

Abstract
Dynamic field gradient focusing uses an electric field gradient generated by controlling the voltage profile of an electrode array to separate and concentrate charged analytes according to their individual electrophoretic mobilities. This study describes a new instrument in which the electrodes have been placed within the separation channel. The major challenge faced with this device is that when applied voltages to the electrodes are larger than the redox potential of water, electrolysis will occur, producing hydrogen ions (H+) plus oxygen gas on the anodes and hydroxide (OH−) plus hydrogen gas on the cathodes. The resulting gas bubbles and pH excursions can cause problems with system performance and reproducibility. An on-column, degassing system that can remove gas bubbles “on-the-fly” is described. In addition, the use of a high capacity, low-conductivity buffer to address the problem of the pH shift that occurs due to the production of H+ on the anodes is illustrated. Finally, the successful separation of three, low-molecular-weight dyes (amaranth, bromophenol blue and methyl red) is described.
Keywords: Dynamic field gradient focusing, Electrode reaction, Low molecular weight, Membrane less
1 Introduction
The ability to separate and analyze low-molecular-weight compounds is crucial in many applications, including the environmental monitoring of pollutants [1, 2] and impurity profiling in pharmaceutics [3, 4]. The most widely used analytical technique for these types of analysis is RPLC [5, 6]. As powerful as they are, the HPLC methods cannot always deliver the resolution and speed desired for quantitative analysis, e.g. chiral separations (several reviews on this subject are found in [7–9]) and isoform fractionation [10], and hence alternative methods and instrumentation are needed.
CZE has recently attracted interest because it is an orthogonal separation strategy [9] which can be used as an alternative approach to or in conjunction with RPLC [11]. Although numerous applications of CZE for monitoring low-molecular-weight species, including pollutants [12–14] and pharmaceuticals [15, 16], have been published, preconcentration steps are required prior to CZE of trace compounds [17–19].
An alternative to isocratic CZE is the use of ITP which utilizes a discontinuous buffer system to concentrate charged analytes between a trailing electrolyte and a leading electrolyte [18, 20]. ITP produces contiguous bands which may be baseline separated by coupling on to another method [17] or by the use of spacers [21, 22].
Recently, a new class of separation techniques, known as electric field gradient focusing (EFGF), which do not require pre-concentration, coupling to other methods or spacers, has been developed. In EFGF, charged analytes are simultaneously separated and concentrated according to their individual electrophoretic mobilities using an electric field gradient and an opposing hydrodynamic flow. Analytes migrate to a discrete position, a.k.a., focal point, at which the net force of the flow and the electric field acting upon the analyte sum to zero. Several different EFGF devices have been investigated with each generating an electric field gradient in a unique fashion, e.g. shaped channels [23–28], mismatched buffers to generate conductivity gradients [29–31], temperature gradient focusing (TGF) [32–39], ion-depleted regions [40, 41] or controlling the voltage on discrete electrodes [42–51].
This last technique, known as dynamic field gradient focusing (DFGF), has several advantages over the other EFGF methods. In DFGF, the electric field gradient is generated by specifying the voltage on individual electrodes in a computer-controlled electrode array. This provides dynamic control of the electric field which gives the operator the option to manipulate the electric field profile “on-the-fly” to increase resolution or to individually elute species past a detector, providing the operator greater control of the system compared with other analytical separation techniques. By incorporating a whole-column imaging detector [52–55], the manipulation of the electric field profile could be completely automated or, conversely, once a method has been optimized, the instrument could be operated like any other automated chromatography system.
One of the problems with DFGF as well as most EFGF devices is the need for a semi-permeable membrane. This membrane isolates the separation channel from the electrodes allowing the passage of current-carrying ions, while at the same time retaining target analytes. When the species being analyzed are large, i.e. several orders of magnitude greater than the current-carrying ions, the semi-permeable membrane is able to accomplish this without retarding the passage of these ions through the membrane. When the target analytes are less than an order of magnitude greater than the membrane pores, it is difficult to keep them from crossing this membrane. In addition, under these conditions, the current-carrying ions experience resistance as they pass through the membrane, leading to distortions in the electric field [43] which, if severe enough, can render the device unusable. Up until now, this has limited the application of DFGF to the analysis of large-molecular-weight species.
Of the EFGF devices, only TGF and a recently developed device which utilizes an embedded bipolar electrode [40, 41] do not require a semi-permeable membrane. In these two cases, the electrodes are placed in large reservoirs at the ends of the separation channel. These devices are not limited to the separation of large molecules, and in fact, the analysis of small-molecular-weight species in TGF has been demonstrated in a number of publications [33–39, 56–58]. However, EFGF devices which utilize shaped channels or conductivity gradients, specifically those generated by mismatched buffers, require a semi-permeable membrane.
Operating a DFGF without a membrane would require that the electrodes be placed within the separation channel, introducing several problems: (i) the shape of the electric field, (ii) the production of electrolysis products, including gases, hydrogen ions and hydroxides, and (iii) the reaction/degradation of the target analytes on the surface of the electrodes.
