

Abstract
Human milk oligosaccharides (HMOs)a perform a number of functions including serving as prebiotics to stimulate the growth of beneficial intestinal bacteria, as receptor analogs to inhibit binding of pathogens, and as substances that promote postnatal brain development. There is further evidence that HMOs participate in modulating the human immune system. Because the absorption, catabolism and biological function of oligosaccharides (OS) have strong correlations with their structures, structure elucidation is key to advancing this research. Oligosaccharides are produced by competing enzymes that provide the large structural diversity and heterogeneity that characterizes this class of compounds. Unlike the proteome, there is no template for oligosaccharides making it difficult to rapidly identify oligosaccharide structures. In this research, the annotation of the neutral free oligosaccharides in milk is performed to develop a database for the rapid identification of oligosaccharide structures. Our strategy incorporates high performance nanoflow liquid chromatography and mass spectrometry for characterizing HMO structures. HPLC-Chip/TOF MS provides a sensitive and quantitative method for sample profiling. The reproducible retention time and accurate mass can be used to rapidly identify the OS structures in HMO samples. A library with 45 neutral OS structures has been constructed. The structures include information regarding the epitopes such as Lewis type as well as information regarding the secretor status.
Keywords: human milk oligosaccharides, HPLC-Chip/TOF, MALDI FT-ICR, IRMPD, CID, exoglycosidase, structure library
Introduction
Free oligosaccharides (OS) are the third most abundant solid component in human milk after lactose and lipids at about 7–12 g/L in mature milk.1–4 The studies of human milk oligosaccharides (HMOs) indicate that nutrients are not the only benefits the infants get from their mothers’ milk. The use of defatted HMOs increased the platelet-neutrophil complexes (PNCs) levels.2 HMOs are involved in intestinal absorption and renal excretion that may also enhance the mineral absorption and promote the postnatal brain development.2, 5, 6 In addition, by binding to certain pathogenic microorganisms, HMOs can inhibit the adherence of pathogens with epithelial cell surface glycans and therefore limit the virulence of some pathogens.2, 6–9 HMOs with specific epitopes inhibit the adhesion of certain microorganisms like Escherichia coli, Pneumococci, and Vibrio cholerae with receptors. This lowers the risk for newborn infants of getting diseases like diarrhea, otitis media and meningitis.2, 6 HMOs also serve as prebiotics by stimulating the growth of beneficial intestinal bacteria like bifidobacteria and lactobacilli (probiotics) in neonates.2, 4–6, 10, 11 The development of balanced intestinal microflora may play an important role for modulating the postnatal immune system.2, 6
The large diversity of structures suggests a multitude of functions. Absorption, catabolism and biological functions of OS all correlate with structures.2, 6, 12 Thus, knowledge of HMO structures is essential for determining biological functions. Shown in Figure 1a is the structure of a common milk oligosaccharide (right structure) along with the monosaccharide symbolic structures (left structure). The structures of HMOs start with the glucose (Glc) at the reducing end and a β1-4 galactose (Gal) bound to the glucose to form a lactose core. Attached to the core structure N-acetylglucosamine (GlcNAc) is a β1-3 linkage leading to a linear chain (right structure, Figure 1b). When two GlcNAc are added on both β1-3 and β1-6 position, it leads to a branched chain (left structure, Figure 1b). After addition of the GlcNAc, another galactose is added either at β1-4 or β1-3. The resulting N-acetylglucosamine and galactose disaccharide may repeat as many as 25 times. The structures for HMOs can be further diversified by fucosylation and sialylation on vital positions.7 Fucosylation on the reducing end always yields Fuc(α1-3), while on the non-reducing end the Fuc(α1-2) is dominant, which is found in secretors.12 Another Lewis gene-dependent fucosyltransferase forms either α1-3 or α1-4 fucose on GlcNAc. Sialylation occurs on the non-reducing terminal attaching either α2-3 or α2-6 sialic acid (NeuAc) to galactose, while on GlcNAc it only forms α2-6 linkages(Figure 1b).7, 13
Figure 1.

(a) An illustration of an HMO structure with the key for interpreting symbols. (b) Examples of “branched” and “linear” HMO structures.
Numerous techniques have been used to analyze the structures of HMOs including high performance liquid chromatography (HPLC), high pH anion-exchange chromatography (HPAEC), capillary electrophoresis (CE) for sample separation, and nuclear magnetic resonance (NMR) and mass spectrometry (MS) for structure characterization.13–26 So far, more than 200 oligosaccharides from human milk have been reported and more than 90 different oligosaccharide structures have been published.7, 27–29 However, those who want to study milk OS need to repeat the complicated sample separation and time-consuming data analysis to confirm the HMO structures in their own samples.
In this research, an oligosaccharide library is constructed based on the retention time and accurate mass obtained from nano-LC coupled to MS in order to provide a facile and sensitive method for identifying OS structures from biological mixtures.30–32 The instrument employs microchip-based nanoLC technology with a column packed with porous graphitized carbon (PGC).33–35 An orthogonal time-of-flight (TOF) mass spectrometer is used as the detector and coupled to the nano-LC via nano electrospray ionization (nano-ESI). Structural analysis is also performed by MALDI FT-ICR MS with collision induced dissociation (CID)36–38 and infrared multiphoton dissociation (IRMPD).39–41 Together with controlled exoglycosidase digestion, the specific linkages between monosaccharides were elucidated.42, 43 Because the retention time (RT) is highly reproducible on the HPLC-Chip/TOF MS, the oligosaccharide structures from milk samples can be rapidly identified by simply matching the retention times and accurate masses from the library.27, 44
Experimental
Reagents and Materials
OS were extracted from human milk obtained from the milk banks in San Jose, CA and Austin, TX. The extraction method was the same as in our previous publication.27, 45 Sodium borohydride (98%) and 2,5-dihydroxybenzoic acid (DHB) were purchased from Sigma-Aldrich (St. Louis, MO). Nonporous graphitized carbon cartridges (GCC, 150mg bed weight, 4mL cartridge volume) were bought from Alltech (Deerfield, IL). Standard HMOs were purchased from Dextra Laboratories (Earley Gate, UK). α(1-2)-Fucosidase was from EMD Calbiochem (La Jolla, CA). β(1-3)-Galactosidase was from New England Biolab (Beverly, MA). β(1-4)-Galactosidase was from ProZyme (San Leandro, CA). α(1-3,4)-Fucosidase was from Sigma-Aldrich (St. Louis, MO). All other reagents were of analytical or HPLC grade.
