<0.05 for Throm/14days) (Figure 4A). Similarly, in the absence of hirudin or PP2, EB in the brains decreased from 2588.6 ± 188.5 ng/ml to 1441.0 ± 69.1 ng/ml after 7 days (##p<0.05 for Throm/7days) and to 945.7 ± 50.6 ng/ml after 14 days (##p<0.05 for Throm/14days) (Figure 4B).
Lastly, PP2 given once a day from days 2 to 6, blocked the alleviation of NF extravasation at 7 days after the thrombin injections [Figure 4A; 545.7 ± 17.0 ng/ml (Throm/PP2-days2-6/7days) vs. 353.9 ± 23.8 ng/ml (Throm/7days); ‡‡ p<0.01]. Similarly, PP2 given once a day from days 2 to 6, blocked the alleviation of EB extravasation a 7 days after thrombin injections [Figure 4B; 2217.1 ± 71.5 ng/ml (Throm/PP2-days2-6/7days) vs. 1441.0 ± 69.1 ng/ml (Throm/7days); ‡‡ p<0.01].
The effects of thrombin on brain water content
Thrombin injections into the lateral ventricle significantly increased brain water content 1 day later [79.6 ± 0.1% (Throm/1day) vs. 79.0 ± 0.1% (Cont); ** p<0.01]. Acute administration (day 0) of both hirudin and PP2 blocked this effect [Figure 5; 79.0 ± 0.1% (Throm/Hir/1day), ## p<0.01; 78.9 ± 0.1% (Throm/PP2/1day ), ## p<0.01, respectively]. The water content at seven days following thrombin injections decreased to 79.2 ± 0.1% (# p<0.05 for Throm/7days) and decreased further to 78.7 ± 0.1% (## p<0.01 for Throm/14days) at 14 days following thrombin injections (Figure 5). However, PP2 given once a day from days 2 to 6, postponed resolution of the brain edema [Figure 5; 79.6 ± 0.1% (Throm/PP2-days2-6/7days) vs. 79.2 ± 0.1% (Throm/7days); ‡ p<0.05] at 7 days after the thrombin injections (Figure 5).
Figure 5.

Brain edema (water content) increased at 1 day after thrombin (Throm) injections (20U, i.c.v.), and decreased by 7 and 14 days. The thrombin inhibitor hirudin (Hir, 20U, i.c.v.) blocked elevation thrombin-induced brain water content at 1 day after co-injection into the cerebral ventricle. Administration of PP2 (src family kinase inhibitor) at day 0 blocked the increase in brain water content observed at 1 day after thrombin injection, whereas delayed PP2 administration (days 2-6) prevented the resolution of brain water content at 7 days post-thrombin injection. Each column and vertical bar represents the mean ± standard error of the mean. ** p<0.01 vs. Cont; #p<0.05, ##p<0.01 vs. Throm/1day, ‡ p<0.05 vs. Throm/7days (one-way ANOVA followed by Tukey's post hoc test).
Discussion
The acute cerebral edema that occurs following intracerebral hemorrhage (ICH) is mediated by thrombin 1. Once ICH occurs in humans or animal models, thrombin is activated through the coagulation cascade and diffuses into the brain parenchyma. Thus, the intraventricular injections used here provide a model for the diffusion of thrombin into brain following ICH. Direct thrombin injections into the brain have been used widely to model this aspect of ICH 3, 4, 10, 11. One milliliter (1 ml) of whole blood produces ~260-360 U of thrombin and a 50 μl clot (used experimentally in rats) produces up to ~15 U of thrombin 1. Therefore, in this study we injected 20U of thrombin into the cerebral ventricle of the rat to get an approximate acute concentration of 35 U/ml of thrombin in the cerebrospinal fluid (CSF), based on an estimated volume of CSF in a 300 g rat of ~580 μl 15. This dose was slightly more than the threshold (30 U/ml) above which thrombin began to produce neuronal cell death in vitro as described in our previous studies 11.
The intraventricular thrombin injections caused reductions in BMVEC and perivascular astrocyte immunoreactivity, and disrupted the BBB as manifested by increased BBB permeability and increased cerebral water content 1 day later. The decreased BMVEC RECA-1 and astrocyte GFAP immunoreactivity could represent decreases of these proteins in injured cells, or death of the BMVEC and astrocytes. Whichever it is, thrombin induced injury to the BBB was blocked by acute administration of hirudin and PP2. Hirudin is a direct peptidomimetic thrombin inhibitor, demonstrating that it is thrombin signaling that mediates the increased edema. However, thrombin inhibitors may not be good treatment targets for ICH since these might affect the clotting and hemostatic functions of thrombin needed to halt progression of ICH 16. We recently reported that non-specific src family kinase inhibitors (like PP2), which are less likely to affect coagulation, decreased glucose hypermetabolism and cell death around ICH and improved behavioral deficits following ICH 11, 17. Thus, we tested and have shown here that blocking src family kinases with PP2 totally blocked the reductions in BMVEC and peri-vascular astrocyte immunoreactivity, and blocked increased BBB permeability and edema produced by thrombin. This can be explained by the fact that thrombin binding to thrombin receptors, called protease-activated receptors (PARs) 18, activates src kinase family members 19, 20. Thus, the current data suggest that thrombin induced edema is mediated by the following pathway: thrombin → PAR → src family kinase activation → BBB break down → increased BBB permeability and brain edema. Src family kinase members could mediate BBB permeability changes and edema by phosphorylating metalloproteinases, tight junction proteins and other BBB proteins 21, 22, and also via increased induction of VEGF 23.