The presence of the discrete electrodes can have a large impact on the shape of the electric field profile. As the electrodes in this newly developed DFGF device will be placed in the separation channel, voltage degradation, which in this case refers to the difference between the voltages applied to the electrodes and the voltages measured within the separation channel, will not occur. A previous publication out of our group [42] examined the impact of electrode placement on the shape of the electric field and its influence on system performance. When electrodes were placed at large distances from the separation channel, there was substantial voltage degradation that negatively impacted the shape of the electric field. As the electrodes were moved closer to the separation channel, voltage degradation became less of an issue and the presence of the discrete electrodes became more pronounced, causing the electric field to become stair-stepped. This stair-stepping can ultimately limit the maximum peak capacity of the system to the number of electrodes. However, the peak capacity could be increased by incorporating more electrodes into the separation channel.
When current is applied to platinum electrodes placed within aqueous solutions, hydrolysis occurs if the applied voltages are larger than the redox potential of water. Oxidation at the anodes results in the generation of hydrogen ions (H+) and oxygen gas, while reduction at the cathode produces hydroxide (OH−) and hydrogen gas according to the following equations:
| (1) |
| (2) |
As the DFGF system is typically run with a series of anodes and only one cathode [44], the production of H+ will result in the pH decreasing as the buffering capacity of the system is diminished. Simply increasing the concentration of the buffering ion will not solve this problem, because this would increase the current and would inevitably increase the production rate of the electrolysis products [59].
In addition to changes in pH, gas bubbles will be produced at all electrodes, hindering electrophoretic focusing of the analytes and obstructing current and fluid flow. Although chemical techniques such as an oxygen scavenger [60, 61] or a redox pair (i.e. quinone/hydroquinone) [62, 63] could be used to mitigate these issues, their incorporation increases the complexity of the running buffer and could interfere with the separation.
This study describes the development of a membrane-less DFGF device which has electrodes placed directly in the separation channel. Experimental results are used to examine the removal of electrolysis gases and the pH shift caused by electrolysis within the separation channel. Finally, the separation of a three component sample will be shown to illustrate the capabilities of this DFGF device for the separation of low-molecular-weight species.
2 Materials and methods
2.1 Reagents
Alexa Fluor® 350 carboxylic acid, succinimidyl ester reactive dye was purchased from Invitrogen (Carlsbad, CA, USA). Bromophenol blue (BPB) was obtained from Bio-Rad Laboratories (Hercules, CA, USA). Amaranth (AM), methyl red (MR), neutral red and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2 DFGF apparatus and operation
A schematic of the focusing chamber is shown in Fig. 1A. The DFGF apparatus is constructed of three 5.08 cm×8.02 cm×1.27 cm pieces of acrylic. The top acrylic block contains a 3.56 cm×0.1 cm×200 μm separation channel which is packed with Bio-Rad P-2 resin (Bio-Rad Laboratories) to reduce convective dispersion. Unlike previous generations of DFGF chambers, the 25 computer-controlled platinum electrodes, rather than being located in a separate electrode housing, are located within the separation channel. These electrodes are used to generate the electric field gradient and are controlled using a previously described voltage controller [42, 64]. The bottom block contains a 3.81 cm×0.64 cm×1.11 cm channel. A vacuum (~635 mm Hg) is applied to this channel and serves as an on-column degasser to remove gases generated from electrolysis of water on the electrode surface. The separation channel and vacuum chamber are isolated from each other using a 250-μm thick Teflon sheet (C.S. Hyde Company, Lake Villa, IL, USA) supported by a 3.56 cm×0.1 cm×0.32 cm piece of porous ceramic (Kerafol, Germany), which prevents bowing of the Teflon when a vacuum is applied. The Teflon has a high permeability for oxygen and can be used to remove gases without allowing for the passage of liquid or target analytes.
Figure 1.
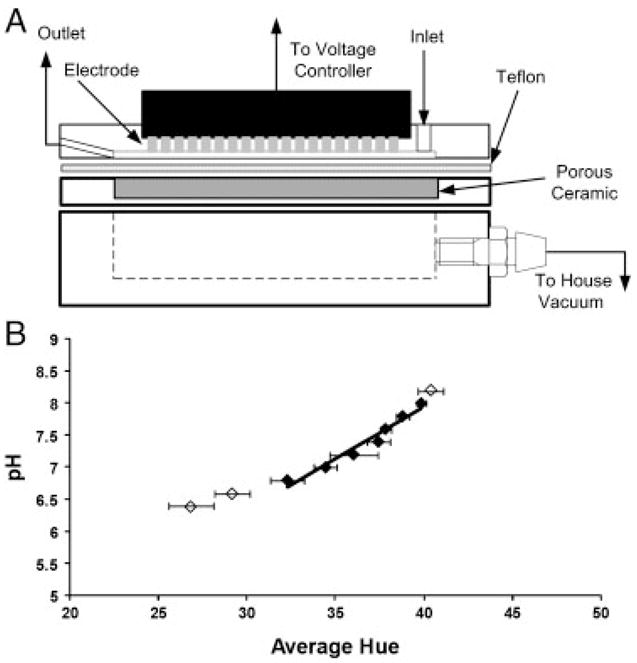
(A) Schematic of the membrane-less DFGF device. The electrodes in this apparatus are placed within the separation channel. To remove gas produced on the electrodes, an on-column degassing system, which uses a 250-μm thick Teflon sheet with an applied vacuum, is employed. (B) Calibration curve to determine pH based on the measured hue within the separation channel. Only the (◆) symbols were included in the correlation as the (◇) values line outside of the indicating range for neutral red. The solid line represents the least squares linear fit and has a standard error of ~0.09 pH units.