Oligosaccharide Reduction and Purification
The extracted HMOs (50mg in 250μL deionized water) were reduced by 250μL of 2.0 M sodium borohydride in water bath at 42°C for 17 hours. The reaction product was desalted and purified by solid phase extraction (SPE) using GCC.46 Prior to use, the GCC was conditioned by 6mL 80% acetonitrile (ACN) in water (v/v) with 0.1% trifluoroacetic acid (TFA, v/v) and then 6mL deionized water. After loading the OS sample on the GCC, the cartridges were washed with 2.5mL deionized water for 8 times to remove the salts. The OS were eluted with 6mL 20% ACN in water (v/v) and 6mL 40% ACN in water (v/v) with 0.05% TFA. After the GCC fractions were combined, the sample was dried in vacuo and reconstituted with nanopure water before MS analysis.
HPLC-Chip/TOF MS Analysis
The HMOs pooled sample was analyzed by the Agilent 6200 HPLC-Chip/TOF MS instrument (Agilent Technologies, Santa Clara, CA) equipped with a capillary pump as the loading pump for sample enrichment, a nano-pump as the analytical pump for sample separation, a microwell-plate autosampler maintained at 6°C by the thermostat, HPLC-Chip cube as interface and Agilent 6210 TOF MS. The micro-Chip consisted of an enrichment column with a volume of 40nL and an analytical column 43 × 0.075 mm i.d., which were both packed with PGC with a 5 μm pore size. Both pumps use binary solvent: A 3.0% ACN/water (v/v) with 0.1% formic acid and B 90% ACN/water (v/v) with 0.1% formic acid. 4 μL/min flow rate of solvent A was used for sample loading with 1 μL injection volume. A 45 minute gradient delivered by a nanoflow pump with a flow rate of 0.3 μL/min was used for separation: 2.5–20.0 min, 0–16% B; 20.0–30.0 min, 16–44% B; 30.0–35.0 min, 44–100% B; 35.0–45.0 min, 100% B and a 20 minute equilibration time at 0% B. The data was collected in the positive mode and calibrated by a dual nebulizer electrospray source with internal calibrant ions with a wide mass range – m/z 118.086, 322.048, 622.029, 922.010, 1221.991, 1521.972, 1821.952, 2121.933, 2421.914 and 2721.895.
Data analysis was performed on Analyst QS 1.1 software and deconvoluted by Agilent Mass Hunter (Agilent Technologies Inc.). The composition of each HMO was calculated by an in-house software – Glycan Finder, written in Igor Pro (Wavemetrics, Inc.). The output of the Glycan Finder consisted of measured mass, calculated mass with mass error and composition of each single oligosaccharide sorted based on retention times and intensities.
Nineteen standard milk OS obtained from commercial sources were reduced and introduced into the HPLC-Chip/TOF under identical conditions. The reproducibility of the LC retention time is excellent and typically less than 5 seconds on samples run on the same days. The mass error is less than 5ppm. The corresponding structures in the milk pool were determined by matching the retention times and accurate masses against the standards.
Separation of HMOs by HPLC
The reduced HMOs were separated on the Agilent Hewlett-Packard Series 1100 HPLC instrument with hypercarb PGC column (100×2.1 mm, 5μm particle size, Thermoquest, Hypersil Division) detected at 206nm and 254nm. The sample was eluted by solvent nanopure water (A) and ACN (B) with the flow rate of 0.25mL/min and a gradient of 0.0–25.0min, 0–15% B; and 25.0–50.0 min, 15–40% B; 50.0–70.0 min, 40–100% B. 80 fractions were collected, dried and reconstituted with 25μL nanopure water before analyzed by MALDI FT-ICR MS.
Analyze HPLC Fractions by MALDI FT-ICR
The HiRes MALDI FT-ICR (IonSpec, Irvine, CA) has an external MALDI source with a pulsed 355nm Nd:YAG laser, a hexapole ion guide, an ultra-high vacuum system maintained by two turbo pumps, one cryopump, and a 7.0 Tesla shielded superconducting magnet. DHB was used as matrix (8mg/160μL in 50% ACN/water (v/v)) in both positive and negative modes. The HMOs solution (0.5μL) was spotted on a 100-sample stainless steel probe followed by adding 0.25μL, 0.01 M NaCl solution as a cation dopant and 0.5μL matrix solution. In the negative mode experiment, no NaCl solution was added. The sample was dried in the vacuum chamber before putting into the ion source.
The IRMPD was performed to examine the structures for HMOs. The precursor ion was isolated in the ICR cell by using the arbitrary-wave form generator and the frequency synthesizer. A continuous-wave Parallax CO2 laser (10.6μm wavelength) was used for photon dissociation as described in previous publications.39–41
CID was also performed by sustained off resonance irradiation (SORI) using an arbitrary waveform generator. The precursor ion was excited at about 1000 Hz higher than its cyclotron frequency for 1000ms at 2–8 V. The collision energy was adjusted according to the size of the OS and the degree of fragmentation.
Exoglycosidase Digestion
The detailed procedure and condition for digestion was reported in previous publications.42, 43 Typically, buffer solutions were prepared by adding the glacial acetic acid into the 0.1 M ammonium acetate solution until the specific pH value was reached. For certain enzymes, the commercial buffer with the package was used directly for the digestion. 1μL enzyme solution was added into 1μL oligosaccharide solution with another 3μL buffer solution and incubated at 37°C for certain periods of time depending on the types of enzyme used. The mole ratio of the protein to oligosaccharides is approximately 1:100~200, and varies according to the concentrations of the enzyme provided by different manufacturers. The volume of enzyme added can be changed based on the concentration of the OS sample. The only complication is when an α-fucose is adjacent to a β-galactose, which blocks the release of the β-galactose due to steric hindrance.42 The α-fucosidase needs to be applied first before further digestion with the β-galactosidase. β-galactose without the adjacent α-fucose is referred to as a “free galactose” in the following discussion. The workflow for elucidating the structures is shown in Supplementary Figure 1.
Results and Discussion
MALDI MS Analysis of Reduced HMOs
The reduced HMOs were initially analyzed by MALDI FT-ICR MS. In the positive mode (Supplementary Figure 2a) the spectrum showed mainly the sodiated ions for reduced neutral HMOs (2.0 Da larger than non-reduced HMOs). The compositions were calculated using an in-house software “oligosaccharide calculator” written in Igor (using a mass accuracy of less than 5ppm). The ions generated by anionic OS were suppressed by neutral OS in the positive mode. However, the negative ion mode spectrum (Supplementary Figure 2b) gives stronger signals for anionic OS. The results from the MALDI showed that most of the HMOs are fucosylated with up to five fucoses. Most of the anionic HMOs contain only a single sialic acid (N-acetylneuraminic acid).47 For this study, we focus on the neutral oligosaccharides elucidation, as the method for anionic analysis differs significantly. Anionic structures will be the focus of a future publication.
HPLC-Chip/TOF MS Analysis
HPLC separation with PGC provides a robust and reproducible method for separation of oligosaccharides.27, 48, 49 In this research, HPLC-Chip/TOF MS is used for the nano-LC separation of the OS with the PGC stationary phase packed into microchip columns. We have previously shown that the HPLC-Chip/TOF MS yields high retention time reproducibility and effective separation of oligosaccharide isomers.27 To simplify the chromatogram, HMOs are reduced to eliminate the ambiguity due to the separation of anomers under the HPLC conditions.