The data show that although the BBB is severely damaged by one day after thrombin injection, there is significant repair by 7 days and apparent complete recovery of normal BBB permeability and brain water content by 14 days. By examining the time course of proliferating cells with BrdU, we show that the birth of BMVEC and peri-vascular astrocytes correspond with the functional repair of the BBB - as manifested by decreased brain edema and decreased BBB permeability.
Since this observation only reveals a correlation, we sought to demonstrate a causal relationship between birth of BMVEC and astrocytes and BBB repair. To test this, we administered PP2 throughout the time when BBB repair was occurring (days 2 through 6) following the thrombin injections. We used PP2 since src family kinase members play a major role in regulating the cell cycle. Src mutations result in uncontrolled cell growth – i.e. cancer. Inhibiting src family kinase members decreases astrocyte proliferation 24. Moreover, blocking src family kinases (c-Fyn) prevents BMVEC proliferation and formation of tube like structure formation of murine brain capillary endothelial cells 25. Thus, we postulated that PP2 blockade of the birth of new BMVEC and peri-vascular astrocytes would prevent BBB repair. Indeed, the data show that administration of PP2 for days 2 through 6 following thrombin injections prolongs BBB permeability and prevents the resolution of brain edema that would normally occur by 7 days. Thus, the data support the hypothesis that birth of BMVEC and perivascular astrocytes contribute to BBB repair.
Our hypothesis that thrombin injury induced the birth of new astrocytes is in agreement with other studies that have described astrocytic proliferation in CNS disorders 26-28. In particular, activation of PAR1 triggers astrogliosis after brain injury 27, and low doses of thrombin activate PAR1 to mediate proliferation of astrocytes via MAPK signaling pathways 29. Thus, the thrombin induced astrocyte proliferation pathway appears to be: thrombin → PAR1 → Src kinsase → MAPK → astrocyte proliferation. Thrombin also promotes astrocyte survival at low concentrations via a separate pathway: thrombin → PAR1 → JNK → release of the chemokine GRO/CINC-1 30.
The primary evidence for BMVEC and astrocyte proliferation in this study is the incorporation of BrdU into the cells. Although BrdU can label cells that are undergoing DNA repair and cells that re-enter the cell cycle and eventually die 31, the BrdU labeled BMVEC and astrocytes in this study likely represent newborn cells, since RECA-1-stained BMVEC and GFAP-stained astrocytes decreased 1 day following thrombin injections, and BrdU+/RECA-1+ or BrdU+/GFAP+ double-labeled BMVEC and astrocytes increased and persisted at 7 and 14 days following the thrombin injections.
In addition to the proliferating BMVEC and astrocytes, we detected a number of other BrdU+ cells located in the brain parenchyma close to the vessel wall. These could be newborn pericytes, inflammatory cells or other cells that might respond to thrombin-induced injury to the neurovascular unit and disruption of the BBB. Further studies are needed to confirm the identity(ies) of these other newborn cells.
BMVEC and astrocytes play a key role in the formation and maintenance of the BBB, and since the injury and proliferation of these cells in our study paralleled the repair of the BBB, the newborn BMVEC and astroyctes likely play an important role in the repair process. In culture, astrocytes contribute to tighter tight junctions 32 and astrocytic end feet have an important functional relationship with BMVEC 6, 9, 33-35. Astrocytes are important for correct assembly of BMVEC and pericytes into tube-like structures in vitro 36. Our data support the idea that birth of new BMVEC and astrocytes is necessary to re-establish a functional barrier and decrease BBB permeability and cerebral edema in vivo.