A syringe pump (KD Scientific, Holliston, MA, USA) fitted with a 250-μL glass Hamilton syringe was used to pump running buffer through the separation channel at a rate of 0.04 μL/min. The dye sample of 0.0375 mg/mL BPB, 0.075 mg/mL AM and 0.075 mg/mL MR was introduced by completely filling the separation channel with sample prior to applying the electric field. Voltages were applied to the electrodes to generate an electric field gradient of 3.9 V/cm2 with a top end electric field strength of 14.2 V/cm. The dyes were then allowed to focus for up to 11.5 h.
2.3 pH measurement
Determination of in situ pH in this device cannot be done by simply measuring the pH of the outlet fluid. This pH provides little information on the pH within the separation channel. Even the incorporation of an on-column probe would only provide point pH information within the column and the use of multiple probes is not practical. To mitigate the need for a pH probe, optical techniques can be used to measure the pH. Although the most common technique, specifically in biological systems [65–68] and in microchips [69, 70], is the use of pH-sensitive fluorescent dyes, these have problems with quenching and degradation [71]. In addition, they require expensive equipment for excitation and detection. Instead, we have chosen to modify the chromatographic packing with a pH indicating dye [71–74]. These dyes do not suffer from quenching, and instead of needing expensive equipment, this approach only requires a digital camera and digital image analysis software.
Neutral red, a visible pH indicator with a pH transition range of 6.8–8.0 [75], was bound to the surface of the Bio-Rad P-2 resin following the method outlined by Hardin and Ivory [74]. Briefly, 2.5 mL of settled resin was combined with 10 mL of 0.002% neutral red (w/v) solution and gently mixed at room temperature for 1 h. The resin was washed with buffer five times or until very little color was noticeable in the supernatant. This packing was then loaded into the separation channel. Overtime, the dye tended to leach from the surface of the resin. To overcome this experimentally, a small amount of netural red (0.1 mM) was added to the running buffer [74] to ensure that sufficient dye remained on the surface of the resin. This can be done because neutral red will not focus within the separation channel.
A digital image of the separation channel was taken using a Nikon D60® (Nikon, Tokyo, Japan) camera. The image was analyzed with Adobe Photoshop 5.5® (Adobe, San Jose, CA, USA) to determine the average hue. The pH was then determined by evaluating the average hue across the separation channel as a function of pH. Figure 1B shows that the relationship between pH and hue is linear within the indicating range of pH 6.8–8.0. Least square analysis was used to determine a linear fit of the data.
To examine the impact of a focused peak on the local pH, Alexa Fluor 350®, a fluorescent dye, was used because it had no noticeable color under visible light and would not interfere with hue measurements. The dye was pumped into the separation channel, focused for 3 h, and then the pH of the separation channel was calculated as described above. To determine the location of the focused dye, the system was exposed to UV light at 365 nm. Under this wavelength, the dye was visible and digital images were taken. These images were analyzed using ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA) to produce plot profiles.
3 Results and discussion
3.1 On-column removal of gas bubbles
For this device, we chose to use a channel length of 3.56 cm. Compared with our traditionally used channel lengths of 12.7 cm [64], 6.35 cm [45] or 5.08 cm [42–44], the shorter length of this device allows us to operate at lower voltages which will decrease the production rate of the electrolysis products, while still achieving usable electric field strengths.
Although operating with the shorter channel length helps to limit the production of electrolysis products, the generation of electrolysis gases still occurs. Rather than use an oxygen scavenging chemical to remove gas bubbles produced on the electrodes, we chose to use on-column degassing. To do this, a 250-μm thick Teflon sheet was placed below the separation channel and a vacuum was applied. Teflon has a high permeability of oxygen and does not allow the passage of the target analytes or liquid. By applying a vacuum to the device, oxygen gas produced within the separation channel can be removed “on-the-fly”.
To test the performance of the on-column degassing system, the maximum electric field achievable before substantial gas bubble formation with and without gas removal was tested. Substantial gas bubble formation was determined qualitatively as the amount of gas generation that would hinder focusing experiments. Without on-line gas removal, bubbles on the anodes became noticeable at 8.7 V/cm, and by 11.8 V/cm the device was no longer usable for performing separation experiments. With the vacuum turned-on and bubbles being removed on-the-fly, the first noticeable bubbles on the anodes did not appear until 16.5 V/cm with the channel becoming completely obstructed at 18.5 V/cm.
One problem encountered is that as the system only contains one cathode, all current entering the device must exit at a single electrode. This means that the current density at the cathode is higher than on any of the other electrodes. As a result, bubbles produced on the cathode are difficult to control and remove. It is the production of electrolysis gas on the cathode that limits the electric field strengths which can be used. To alleviate this problem, the cathode should be located beyond the outlet of the separation channel and if possible designed to be flushed to remove electrolysis products. This has not been incorporated into the design of this device, but will be considered for future iterations.
3.2 Minimizing the pH excursion
As the pH changes within the separation channel due to the production of H+, the net charge of the analytes, and thus their electrophoretic mobilities, will change, leading to reproducibility problems and a subsequent decrease in system performance. To examine the extent of the experimental pH excursion, the pH measurement technique described above was used. A typical DFGF buffer of 25 mM Tris acetate at pH 8.0 was pumped through the separation channel at 0.04 μL/min. A top-end electric field strength of 14.2 V/cm was used and digital images were taken at 3 and 10 h. The results of the pH analysis are shown in Fig. 2A.
Figure 2.