Figure 2a shows the base peak chromatogram (BPC) of the HMOs from a pooled sample collected from five donors. Nineteen standard milk OS were obtained from commercial sources and analyzed with the HPLC-Chip/TOF MS. Figure 2b shows the BPC for a mixture consisting of eight commercially obtained standards labeled with the corresponding compound name. Isomers eluted from nano-LC typically have different retention times. OS that overlap in nano-LC separation were mostly of different masses. Figure 2c is the extracted ion chromatogram (EIC) for isomers with the neutral mass 1585.6 (m/z 793.8 doubly charged, MS inset). The EIC and deconvoluted data showed seven different isomers with their retention times and relative abundances (Table 1). The reproducibility of the retention times from Chip-LC and the accurate mass from the high performance TOF MS provide a sensitive and efficient method for identifying oligosaccharide structures. By matching the RT against the standard OS (Figure 2a and 2b), nineteen neutral OS structures were determined in this manner from the pooled sample directly.
Figure 2.

LC/MS chromatograms from Chip/TOF MS. (a) Base peak chromatogram (BPC) of pooled HMOs from five individual. (b) BPC of milk OS standards consisting of eight compounds; (c) EIC for 1585.586 isomers (with MS inset).
Table 1.
Seven isomers with the same neutral mass 1585.6 detected by HPLC-Chip/TOF from pooled HMOs sample.
| Mass (exp) | Mass (cal) | Error(Da) | Hex | Fuc | HexNAc | NeuAc | RT(min) | Abund. |
|---|---|---|---|---|---|---|---|---|
| 1585.585 | 1585.586 | −0.001 | 5 | 1 | 3 | 18.85 | 747173 | |
| 1585.583 | 1585.586 | −0.003 | 5 | 1 | 3 | 19.89 | 6403774 | |
| 1585.585 | 1585.586 | −0.001 | 5 | 1 | 3 | 20.76 | 391731 | |
| 1585.588 | 1585.586 | 0.002 | 5 | 1 | 3 | 21.36 | 1651982 | |
| 1585.585 | 1585.586 | −0.002 | 5 | 1 | 3 | 22.27 | 2861336 | |
| 1585.589 | 1585.586 | 0.003 | 5 | 1 | 3 | 22.92 | 1326146 | |
| 1585.586 | 1585.586 | 0.000 | 5 | 1 | 3 | 23.47 | 1846421 |
the abundance is from HPLC-Chip/TOF counts per second (cps)
The most abundant OS in the pooled sample correspond to lacto-N-tetraose (LNT) (m/z 710.3, RT 15.1min) and lacto-N-fucopentaose I (LNFP I) (m/z 856.3, RT 14.6min). High mass accuracy was obtained by using a dual ESI source assembly, which provides continuous calibration and guarantees high mass accuracy during the entire process. All HMOs found by HPLC-Chip/TOF MS analysis are listed in Supplementary Table 1 with their accurate masses, retention times, monosaccharide compositions and relative abundances all assigned. Over 200 oligosaccharides were found, consistent with our previous report.27
Characterization of Unknown HMO Structures by Tandem MS
To characterize unknown structures, the pooled sample was first separated using standard off-line HPLC into smaller sample pools. From the HPLC separation, 80 fractions were collected and analyzed by MALDI FT-ICR as discussed in the methods section. Less than half, or 34 fractions, were found to have OS. Each fraction was further analyzed by HPLC-Chip/TOF MS to determine the number of isomers in each fraction.
The HPLC fractions allow the tandem MS and exoglycosidase examination of enriched components. Structural information was obtained by tandem MS (IRMPD or CID). The sequence and connectivity of the saccharide residues are readily determined. The tandem MS of the 19 HMO standards were used to obtain the characteristic fragmentation behavior of the OS. Isomers with, for example, different branching arrangements will have different fragmentation pathways that generate unique diagnostic peaks. Diagnostic peaks provide structural information, like fingerprints, that when combined allow structural elucidation of the compound.
Supplementary Figure 3 shows the tandem MS (employing IRMPD in the FT ICR MS) for two LNFP isomers in the positive mode. Both spectra have a y type ion50 [3Hex+1HexNAc+Na]+ (m/z 732.3) due to the loss of the fucose. LNFP II can generate a b type ion [2Hex+1HexNAc+1Fuc+Na]+ (m/z 696.2) due to the loss of glucose on the reducing end and the sequential loss of a Hex to form [1Hex+1HexNAc+1Fuc+Na]+ (m/z 534.2). However, in LNFP V these two ions are not found since the fucosylation is on the reducing end, and there is no possibility of losing a glucose reducing end without losing the fucose first. Also, LNFP V can form a y ion [2Hex+1Fuc+Na]+ (m/z 513.2) due to the loss of [1Hex+1HexNAc] from the nonreducing end.
Other tandem MS examples include the IRMPD of DFLNH b and DFpLNH II (Figure 3). DFpLNH II with the linear structure fragments starting from the reducing end to form a b type ion [2Hex+2HexNAc+2Fuc+Na]+ (m/z 1045.4), which sequentially loses one of the fucoses to form [2Hex+2HexNAc+1Fuc+Na]+ (m/z 899.3). The same highly intense ion is not observed in DFLNH b, which has a β1-3 and β1-6 branch at the lactose core. Another difference between branched and linear structure is the relative abundances of m/z 753.3 and m/z 732.3. Since the ion with m/z 899.3 can lose another fucose to generate m/z 753.3, it is more abundant than m/z 732.3 in DFpLNH II. While with DFLNH b there are more fragmentation pathways that form m/z 732.3 causing this ion to be more abundant. In general, HMOs with linear and branched chains can be readily identified in this manner.
Figure 3.

Infrared multiphoton dissociation using FT ICR MS of two DFLNH isomers, one linear and one branch, illustrating significantly different fragmentation patterns. In general, isomers have distinct fragmentation that allows differentiation of isomers. Arrows point to diagnostic peaks.
The strategy for finding unknown structures is illustrated as follows. All HPLC fractions were examined by both MALDI FT-ICR and Chip/TOF (Figure 4). For example, HPLC Fraction 21 contains three major neutral OS, m/z 732.3 (RT 15.1 min, [3Hex+1HexNAc+Na]+), m/z 1243.4 (RT 15.7 min, [4Hex+2HexNAc+1Fuc+ Na]+), m/z 1389.5 (RT 16.1 min, [4Hex+2HexNAc+2Fuc+Na]+). Among the three OS, m/z 732.3 is determined to be LNT by comparison of the retention time against the standard LNT. Chip/TOF results showed only one major isomer for both m/z 1243.4 and m/z 1389.5 (the corresponding protonated ions were shown in Figure 4b). The two ions were examined by IRMPD, which also confirmed their monosaccharide composition (Figure 5). By searching the diagnostic peaks and comparing the fragmentation pattern with standard OS, m/z 1243.4 was determined to have a branched structure. From the published structures, only three have branched structures with this composition, namely MFLNH I, MFLNH III and MFLNnH (Figure 5a). MFLNH III is available as a standard with retention time at 17.3 min. The possible structures for this isomer was narrowed from eight possible isomers to two branched chain isomers, MFLNH I and MFLNnH. However, the possibility of a yet unknown structure cannot be dismissed at this time.