The cell source of the newborn BMVEC and astrocytes was not examined in this study. Recent studies suggest that a number of “stem cells” or “progenitor cells” exist throughout the mammalian brain, and some of these are associated with vascular niches 37. Such progenitor cells could serve as a source of newborn BMVEC, astrocytes and other cells of the neurovascular unit that would play a major role in re-establishing the BBB, as appeared to be the case in this study 38. Indeed, thrombin injections into the brain stimulate the birth of new neurons 39, so it is reasonable to believe that the birth of other cell types also occurs following ICH and thrombin activation. Thus, the “progenitor cells” associated with the neurovascular niche are well situated to give birth to new cells needed to repair injury to the BBB. Since delayed inhibition of src family kinases prevents repair of the BBB and is known to prevent proliferation of astrocytes and endothelial cells, we propose that src family kinases mediated proliferation of newborn BMVEC and peri-vascular astrocytes located in the neurovascular niche repair the damaged BBB. In addition, src- mediated birth of all cells that constitute the neurovascular unit and regulate the barrier (BMVEC, astrocytes, pericytes and neurons) may be required for the complete repair of the BBB following ICH/thrombin-induced injury. This process may also occur following many other types of brain injury.
There are several limitations of the study. The loss of RECA-1 and GFAP immunostaining following thrombin injections could represent decreases of these proteins in surviving cells. Alternatively, the loss of the RECA-1 and GFAP staining could represent death of endothelial cells and astrocytes. The uncertainty in the interpretation of this finding does not affect the major conclusions of the study: acute BBB breakdown is blocked by a src family kinase inhibitor; and repair of the BBB is mediated by src mediated proliferation of cells – likely including endothelial cells and astrocytes. The double-labeled BrdU-GFAP and BrdURECA-1 stained cells were not confirmed using orthogonal confocal microscopy. This is not likely to be major problem since the morphology of the double labeled cells often was similar to the morphology of the stained nuclei. Lastly, chronic PP2 administration eliminated these peri-vascular double-labeled cells supporting the interpretation that these are newborn endothelial cells and astrocytes (not shown).
Since a non-specific src family kinase inhibitor was used (PP2), the current study does not address which specific src family members might mediate the effects reported here: src itself, Fyn, Lyn or others. It is even possible that different src family members might mediate BBB breakdown and BBB repair.
Supplementary Material
Acknowledgements
This study was supported by NIH grant NS054652 (FRS). The authors have no conflicts of interest.
Footnotes
Supplementary Files
Supplementary files (Supplementary Text) are viewable in the online version.
References
- 1.Hua Y, Keep RF, Hoff JT, Xi G. Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke. 2007;38:759–762. doi: 10.1161/01.STR.0000247868.97078.10. [DOI] [PubMed] [Google Scholar]
- 2.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 3.Hua Y, Schallert T, Keep RF, et al. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33:2478–2484. doi: 10.1161/01.str.0000032302.91894.0f. [DOI] [PubMed] [Google Scholar]
- 4.Guan JX, Sun SG, Cao XB, et al. Effect of thrombin on blood brain barrier permeability and its mechanism. Chin Med J (Engl) 2004;117:1677–1681. [PubMed] [Google Scholar]
- 5.Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1:223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- 6.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 7.Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol. 2002;38:323–337. doi: 10.1016/s1537-1891(02)00200-8. [DOI] [PubMed] [Google Scholar]
- 8.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 9.Abbott N. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:527. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujimoto S, Katsuki H, Ohnishi M, et al. Thrombin induces striatal neurotoxicity depending on mitogen-activated protein kinase pathways in vivo. Neuroscience. 2007;144:694–701. doi: 10.1016/j.neuroscience.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 11.Liu DZ, Cheng XY, Ander BP, et al. Src kinase inhibition decreases thrombin-induced injury and cell cycle re-entry in striatal neurons. Neurobiol Dis. 2008;30:201–211. doi: 10.1016/j.nbd.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanley DF. Intraventricular hemorrhage: severity factor and treatment target in spontaneous intracerebral hemorrhage. Stroke. 2009;40:1533–1538. doi: 10.1161/STROKEAHA.108.535419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paxinos G, Watson C. The Rat Brain in Stereotatic Coordinates. Fourth edition Academic Press; London: 1998. [Google Scholar]
- 14.Hawkins BT, Egleton RD. Fluorescence imaging of blood-brain barrier disruption. J Neurosci Methods. 2006;151:262–267. doi: 10.1016/j.jneumeth.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Lai YL, Smith PM, Lamm WJ, Hildebrandt J. Sampling and analysis of cerebrospinal fluid for chronic studies in awake rats. J Appl Physiol. 1983;54:1754–1757. doi: 10.1152/jappl.1983.54.6.1754. [DOI] [PubMed] [Google Scholar]