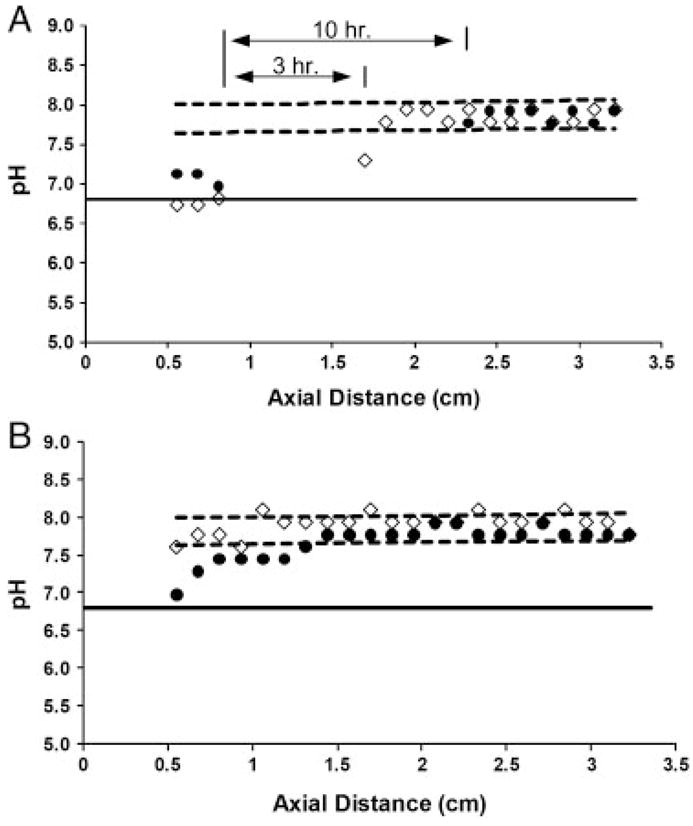
(A) Measured pH profile along the separation channel for 25 mM Tris acetate buffer at 3 h (diamonds) and 10 h (circles). The region between the 3 and 10 h lines indicates where the pH dropped below the indicating range for neutral red. As a result, pH values in these regions could not be measured with any accuracy. The dashed lines indicate 95% confidence intervals based on our ability to accurately measure pH 8.0. (B) Experimentally measured pH profile for 100 mM bis-Tris EACA at pH 8.0 at 3 h (diamonds) and 10 h (circles). The dashed lines represent 95% confidence intervals.
After only 3 h, the observed pH shift near the inlet of the separation channel is lower than the initial pH and shows a shift of ~1.2. At 0.9 cm, the pH drops far below 6.8 and the pH cannot be determined with any certainty because it lies outside of the linear region of the calibration curve (Fig. 2A). The pH stays below 6.8 for 1 cm before returning to a value of nearly 8.0 near the outlet, which is due to the presence of the cathode. After 10 h, the pH excursion has grown to encompass nearly half of the separation channel.
A possible solution for decreasing the pH excursion is to operate at higher flow rates. The increased flow would introduce more “fresh” buffer into the system while removing more “exhausted” buffer. This, however, is not a viable solution to the pH shift problem, because the flow rate can only be increased so far before analytes are no longer able to focus in the separation channel. Instead, the buffering capacity needs to be increased; however, simply increasing the concentration of the running buffer will not work, because the increased conductivity will lead to an increase in the current and, subsequently, to an increase in the production of electrolysis products.
Instead of simply increasing the concentration of the running buffer, a system with high buffer capacity and low conductivity needs to be employed. Bier et al. [76] developed a new buffer system to be used with continuous flow electrophoresis or IEF which fits these requirements. They made a list of several buffer pairs which were specifically designed so that they were largely neutral (90–99%) and had overlapping pK ranges such that they titrated each other. These buffer pairs, which were marketed as Optifocus™ buffer pairs by Protein Technologies (Tucson, AZ, USA) and later by Bio-Rad Laboratories under the trade name Rotolytes ™, were relatively stable to the passage of current and could achieve higher buffering capacities while still being at or below the conductivities of traditional electrophoresis buffers.
For our system, we chose 100 mM bis-Tris titrated to pH 8.0 with å-amino caproic acid (EACA). This buffer has greater buffering capacity but a twofold lower conductivity when compared with 25 mM Tris acetate (pH 8.0). The result of using this buffer was shown experimentally (Fig. 2B). After 3 h, the pH measured within the separation channel was within the 95% confidence intervals and indicates that the pH shift is smaller than the error associated with our ability to measure pH 8.0. Even after 10 h, the pH lies only slightly outside of the confidence interval and shows a pH shift of only 0.5–0.8, a marked improvement over the Tris acetate buffer. In practice, separations will typically be run for only 2–3 h such that substantial pH shifts should not be occurring.
One further consideration for this system is the effect of a focused analyte on the pH. As the species concentrates in the device, the local pH will decrease, provided that the conductivity of the focused band is greater than the background electrolyte. To show the effect of a focused band, Alexa Fluor 350® was injected into the separation channel and allowed to focus for 3 h. Alexa Fluor 350® was used because it shows no visible color, which would interfere with the pH measurement, but can be visualized when exposed to UV light. The results are shown in Fig. 3. For an initial concentration of 0.1 mg/mL, which corresponds to 0.7 μg of injected dye, the effect of the focused peak on the pH was minimal and the pH measured along the separation channel fell within the 95% confidence interval (Fig. 3A). When the concentration was increased to 0.5 mg/mL (3.5 μg), a pH shift occurred in the region of the separation channel in which the dye had focused (Fig. 3B). Although the shift was relatively small (~0.5 pH units), this could still cause problems, suggesting that the initial loads of samples should be chosen so as to minimize the effect of the focused peak on the local pH.