Figure 4.

(a) MALDI-MS of HPLC fraction 21 in the positive mode; (b) LC/MS of fraction 21 shown as BPC and EIC. Middle panel corresponds to m/z 1243.4 and lower panel to m/z 1389.5. Differences in m/z correspond to sodiated versus protonated species.
Figure 5.

IRMPD of (a) m/z 1243.4 and (b) m/z 1389.5 from HPLC fraction 21. Possible structures are inset. Structures with arrows are those consistent with tandem MS.
Employing the same method, m/z 1389.5 was also determined to have a branched structure (Figure 5b). The two species m/z 1389.5 and m/z 1243.4 differed only by a fucose but had very similar fragmentation patterns. The possible structures for m/z 1389.5 can also be narrowed from six isomers to three branched isomers, DFLNHa, DFLNHc and DFLNnH.
Employment of Exoglycosidase Reactions for Structural Elucidation
The tandem MS spectra of reduced HMOs in the positive mode yielded b, y or c, z type ions50 with little or no cross-ring fragments (a and x ions). The linkages between monosaccharides cannot be determined by tandem MS alone. Exoglycosidase digestion can however selectively cleave monosaccharides from the nonreducing end, because exoglycosidases are highly specific for the linkages (including the anomeric character) and the monosaccharides.51 By using the various exoglycosidase in a strategic manner while employing MALDI MS to monitor the products, the linkages between monosaccharides can be determined. However, the reaction time is of some importance as the specificity decreases when the reaction is allowed to continue for too long. Nonetheless, under the right conditions the reaction is highly specific and will not cleave other linkages or other saccharide residues. Since different enzymes will retain activity and specificity even when other enzymes are present, different enzymes can be added stepwise without removal while the reaction is monitored by mass spectrometry. In so doing, the complete structure can be elucidated by combining the results from the MS, the tandem MS and the exoglycosidase digestion.
Figure 6 is the sequential exoglycosidase digestion of HPLC fraction 21 by α(1-2) fucosidase and β(1-3) galactosidase, respectively. Figure 6a is the MALDI MS of fraction 21 with m/z 1243.4 and 1389.5. After 5 hours digestion by α(1-2) fucosidase, the m/z 1389.5 is nearly all consumed generating m/z 1243.4 without further digestion (Figure 6b). The result indicates that m/z 1389.5 has one Fuc(α1-2), but its digested product and the original m/z 1243.4 both have no Fuc(α1-2). The possible structures can be further narrowed to DFLNHa and DFLNHc for m/z 1389.5 and MFLNnH for m/z 1243.4 (the inset structures in Figure 6b). To determine the structures further, a β(1-3) galactosidase was added directly into the reaction mixture and incubated for another 2 hours. Figure 6c indicates that the new peak m/z 1243.4, which results from the cleavage of 1389.5 and uncleaved 1243.4 (Figure 6b), has a free Gal(β1-3), because it yields a m/z 1081.4 product. Note that the m/z 1081.4 is as abundant as the original 1389.5 (Figure 6a) suggesting that the original m/z 1389.5 contains both Fuc(α1-2) and a free Gal(β1-3), which corresponds well to DFLNHa (inset structure in Figure 6c). To confirm further that the structure of m/z1389.5 is DFLNHa and determine the structure of m/z1243.4, more digestion experiments were performed. For clarity, Figure 6a is duplicated in Figure 7a. The incubation of the mixture with a β(1-4) galactosidase for one hour (Figure 7d) generated a new peak m/z 1081.4. The result indicated one free Gal(β1-4) is present in the species corresponding to m/z 1243.4. Note that the same enzyme left m/z 1389.5 intact. Previously, we have shown that Fuc(α1-2) is not present in m/z 1243.4, suggesting that the fucose is either α1-3 or α1-4 linked. Reaction of the compound with α(1-3,4) fucosidase for an hour (Figure 7b) yielded m/z 1097.4 and m/z 1243.4, confirming both the presence of the fucose residues and the absence of Fuc(α1-2) for the original peak m/z 1243.4. The resulting compound was further digested using a β(1-3) galactosidase (Figure 7c) and a β(1-4) galactosidase (Figure 7e) indicated that both free β1-3 and β1-4 galactose are present in m/z 1097.4 in Figure 7b, which in turn corresponds to LNH. On the other hand, the original m/z 1389.5 is DFLNHa, which is further confirmed by comparing the results obtained in Figure 7d and Figure 7e. The compound corresponding to m/z 1389.5 is identified to be DFLNHa with a Lewis x epitope.
Figure 6.

Sequential exoglycosidase digestion of HPLC fraction 21. (a) MALDI-MS of fraction 21 before digestion; (b) Digested by α(1-2) fucosidase for 5 hours; (c) Sequentially digested by β(1-3) galactosidase for 2 hours.
Figure 7.

Confirmation of structures and Lewis epitopes in fraction 21 by multiple steps of exoglycosidase digestion.
In order to ensure that the fucose on m/z 1243.4 is not on the reducing end with a α1-3 linkage (linkage rules mentioned in introduction), CID was performed (Supporting Figure 4). By adjusting the proper collision energy, CID of m/z 1243.4 yields the b type ion m/z 1061.4 [M+ Na-182]+ by losing the reducing end. Therefore, the species m/z 1243.4 is shown to be a new structure annotated in Figure 7a with a Lewis a epitope and not MFLNnH
Conclusions
The library of neutral HMOs is presented in Table 2. It includes 45 structures, with 13 that are new. The larger oligosaccharides are typically highly fucosylated and are often difficult to elucidate completely. In some cases, partial structures are provided when the entire structure cannot be fully elucidated. This library includes structures, accurate masses, and LC retention times. This library provides a valuable template for the rapid identification of HMOs structures. We can now identify any compound in the library from biological samples by simply comparing their retention times and accurate masses to the database.
Table 2.