- 16.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 17.Ardizzone TD, Zhan X, Ander BP, Sharp FR. SRC kinase inhibition improves acute outcomes after experimental intracerebral hemorrhage. Stroke. 2007;38:1621–1625. doi: 10.1161/STROKEAHA.106.478966. [DOI] [PubMed] [Google Scholar]
- 18.Biscardi JS, Ishizawar RC, Silva CM, Parsons SJ. Tyrosine kinase signalling in breast cancer: epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000;2:203–210. doi: 10.1186/bcr55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Reiser G. Thrombin signaling in the brain: the role of protease-activated receptors. Biol Chem. 2003;384:193–202. doi: 10.1515/BC.2003.021. [DOI] [PubMed] [Google Scholar]
- 20.Xue M, Hollenberg MD, Yong VW. Combination of thrombin and matrix metalloproteinase-9 exacerbates neurotoxicity in cell culture and intracerebral hemorrhage in mice. J Neurosci. 2006;26:10281–10291. doi: 10.1523/JNEUROSCI.2806-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guerrero J, Santibanez JF, Gonzalez A, Martinez J. EGF receptor transactivation by urokinase receptor stimulus through a mechanism involving Src and matrix metalloproteinases. Exp Cell Res. 2004;292:201–208. doi: 10.1016/j.yexcr.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 22.Kale G, Naren AP, Sheth P, Rao RK. Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2, and ZO-3. Biochem Biophys Res Commun. 2003;302:324–329. doi: 10.1016/s0006-291x(03)00167-0. [DOI] [PubMed] [Google Scholar]
- 23.Paul R, Zhang ZG, Eliceiri BP, et al. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med. 2001;7:222–227. doi: 10.1038/84675. [DOI] [PubMed] [Google Scholar]
- 24.MacFarlane SN, Sontheimer H. Modulation of Kv1.5 currents by Src tyrosine phosphorylation: potential role in the differentiation of astrocytes. J Neurosci. 2000;20:5245–5253. doi: 10.1523/JNEUROSCI.20-14-05245.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsuda S, Ohtsuru A, Yamashita S, et al. Role of c-Fyn in FGF-2-mediated tube-like structure formation by murine brain capillary endothelial cells. Biochem Biophys Res Commun. 2002;290:1354–1360. doi: 10.1006/bbrc.2002.6345. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Rudin M, Kozlova EN. Glial cell proliferation in the spinal cord after dorsal rhizotomy or sciatic nerve transection in the adult rat. Exp Brain Res. 2000;131:64–73. doi: 10.1007/s002219900273. [DOI] [PubMed] [Google Scholar]
- 27.Nicole O, Goldshmidt A, Hamill CE, et al. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J Neurosci. 2005;25:4319–4329. doi: 10.1523/JNEUROSCI.5200-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishino A, Suzuki M, Yoshimoto T, et al. A novel aspect of thrombin in the tissue reaction following central nervous system injury. Acta Neurochir Suppl (Wien) 1994;60:86–88. doi: 10.1007/978-3-7091-9334-1_22. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Ubl JJ, Stricker R, Reiser G. Thrombin (PAR-1)-induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am J Physiol Cell Physiol. 2002;283:C1351–1364. doi: 10.1152/ajpcell.00001.2002. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Luo W, Stricker R, Reiser G. Protease-activated receptor-1 protects rat astrocytes from apoptotic cell death via JNK-mediated release of the chemokine GRO/CINC-1. J Neurochem. 2006;98:1046–1060. doi: 10.1111/j.1471-4159.2006.03950.x. [DOI] [PubMed] [Google Scholar]
- 31.Kuan CY, Schloemer AJ, Lu A, et al. Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci. 2004;24:10763–10772. doi: 10.1523/JNEUROSCI.3883-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubin LL, Hall DE, Porter S, et al. A cell culture model of the blood-brain barrier. J Cell Biol. 1991;115:1725–1735. doi: 10.1083/jcb.115.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verkman AS. Mammalian aquaporins: diverse physiological roles and potential clinical significance. Expert Rev Mol Med. 2008;10:e13. doi: 10.1017/S1462399408000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noell S, Fallier-Becker P, Beyer C, et al. Effects of agrin on the expression and distribution of the water channel protein aquaporin-4 and volume regulation in cultured astrocytes. Eur J Neurosci. 2007;26:2109–2118. doi: 10.1111/j.1460-9568.2007.05850.x. [DOI] [PubMed] [Google Scholar]
- 35.Lok J, Gupta P, Guo S, et al. Cell-cell signaling in the neurovascular unit. Neurochem Res. 2007;32:2032–2045. doi: 10.1007/s11064-007-9342-9. [DOI] [PubMed] [Google Scholar]
- 36.Ramsauer M, Krause D, Dermietzel R. Angiogenesis of the blood-brain barrier in vitro and the function of cerebral pericytes. Faseb J. 2002;16:1274–1276. doi: 10.1096/fj.01-0814fje. [DOI] [PubMed] [Google Scholar]
- 37.Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci. 2006;26:13007–13016. doi: 10.1523/JNEUROSCI.4323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 39.Yang S, Song S, Hua Y, et al. Effects of thrombin on neurogenesis after intracerebral hemorrhage. Stroke. 2008;39:2079–2084. doi: 10.1161/STROKEAHA.107.508911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.