Figure 3.
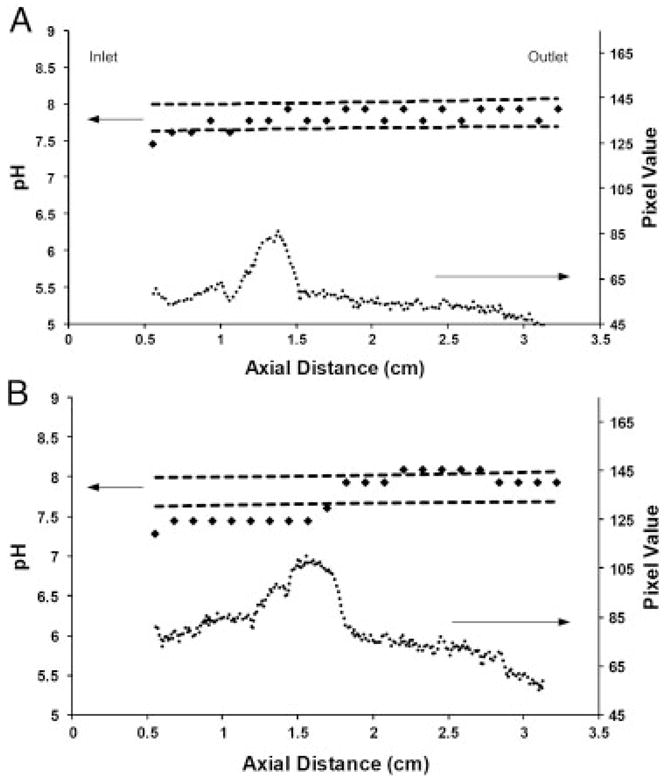
(A) Experimentally measured pH (diamonds) with focused Alexa Fluor 350® injected at a concentration 0.1 mg/mL. The presence of the focused dye (circles) has very little impact on the pH profile. The dashed lines are 95% confidence intervals. (B) Experimentally measured pH (diamonds) with focused Alexa Fluor 350® injected at a concentration of 0.5 mg/mL. The focused dye causes a 0.5 pH unit shift in the separation channel. The dashed lined represent the 95% confidence intervals.
3.3 Three component separation
A sample of three low-molecular-weight dyes (AM, BPB and MR), which could not have been separated in the previous DFGF devices, was injected in 300 mM bis-Tris EACA (pH 8.6) buffer and allowed to focus. The use of dyes allowed for easy detection and a digital camera (Nikon D60) could be used to document the results. The electric field strength used in this separation is below the value at which noticeable gas bubbles would form with the degassing system in place so that gas accumulation does not occur. In addition, the total dye loads used were below 1 μg to ensure that when they focused that they did not cause a local pH excursion.
The results are shown in Fig. 4. Under these conditions, the three dyes are fully resolved with peak widths approximately equal to the distance between two electrodes (~1.27 mm). In practice, this is likely the tightest that each band will become due to the presence of stair-stepping in the electric field. The dyes could be held within the separation channel under focusing conditions for more than 10 h with no noticeable change in concentration or focusing position. An implication of this is that it is unlikely that the dyes are reacting/degrading to a large extent on the electrode surface. One would expect that over a time period >10 h a noticeable change in focusing position due to a redox reaction or a change in color intensity due to degradation would be noticeable. However, this is not seen experimentally. To determine quantitatively, voltametry along with mass spectrometry could be used.
Figure 4.

Separation of AM, BPB and MR in a membrane-less DFGF apparatus. The three dyes are well resolved and have band widths approximately equal to the distance between two electrodes (1.27 mm). The dyes were held within the separation channel under focusing conditions for greater than 10 h without noticeable signs of degradation or redox reactions on the electrodes. The electrodes within the separation channel are easily recognized.
4 Concluding remarks
This study describes the development of a membrane-less DFGF device for the separation of low-molecular-weight species. This device has the electrodes placed directly within the separation channel which introduces several problems, including the generation of electrolysis gases and changes to the system pH caused by electrolysis.
Electrolysis gases produced on the electrodes were removed using an on-column degassing system which utilized a 250-μm thick Teflon sheet across which a vacuum was applied. With on-column degassing in place, a twofold increase in the electric field strength could be achieved. Although the field strengths (~16.5 V/cm) are moderate by current electrophoresis standards, the important conclusion is that on-the-fly degassing is possible in this DFGF device. Further improvements to the degassing system will be made by using thinner Teflon sheets and by examining other materials which have higher oxygen gas permeability. In addition, the cathode, which produces more gas than any of the other electrodes and ultimately limits the achievable electric field strengths, will be placed beyond the outlet of the separation channel and designed to be flushed to remove electrolysis products. In addition to these improvements, the operating electric field can be increased by moving to even smaller separation channels.