Partial library includes a total number of 45 structures. Thirteen are new OS with possible or partial structures for several large or highly fucosylated OS.
| N.O. | Name | Mass (exp) | Mass (cal) | Error | Composition | RT | Abund. | Structure | [Lewis type] | Secretor Marker | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3′FL | 490.189 | 490.190 | 0.000 | 2100 | 1.31 | 1890133 | 52 | |||
| 2 | LNDFK I | 1001.378 | 1001.380 | −0.002 | 3210 | 10.89 | 419803 | 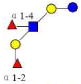 |
[b] | S | 53 |
| 3 | LNDFH II | 1001.381 | 1001.380 | 0.002 | 3210 | 11.01 | 380946 | 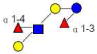 |
[a] | 54 | |
| 4 | LNFP II | 855.320 | 855.322 | −0.002 | 3110 | 11.22 | 1077776 | [a] | 55 | ||
| 5 | A-hepta | 1204.468 | 1204.459 | 0.009 | 3220 | 11.24 | 71112 | 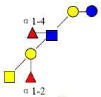 |
S | 56 | |
| 6 | 2′FL | 490.191 | 490.190 | 0.001 | 2100 | 11.69 | 2952268 | S | 57 | ||
| 7 | DFpLNH II | 1366.522 | 1366.512 | 0.010 | 4220 | 13.67 | 1013387 | 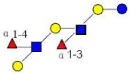 |
[a] | 58 | |
| 8 | LNFP III | 855.323 | 855.322 | 0.001 | 3110 | 14.47 | 196187 | 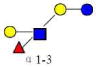 |
[x] | 59 | |
| 9 | LDFT | 636.248 | 636.248 | 0.000 | 2200 | 14.47 | 520323 | 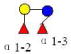 |
S | 53 | |
| 10 | LNFP I | 855.323 | 855.322 | 0.001 | 3110 | 14.61 | 4207193 | 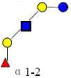 |
S | 60 | |
| 11 | TFLNH | 1512.576 | 1512.570 | 0.006 | 4320 | 14.76 | 1390028 | 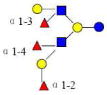 |
[x,b] | 61,62 | |
| 12 | DFLNH b | 1366.519 | 1366.512 | 0.007 | 4220 | 14.98 | 315922 | 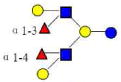 |
[x,a] | 63 | |
| 13 | LNFP V | 855.322 | 855.322 | 0.000 | 3110 | 15.05 | 158407 | 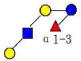 |
64 | ||
| 14 | LNT | 709.266 | 709.264 | 0.002 | 3010 | 15.09 | 25707820 | 65 | |||
| 15 | LNnT | 709.265 | 709.264 | 0.001 | 3010 | 15.20 | 3027201 | 66 | |||
| 16 | MFpLNH IV | 1220.474 | 1220.454 | 0.020 | 4120 | 15.64 | 2147730 | 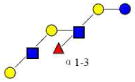 |
62 | ||
| 17 | 4120a | 1220.459 | 1220.454 | 0.005 | 4120 | 16.02 | 3014724 | 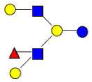 |
[a] | ||
| 18 | DFLNHa | 1366.514 | 1366.512 | 0.002 | 4220 | 16.30 | 13218526 | 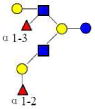 |
[x] | S | 58 |
| 19 | MFLNH III | 1220.464 | 1220.454 | 0.010 | 4120 | 17.29 | 806057 | 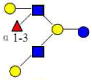 |
[x] | 63 | |
| 20 | MFLNH I | 1220.458 | 1220.454 | 0.004 | 4120 | 17.70 | 3770129 | 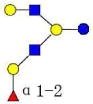 |
S | 58 | |
| 21 | DFLNO I | 1731.644 | 1731.644 | 0.000 | 5230 | 17.95 | 1453837 | 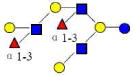 |
[x] | 67 | |
| 22 | Tetra-iso-LNO | 2023.763 | 2023.760 | 0.003 | 5430 | 18.33 | 3224591 | 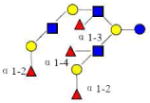 |
[b] | S | 68 |
| 23 | IFLNH III | 1220.455 | 1220.454 | 0.001 | 4120 | 18.44 | 1588579 | 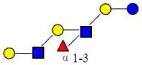 |
16 | ||
| 24 | TFiLNO | 1877.702 | 1877.702 | 0.000 | 5330 | 18.50 | 4815678 | 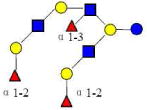 |
S | 69 | |
| 25 | LNH | 1074.402 | 1074.396 | 0.006 | 4020 | 18.82 | 862591 | 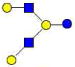 |
70 | ||
| 26 | DFLNnO II | 1731.642 | 1731.644 | −0.002 | 5230 | 18.93 | 6872706 | 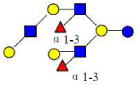 |
[x] | 67 | |
| 27 | LNnH | 1074.395 | 1074.396 | −0.002 | 4020 | 19.47 | 20764678 | 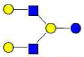 |
71 | ||
| 28 | 5330a | 1877.699 | 1877.702 | −0.003 | 5330 | 19.61 | 5121388 | 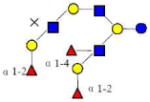 |
[b] | S | |
| 29 | 5230a | 1731.642 | 1731.644 | −0.002 | 5230 | 19.76 | 1740807 | 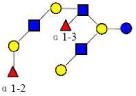 |
S | ||
| 30 | 5130a | 1585.583 | 1585.586 | −0.003 | 5130 | 19.89 | 6403774 | 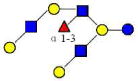 |
|||
| 31 | 4320a | 1512.568 | 1512.570 | −0.002 | 4320 | 20.11 | 1779607 | 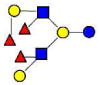 |
[y,a] | S | |
| 32 | DFLNnO I or DFLNO II | 1731.642 | 1731.644 | −0.002 | 5230 | 20.25 | 7221123 | 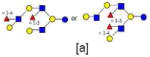 |
[a] | 67 | |
| 33 | 6440a | 2388.891 | 2388.892 | −0.001 | 6440 | 20.25 | 750700 | 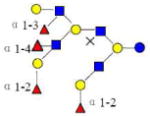 |
[x,b] | S | |
| 34 | 6340a | 2242.835 | 2242.834 | 0.001 | 6340 | 20.33 | 645652 | 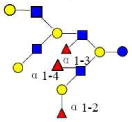 |
[b] | S | |
| 35 | 6340b | 2242.835 | 2242.834 | 0.001 | 6340 | 20.65 | 3053118 | 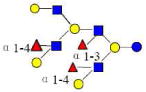 |
[a] | ||
| 36 | p-LNH | 1074.396 | 1074.396 | −0.001 | 4020 | 22.70 | 594027 | 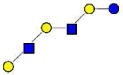 |
58 | ||
| 37 | 5130b | 1585.585 | 1585.586 | −0.001 | 5130 | 20.76 | 391731 | 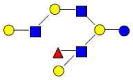 |
[a] | ||
| 38 | DFLNHc | 1366.510 | 1366.512 | −0.002 | 4220 | 20.78 | 1839355 | 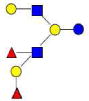 |
[b] | S | |
| 39 | 6340c | 2242.836 | 2242.834 | 0.001 | 6340 | 21.22 | 332242 | 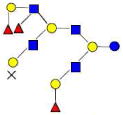 |
[y] | S | |
| 40 | 6240a | 2096.780 | 2096.776 | 0.004 | 6240 | 21.34 | 3805063 | 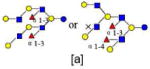 |
|||
| 41 | F-LNO | 1585.588 | 1585.586 | 0.002 | 5130 | 21.36 | 1651982 | 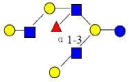 |
72 | ||
| 42 | 5230b | 1731.649 | 1731.644 | 0.005 | 5230 | 21.48 | 3389544 | 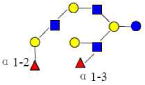 |
[x] | S | |
| 43 | IFLNH I | 1220.457 | 1220.454 | 0.003 | 4120 | 21.55 | 2491388 | 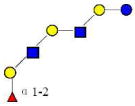 |
S | 16 | |
| 44 | 5130c | 1585.585 | 1585.586 | −0.002 | 5130 | 22.27 | 2861336 | 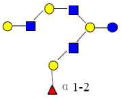 |
S | ||
| 45 | 6140a | 1950.719 | 1950.719 | 0.001 | 6140 | 22.46 | 1738239 | 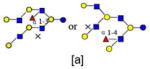 |
[a] |
Monosaccharides composition 3Hex:2Fuc:1HexNAc:0NeuAc represented as 3210
The red names like 4120a are new structures, the blue names like 4320a are possible structures
The abundance is from HPLC-Chip/TOF counts per seconds (cps)
S are secretor Markers which have Fuc(α1-2)
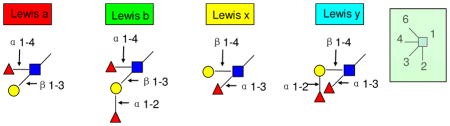
Examination of the neutral HMO structures generated a few general linkage rules that may be useful for studying the biosynthesis of HMOs (Supplementary Figure 5). The most general observation is that all fucoses are linked α and all other residues are linked β. The other observations are:
HMOs can be divided into those with a linear core (Supplementary Figure 5, L) and a branched core (B) structure.
For linear core structures, the GlcNAc attached to the lactose core will always have the β1-3 linkage (L) with no exception observed thus far.
For linear core structures, a single fucose on the reducing end always links via α1-3 (L1) the same as the observation in Stahl, B et al.7, 13
Monofucosylated structure with the fucose attached to a non-reducing terminal Gal(β1-3) is always Fuc(α1-2) (L2) to yield a type I H antigen. The Fuc(α1-2) characterizes the secretor status of the mother.
In the branched core structure, the galactose bound to the GlcNAc(β1-6) will always have the β1-4 linkage with no exception observed so far (B). The galactose on the GlcNAc(β1-3) branch can either be Gal(β1-3) or (β1-4).
In a branched core structure, no fucosylation is found on the reducing end (B3).
For monofucosylated structure, the Fuc(α1-2) always attached to the Gal(β1-3) (B1) but not the Gal(β1-4) (B2).
HMOs contain a number of Lewis epitopes including Lewis a, b, x, and y. From the pooled sample we can make some general conclusions regarding the relative abundances of each epitope. Lewis a (8~13%) and Lewis x (10~15%) are about equally abundant and more abundant than Lewis b and y. Lewis b (4~8%) is found to be more abundant than Lewis y (1~3%) (Supplementary Table 2). This observation is consistent with the proposed biosynthetic pathway (pathways in Supplementary Figure 5). Lewis b is formed from both Lewis a and the type I H antigen, while Lewis y can only come from Lewis x.
Finally, the larger oligosaccharide structures such as TFLNH and 6340b (Table 2) offer some intriguing characteristics like the presence of multiple epitopes. Multiple epitopes may enhance the activity of these compounds. Elucidating their structures offer a greater challenge, although a recent report by Amano et al.73 described strategies to access them.
Supplementary Material
Acknowledgments
Funding provided by the National Institutes of Health (HD059127 and HD061923), UC Discovery, and the California Dairy Research Fund are gratefully acknowledged.
Footnotes
Supporting Information Available: Supplementary Figure 1 (The workflow for analyzing HMOs structures); Supplementary Figure 2 (MALDI FT-ICR MS spectra of pooled HMO sample with the list of monosaccharide compositions in (a)positive mode and (b) negative mode); Supplementary Figure 3 (The tandem MS spectra obtained by IRMPD of two LNFP isomers); Supplementary Figure 4 (Confirmation of the position of fucose in m/z 1243.4 from HPLC fraction 21 by tandem MS); Supplementary Figure 5 (The linkage rules obtained from the analysis of HMO structures); Supplementary Table 1 (The deconvoluted data from HPLC-Chip/TOF with accurate masses, monosaccharide compositions, retention times, and relative abundances assigned for the HMOs); Supplementary Table 2 (The ratio of Lewis epitopes based on partial library). This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Coppa GV, Pierani P, Zampini L, Carloni I, Carlucci A, Gabrielli O. Oligosaccharides in human milk during different phases of lactation. Acta Paediatrica. 1999;88:89–94. doi: 10.1111/j.1651-2227.1999.tb01307.x. [DOI] [PubMed] [Google Scholar]
- 2.Kunz C, Rudloff S, Baier W, Klein N, Strobel S. Oligosaccharides in human milk: Structural, functional, and metabolic aspects. Annual Review of Nutrition. 2000;20:699–722. doi: 10.1146/annurev.nutr.20.1.699. [DOI] [PubMed] [Google Scholar]
- 3.Kunz C, Rudloff S. Health promoting aspects of milk oligosaccharides. International Dairy Journal. 2006;16(11):1341–1346. [Google Scholar]
- 4.Boehm G, Stahl B. Oligosaccharides from milk. Journal of Nutrition. 2007;137(3):847S–849S. doi: 10.1093/jn/137.3.847S. [DOI] [PubMed] [Google Scholar]
- 5.Bode L. Recent advances on structure, metabolism and function of human milk oligosaccharides. Journal of Nutrition. 2006;136(8):2127–2130. doi: 10.1093/jn/136.8.2127. [DOI] [PubMed] [Google Scholar]
- 6.Newburg DS, Ruiz-Palacios GM, Morrow AL. Human milk glycans protect infants against enteric pathogens. Annual Review of Nutrition. 2005;25:37–58. doi: 10.1146/annurev.nutr.25.050304.092553. [DOI] [PubMed] [Google Scholar]
- 7.Boehm G, Stahl B. Functional Dairy Products. CRC Press; Cambridge, England: 2003. p. 203. [Google Scholar]
- 8.Morrow AL, Ruiz-Palacios GM, Altaye M, Jiang X, Guerrero ML, Meinzen-Derr JK, Farkas T, Chaturvedi P, Pickering LK, Newburg DS. Human milk oligosaccharides are associated with protection against diarrhea in breast-fed infants. Journal of Pediatrics. 2004;145(3):297–303. doi: 10.1016/j.jpeds.2004.04.054. [DOI] [PubMed] [Google Scholar]
- 9.Jiang X, Huang PW, Zhong WM, Tan M, Farkas T, Morrow AL, Newburg DS, Ruiz-Palacios GM, Pickering LK. Human milk contains elements that block binding of noroviruses to human histo-blood group antigens in saliva. Journal of Infectious Diseases. 2004;190(10):1850–1859. doi: 10.1086/425159. [DOI] [PubMed] [Google Scholar]
- 10.Locascio RG, Ninonuevo MR, Freeman SL, Sela DA, Grimm R, Lebrilla CB, Mills DA, German JB. Glycoprofiling of bifidobacterial consumption of human milk oligosaccharides demonstrates strain specific, preferential consumption of small chain glycans secreted in early human lactation. Journal of Agricultural and Food Chemistry. 2007;55(22):8914–8919. doi: 10.1021/jf0710480. [DOI] [PubMed] [Google Scholar]