When operated with a running buffer of 25 mM Tris acetate at pH 8.0, an experimentally observed pH shift of greater than 1.2 and encompassing a substantial portion of the separation channel occurs. Simply increasing the buffering capacity by increasing the concentration of the buffering ion does not work because the increased conductivity leads to an increased production of electrolysis products. Instead, 100 mM bis-Tris EACA, a buffer which has both high capacity and low conductivity, was used. It was shown experimentally that after 3 h no pH shift was noticeable and that after 10 h a shift of only 0.5 occurred.
As the controller controls on voltage rather than current, the increased conductivity due to the presence of focused analytes causes an increase in the production of electrolysis products. Even with the high capacity/low conductivity buffer, dye loads of greater than 0.5 mg/mL (3.5 μg) resulted in a pH shift of more than 0.5 in the region of the focused band. At this overloaded concentration, in addition to the pH shift, other problems will arise, including increased band widths and distortions to the electric field profile [25, 42]. Although these problems are mitigated by keeping sample concentrations low (<1–2 μg), such precautions are not always possible. This result points to the need for the controllers to control on current rather than voltage.
To illustrate the capability of DFGF for the analysis of low molecular species, three dyes (AM, BPB and MR) were separated and concentrated. The three dyes formed well-shaped bands that were fully resolved. In addition, the dyes were held in the separation channel for over 10 h without any noticeable signs of undesirable reactions occurring on the surface of the electrodes.
Acknowledgments
The authors thank Washington State University National Institutes of Health Protein Biotechnology Training Program (grant TM2GM08336) and Pfizer for financial support of this project. They also thank Peter Myers from the Department of Chemistry at the University of Liverpool for frequent communication and insight on this project.
Abbreviations
- AM
amaranth
- BPB
bromophenol blue
- DFGF
Dynamic field gradient focusing
- EACA
ε-amino caproic acid
- EFGF
electric field gradient focusing
- MR
methyl red
- TGF
temperature gradient focusing
Footnotes
The authors have declared no conflict of interest.
References
- 1.Noblitt SD, Mazzoleni LR, Hering SV, Collett JL, Henry CS. J Chromatogr A. 2007;1154:400–406. doi: 10.1016/j.chroma.2007.03.069. [DOI] [PubMed] [Google Scholar]
- 2.Siren H, Vantsi S. J Chromatogr A. 2002;957:17–26. doi: 10.1016/s0021-9673(02)00218-2. [DOI] [PubMed] [Google Scholar]
- 3.Szolar OHJ. Anal Chim Acta. 2007;582:191–200. doi: 10.1016/j.aca.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 4.Holzgrabe U, Brinz D, Kopec S, Weber C, Bitar Y. Electrophoresis. 2006;27:2283–2292. doi: 10.1002/elps.200600016. [DOI] [PubMed] [Google Scholar]
- 5.Chung Y, Lee K. Microchem J. 2001;69:143–152. [Google Scholar]
- 6.Williams RC, Alasandro MS, Fasone VL, Boucher RJ, Edwards JF. J Pharm Biomed Anal. 1996;14:1539–1546. doi: 10.1016/0731-7085(96)01740-2. [DOI] [PubMed] [Google Scholar]
- 7.Chankvetadze B. J Chromatogr A. 2007;1168:45–70. doi: 10.1016/j.chroma.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Gubitz G, Schmid MG. J Chromatogr A. 2008;1204:140–156. doi: 10.1016/j.chroma.2008.07.071. [DOI] [PubMed] [Google Scholar]
- 9.Preinerstorfer B, Lammerhofer M, Lindner W. Electrophoresis. 2009;30:100–132. doi: 10.1002/elps.200800607. [DOI] [PubMed] [Google Scholar]
- 10.Chang WWP, Hobson C, Bomberger DC, Schneider LV. Electrophoresis. 2005;26:2179–2186. doi: 10.1002/elps.200410283. [DOI] [PubMed] [Google Scholar]
- 11.Floridia L, Pietropaolo AM, Tavazzani M, Rubino FM, Colombi A. J Chromatogr B Analyt Technol Biomed Life Sci. 1999;726:95–103. doi: 10.1016/s0378-4347(98)00561-1. [DOI] [PubMed] [Google Scholar]
- 12.Craston DH, Saeed M. J Chromatogr A. 1998;827:1–12. [Google Scholar]