- 11.Coppa GV, Gabrielli O. Human milk oligosaccharides as prebiotics. Therapeutic Microbiology: Probiotics and Related Strategies. 2008;131(146):131–145. [Google Scholar]
- 12.Lowe JB. Biochemistry and biosynthesis of ABH and Lewis antigens. Plenum Press; NY: 1995. –75.pp. 115 [Google Scholar]
- 13.Finke B, Stahl B, Pfenninger A, Karas M, Daniel H, Sawatzki G. Analysis of high-molecular-weight oligosaccharides from human milk by liquid chromatography and MALDI-MS. Analytical Chemistry. 1999;71(17):3755–3762. doi: 10.1021/ac990094z. [DOI] [PubMed] [Google Scholar]
- 14.Pfenninger A, Karas M, Finke B, Stahl B, Sawatzki G. Matrix optimization for matrix-assisted laser desorption/ionization mass spectrometry of oligosaccharides from human milk. Journal of Mass Spectrometry. 1999;34(2):98–104. doi: 10.1002/(SICI)1096-9888(199902)34:2<98::AID-JMS767>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 15.Pfenninger A, Karas M, Finke B, Stahl B. Structural analysis of underivatized neutral human milk oligosaccharides in the negative ion mode by nano-electrospray MSn (Part 1: Methodology) Journal of the American Society for Mass Spectrometry. 2002;13(11):1331–1340. doi: 10.1016/S1044-0305(02)00645-1. [DOI] [PubMed] [Google Scholar]
- 16.Pfenninger A, Karas M, Finke B, Stahl B. Structural analysis of underivatized neutral human milk oligosaccharides in the negative ion mode by nano-electrospray MSn (Part 2: Application to isomeric mixtures) Journal of the American Society for Mass Spectrometry. 2002;13(11):1341–1348. doi: 10.1016/S1044-0305(02)00646-3. [DOI] [PubMed] [Google Scholar]
- 17.Stahl B, Thurl S, Zeng JR, Karas M, Hillenkamp F, Steup M, Sawatzki G. Oligosaccharides from Human-Milk as Revealed by Matrix-Assisted Laser-Desorption Ionization Mass-Spectrometry. Analytical Biochemistry. 1994;223(2):218–226. doi: 10.1006/abio.1994.1577. [DOI] [PubMed] [Google Scholar]
- 18.Chai WG, Piskarev VE, Zhang YB, Lawson AM, Kogelberg H. Structural determination of novel lacto-N-decaose and its monofucosylated analogue from human milk by electrospray tandem mass spectrometry and H-1 NMR spectroscopy. Archives of Biochemistry and Biophysics. 2005;434(1):116–127. doi: 10.1016/j.abb.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 19.Nakhla T, Fu DT, Zopf D, Brodsky NL, Hurt H. Neutral oligosaccharide content of preterm human milk. British Journal of Nutrition. 1999;82(5):361–367. doi: 10.1017/s0007114599001609. [DOI] [PubMed] [Google Scholar]
- 20.Thurl S, MullerWerner B, Sawatzki G. Quantification of individual oligosaccharide compounds from human milk using high-pH anion-exchange chromatography. Analytical Biochemistry. 1996;235(2):202–206. doi: 10.1006/abio.1996.0113. [DOI] [PubMed] [Google Scholar]
- 21.Shen ZJ, Warren CD, Newburg DS. High-performance capillary electrophoresis of sialylated oligosaccharides of human milk. Analytical Biochemistry. 2000;279(1):37–45. doi: 10.1006/abio.1999.4448. [DOI] [PubMed] [Google Scholar]
- 22.Sumiyoshi W, Urashima T, Nakamura T, Arai I, Saito T, Tsumura N, Wang B, Brand-Miller J, Watanabe Y, Kimura K. Determination of each neutral oligosaccharide in the milk of Japanese women during the course of lactation. British Journal of Nutrition. 2003;89(1):61–69. doi: 10.1079/BJN2002746. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki M, Suzuki A. Structural characterization of fucose-containing oligosaccharides by high-performance liquid chromatography and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Biological Chemistry. 2001;382(2):251–257. doi: 10.1515/BC.2001.032. [DOI] [PubMed] [Google Scholar]
- 24.Charlwood J, Tolson D, Dwek M, Camilleri P. A detailed analysis of neutral and acidic carbohydrates in human milk. Analytical Biochemistry. 1999;273(2):261–277. doi: 10.1006/abio.1999.4232. [DOI] [PubMed] [Google Scholar]
- 25.Bahr U, Pfenninger A, Karas M, Stahl B. High sensitivity analysis of neutral underivatized oligosaccharides by nanoelectrospray mass spectrometry. Analytical Chemistry. 1997;69(22):4530–4535. doi: 10.1021/ac970624w. [DOI] [PubMed] [Google Scholar]
- 26.Pfenninger A, Chan SY, Karas M, Finke B, Stahl B, Costello CE. Mass spectrometric detection of multiple extended series of neutral highly fucosylated N-acetyllactosamine oligosaccharides in human milk. International Journal of Mass Spectrometry. 2008;278(2–3):129–136. doi: 10.1016/j.ijms.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ninonuevo MR, Park Y, Yin HF, Zhang JH, Ward RE, Clowers BH, German JB, Freeman SL, Killeen K, Grimm R, Lebrilla CB. A strategy for annotating the human milk glycome. Journal of Agricultural and Food Chemistry. 2006;54(20):7471–7480. doi: 10.1021/jf0615810. [DOI] [PubMed] [Google Scholar]
- 28.Newburg DS, Neubauer SH. Handbook of Milk Composition. Academic Press; 1995. [Google Scholar]
- 29.Urashima T, Asakuma S, Messer M. Milk oligosaccharides, Comprehensive Glycoscience. Elsevier; Amsterdam: 2007. pp. 695–724. [Google Scholar]
- 30.Ninonuevo M, An HJ, Yin HF, Killeen K, Grimm R, Ward R, German B, Lebrilla C. Nanoliquid chromatography-mass spectrometry of oligosaccharides employing graphitized carbon chromatography on microchip with a high-accuracy mass analyzer. Electrophoresis. 2005;26(19):3641–3649. doi: 10.1002/elps.200500246. [DOI] [PubMed] [Google Scholar]