- 13.Dabek-Zlotorzynska E, Aranda-Rodriguez R, Graham L. J Sep Sci. 2005;28:1520–1528. doi: 10.1002/jssc.200400053. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Campana AM, Gamiz-Gracia L, Lara FJ, Iruela MD, Cruces-Blanco C. Anal Bioanal Chem. 2009;395:967–986. doi: 10.1007/s00216-009-2867-9. [DOI] [PubMed] [Google Scholar]
- 15.Jouyban A, Kenndler E. Electrophoresis. 2008;29:3531–3551. doi: 10.1002/elps.200800054. [DOI] [PubMed] [Google Scholar]
- 16.Visky D, Jimidar I, Van Ael W, Vennekens T, Redlich D, De Smet M. Electrophoresis. 2005;26:1541–1549. doi: 10.1002/elps.200410225. [DOI] [PubMed] [Google Scholar]
- 17.Prochazkova A, Krivankova L, Bocek P. J Chromatogr A. 1999;838:213–221. doi: 10.1016/s0021-9673(99)00114-4. [DOI] [PubMed] [Google Scholar]
- 18.Eldridge SL, Almeida VK, Korir AK, Larive CK. Anal Chem. 2007;79:8446–8453. doi: 10.1021/ac7016629. [DOI] [PubMed] [Google Scholar]
- 19.Musilova J, Sedlacek V, Kucera I, Glatz Z. J Sep Sci. 2009;32:2416–2420. doi: 10.1002/jssc.200900276. [DOI] [PubMed] [Google Scholar]
- 20.Gebauer P, Mala Z, Bocek P. Electrophoresis. 2009;30:29–35. doi: 10.1002/elps.200800425. [DOI] [PubMed] [Google Scholar]
- 21.Khurana TK, Santiago JG. Lab Chip. 2009;9:1377–1384. doi: 10.1039/b815460k. [DOI] [PubMed] [Google Scholar]
- 22.Nagyova I, Kaniansky D. J Chromatogr A. 2001;916:191–200. doi: 10.1016/s0021-9673(00)01095-5. [DOI] [PubMed] [Google Scholar]
- 23.Humble PH, Kelly RT, Woolley AT, Tolley HD, Lee ML. Anal Chem. 2004;76:5641–5648. doi: 10.1021/ac040055+. [DOI] [PubMed] [Google Scholar]
- 24.Kelly RT, Li Y, Woolley AT. Anal Chem. 2006;78:2565–2570. doi: 10.1021/ac0521394. [DOI] [PubMed] [Google Scholar]
- 25.Koegler WS, Ivory CF. Biotechnol Prog. 1996;12:822–836. [Google Scholar]
- 26.Koegler WS, Ivory CF. J Chromatogr A. 1996;726:229–236. [Google Scholar]
- 27.Liu J, Sun X, Frarnsworth PB, Lee ML. Anal Chem. 2006;78:4654–4662. doi: 10.1021/ac060204j. [DOI] [PubMed] [Google Scholar]
- 28.Sun X, Farnsworth PB, Woolley AT, Tolley HD, Warnick KF, Lee ML. Anal Chem. 2008;80:451–460. doi: 10.1021/ac0713104. [DOI] [PubMed] [Google Scholar]
- 29.Greenlee RD, Ivory CF. Biotechnol Prog. 1998;14:300–309. doi: 10.1021/bp970113v. [DOI] [PubMed] [Google Scholar]
- 30.Lin SL, Tolley HD, Lee ML. Chromatographia. 2005;62:277–281. [Google Scholar]
- 31.Wang QG, Lin SL, Warnick KF, Tolley HD, Lee ML. J Chromatogr A. 2003;985:455–462. doi: 10.1016/s0021-9673(02)01399-7. [DOI] [PubMed] [Google Scholar]
- 32.Balss KM, Ross D, Begley HC, Olsen KG, Tarlov MJ. J Am Chem Soc. 2004;126:13474–13479. doi: 10.1021/ja030667w. [DOI] [PubMed] [Google Scholar]
- 33.Balss KM, Vreeland WN, Phinney KW, Ross D. Anal Chem. 2004;76:7243–7249. doi: 10.1021/ac049046r. [DOI] [PubMed] [Google Scholar]
- 34.Hoebel SJ, Balss KM, Jones BJ, Malliaris CD, Munson MS, Vreeland WN, Ross D. Anal Chem. 2006;78:7186–7190. doi: 10.1021/ac060934r. [DOI] [PubMed] [Google Scholar]
- 35.Kim SM, Sommer GJ, Burns MA, Hasselbrink EF. Anal Chem. 2006;78:8028–8035. doi: 10.1021/ac061194p. [DOI] [PubMed] [Google Scholar]
- 36.Matsui T, Franzke J, Manz A, Janasek D. Electrophoresis. 2007;28:4606–4611. doi: 10.1002/elps.200700272. [DOI] [PubMed] [Google Scholar]
- 37.Munson MS, Danger G, Shackman JG, Ross D. Anal Chem. 2007;79:6201–6207. doi: 10.1021/ac070689r. [DOI] [PubMed] [Google Scholar]
- 38.Ross D, Locascio LE. Anal Chem. 2002;74:2556–2564. doi: 10.1021/ac025528w. [DOI] [PubMed] [Google Scholar]
- 39.Shackman JG, Munson MS, Ross D. Anal Bioanal Chem. 2007;387:155–158. doi: 10.1007/s00216-006-0913-4. [DOI] [PubMed] [Google Scholar]
- 40.Dhopeshwarkar R, Hlushkou D, Nguyen M, Tallarek U, Crooks RM. J Am Chem Soc. 2008;130:10480–10481. doi: 10.1021/ja8036405. [DOI] [PubMed] [Google Scholar]
- 41.Hlushkou D, Perdue RK, Dhopeshwarkar R, Crooks RM, Tallarek U. Lab Chip. 2009;9:1903–1913. doi: 10.1039/b822404h. [DOI] [PubMed] [Google Scholar]
- 42.Burke JM, Ivory CF. Electrophoresis. 2008;29:1013–1025. doi: 10.1002/elps.200700447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burke JM, Ivory CF. Electrophoresis. 2010 doi: 10.1002/elps.200900222. this issue. [DOI] [Google Scholar]
- 44.Burke JM, Huang Z, Ivory CF. Anal Chem. 2009;81:8236–8243. doi: 10.1021/ac901634y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang Z, Ivory CF. Anal Chem. 1999;71:1628–1632. [Google Scholar]
- 46.Myers P, Bartle KD. J Chromatogr A. 2004;1044:253–258. doi: 10.1016/j.chroma.2004.04.069. [DOI] [PubMed] [Google Scholar]
- 47.Tracy NI, Huang Z, Ivory CF. Biotechnol Prog. 2008;24:444–451. doi: 10.1021/bp070251c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tracy NI, Ivory CF. J Sep Sci. 2008;31:341–352. doi: 10.1002/jssc.200700307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tracy NI, Ivory CF. Electrophoresis. 2008;29:2820–2827. doi: 10.1002/elps.200890058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tracy NI, Ivory CF. AIChE J. 2009;55:63–74. [Google Scholar]
- 51.Tunon PG, Wang Y, Myers P, Bartle KD, Bowhill L, Ivory CF, Ansell RJ. Electrophoresis. 2008;29:457–465. doi: 10.1002/elps.200700443. [DOI] [PubMed] [Google Scholar]
- 52.Wu JQ, Pawliszyn J. Anal Chem. 1992;64:224–227. [Google Scholar]
- 53.Wu XZ, Pawliszyn J. Electrophoresis. 2002;23:542–549. doi: 10.1002/1522-2683(200202)23:4<542::AID-ELPS542>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 54.Wu XZ, Wu JQ, Pawliszyn J. Electrophoresis. 1995;16:1474–1478. doi: 10.1002/elps.11501601244. [DOI] [PubMed] [Google Scholar]
- 55.Yao B, Yang HH, Liang QL, Luo G, Wang L, Kangning R, Gao Y, Yiming W, Qiu Y. Anal Chem. 2006;78:5845–5850. doi: 10.1021/ac060445r. [DOI] [PubMed] [Google Scholar]
- 56.Danger G, Ross D. Electrophoresis. 2008;29:3107–3114. doi: 10.1002/elps.200700778. [DOI] [PubMed] [Google Scholar]
- 57.Munson MS, Meacham JM, Ross D, Locascio LE. Electrophoresis. 2008;29:3456–3465. doi: 10.1002/elps.200800210. [DOI] [PubMed] [Google Scholar]
- 58.Shackman JG, Munson MS, Kan CW, Ross D. Electrophoresis. 2006;27:3420–3427. doi: 10.1002/elps.200600078. [DOI] [PubMed] [Google Scholar]
- 59.Lee DY, Chang GD. Anal Chem. 2009;81:3957–3964. doi: 10.1021/ac900281n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aitken CE, Marshall RA, Pulglisi JD. Biophys J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun G, Anderson VE. Electrophoresis. 2004;25:959–965. doi: 10.1002/elps.200305800. [DOI] [PubMed] [Google Scholar]
- 62.Caldwell KD, Gao YS. Anal Chem. 1993;65:1764–1772. doi: 10.1021/ac00061a021. [DOI] [PubMed] [Google Scholar]
- 63.Moini M, Cao P, Bard AJ. Anal Chem. 1999;71:1658–1661. doi: 10.1021/ac9811266. [DOI] [PubMed] [Google Scholar]
- 64.Huang Z. Washington State University. Pullman, WA: 2001. p. 114. [Google Scholar]
- 65.Blank PS, Silverman HS, Chung OY, Hogue BA, Stern MD, Hansford RG, Lakatta EG, Capogrossi MC. Am J Phys. 1992;263:H276–H284. doi: 10.1152/ajpheart.1992.263.1.H276. [DOI] [PubMed] [Google Scholar]
- 66.Jankowski A, Kim JH, Collins RF, Daneman R, Walton P, Grinstein S. J Biol Chem. 2001;276:48748–48753. doi: 10.1074/jbc.M109003200. [DOI] [PubMed] [Google Scholar]
- 67.Martinezzaguilan R, Martinez GM, Lattanzio F, Gillies RJ. Am J Phys. 1991;260:C297–C307. doi: 10.1152/ajpcell.1991.260.2.C297. [DOI] [PubMed] [Google Scholar]
- 68.Hunter RC, Beveridge TJ. Appl Environ Microbiol. 2005;71:2501–2510. doi: 10.1128/AEM.71.5.2501-2510.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klauke N, Monaghan P, Sinclair G, Padgett M, Cooper J. Lab Chip. 2006;6:788–793. doi: 10.1039/b517237c. [DOI] [PubMed] [Google Scholar]
- 70.Bottenus D, Oh YJ, Han SM, Ivory CF. Lab Chip. 2009;9:219–231. doi: 10.1039/b803278e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cho JK, Wong LS, Dean TW, Ichihara O, Muller C, Bradley M. Chem Commun. 2004:1470–1471. doi: 10.1039/b405198j. [DOI] [PubMed] [Google Scholar]
- 72.Wong LS, Birembaut F, Brocklesby WS, Frey JG, Bradley M. Anal Chem. 2005;77:2247–2251. doi: 10.1021/ac049319i. [DOI] [PubMed] [Google Scholar]
- 73.Maruyama H, Arai F, Fukuda T. Lab Chip. 2008;8:346–351. doi: 10.1039/b712566f. [DOI] [PubMed] [Google Scholar]
- 74.Hardin AM, Ivory CF. J Colloid Interface Sci. 2006;302:560–567. doi: 10.1016/j.jcis.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 75.Bishop E. Indicators. Pergamon Press; Oxford, NY: 1972. [Google Scholar]
- 76.Bier M, Ostrem J, Marquez RB. Electrophoresis. 1993;14:1011–1018. doi: 10.1002/elps.11501401161. [DOI] [PubMed] [Google Scholar]