- 31.Yin NF, Killeen K, Brennen R, Sobek D, Werlich M, van de Goor TV. Microfluidic chip for peptide analysis with an integrated HPLC column, sample enrichment column, and nanoelectrospray tip. Analytical Chemistry. 2005;77(2):527–533. doi: 10.1021/ac049068d. [DOI] [PubMed] [Google Scholar]
- 32.Yin HF, Killeen K. The fundamental aspects and applications of Agilent HPLC-Chip. Journal of Separation Science. 2007;30(10):1427–1434. doi: 10.1002/jssc.200600454. [DOI] [PubMed] [Google Scholar]
- 33.Koizumi K. High-performance liquid chromatographic separation of carbohydrates on graphitized carbon columns. Journal of Chromatography A. 1996;720(1–2):119–126. doi: 10.1016/0021-9673(94)01274-1. [DOI] [PubMed] [Google Scholar]
- 34.Antonio C, Pinheiro C, Chaves MM, Ricardo CP, Ortuno MF, Thomas-Oates J. Analysis of carbohydrates in Lupinus albus stems on imposition of water deficit, using porous graphitic carbon liquid chromatography-electrospray ionization mass spectrometry. Journal of Chromatography A. 2008;1187(1–2):111–118. doi: 10.1016/j.chroma.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 35.Guile GR, Rudd PM, Wing DR, Prime SB, Dwek RA. A rapid high-resolution high-performance liquid chromatographic method for separating glycan mixtures and analyzing oligosaccharide profiles. Analytical Biochemistry. 1996;240(2):210–226. doi: 10.1006/abio.1996.0351. [DOI] [PubMed] [Google Scholar]
- 36.Penn SG, Cancilla MT, Lebrilla CB. Collision-induced dissociation of branched oligosaccharide ions with analysis and calculation of relative dissociation thresholds. Analytical Chemistry. 1996;68(14):2331–2339. doi: 10.1021/ac960155i. [DOI] [PubMed] [Google Scholar]
- 37.Tseng K, Hedrick JL, Lebrilla CB. Catalog-library approach for the rapid and sensitive structural elucidation of oligosaccharides. Analytical Chemistry. 1999;71(17):3747–3754. doi: 10.1021/ac990095r. [DOI] [PubMed] [Google Scholar]
- 38.Cancilla MT, Wang AW, Voss LR, Lebrilla CB. Fragmentation reactions in the mass spectrometry analysis of neutral oligosaccharides. Analytical Chemistry. 1999;71(15):3206–3218. doi: 10.1021/ac9813484. [DOI] [PubMed] [Google Scholar]
- 39.Lancaster KS, An HJ, Li BS, Lebrilla CB. Interrogation of N-linked oligosaccharides using infrared multiphoton dissociation in FT-ICR mass spectrometry. Analytical Chemistry. 2006;78(14):4990–4997. doi: 10.1021/ac0600656. [DOI] [PubMed] [Google Scholar]
- 40.Zhang JH, Schubothe K, Li BS, Russell S, Lebrilla CB. Infrared multiphoton dissociation of O-linked mucin-type oligosaccharides. Analytical Chemistry. 2005;77(1):208–214. doi: 10.1021/ac0489824. [DOI] [PubMed] [Google Scholar]
- 41.Xie YM, Lebrilla CB. Infrared multiphoton dissociation of alkali metal-coordinated oligosaccharides. Analytical Chemistry. 2003;75(7):1590–1598. doi: 10.1021/ac026009w. [DOI] [PubMed] [Google Scholar]
- 42.Xie YM, Tseng K, Lebrilla CB, Hedrick JL. Targeted use of exoglycosidase digestion for the structural elucidation of neutral O-linked oligosaccharides. Journal of the American Society for Mass Spectrometry. 2001;12(8):877–884. doi: 10.1016/S1044-0305(01)00267-7. [DOI] [PubMed] [Google Scholar]
- 43.Zhang JH, Lindsay LL, Hedrick JL, Lebrilla CB. Strategy for profiling and structure elucidation of mucin-type oligosaccharides by mass spectrometry. Analytical Chemistry. 2004;76(20):5990–6001. doi: 10.1021/ac049666s. [DOI] [PubMed] [Google Scholar]
- 44.Pabst M, Bondili JS, Stadlmann J, Mach L, Altmann F. Mass plus retention time = structure: A strategy for the analysis of N-glycans by carbon LC-ESI-MS and its application to fibrin N-glycans. Analytical Chemistry. 2007;79(13):5051–5057. doi: 10.1021/ac070363i. [DOI] [PubMed] [Google Scholar]
- 45.Tao N, Depeters EJ, Freeman S, German JB, Grimm R, Lebrilla CB. Bovine milk glycome. Journal of Dairy Science. 2008;91(10):3768–3778. doi: 10.3168/jds.2008-1305. [DOI] [PubMed] [Google Scholar]
- 46.Packer NH, Lawson MA, Jardine DR, Redmond JW. A general approach to desalting oligosaccharides released from glycoproteins. Glycoconjugate Journal. 1998;15(8):737–747. doi: 10.1023/a:1006983125913. [DOI] [PubMed] [Google Scholar]
- 47.Tao N, DePeters EJ, German JB, Grimm R, Lebrilla CB. Variations in bovine milk oligosaccharides during early and middle lactation stages analyzed by high-performance liquid chromatography-chip/mass spectrometry. Journal of Dairy Science. 2009;92(7):2991–3001. doi: 10.3168/jds.2008-1642. [DOI] [PubMed] [Google Scholar]
- 48.Knox JH, Ross P. Carbon-based packing materials for liquid chromatography - Structure, performance, and retention mechanisms. Advances in Chromatography. 1997;37:73–119. [PubMed] [Google Scholar]
- 49.Ross P, Knox JH. Carbon-based packing materials for liquid chromatography: Applications. Advances in Chromatography. 1997;37:121–162. [PubMed] [Google Scholar]
- 50.Domon B, Costello CE. A Systematic Nomenclature for Carbohydrate Fragmentations in Fab-Ms Ms Spectra of Glycoconjugates. Glycoconjugate Journal. 1988;5(4):397–409. [Google Scholar]
- 51.Zechel DL, Withers SG. Glycosidase mechanisms: Anatomy of a finely tuned catalyst. Accounts of Chemical Research. 2000;33(1):11–18. doi: 10.1021/ar970172+. [DOI] [PubMed] [Google Scholar]
- 52.Montreuil J. Structure De Deux Triholosides Isoles Du Lait De Femme. Comptes Rendus Hebdomadaires Des Seances De L Academie Des Sciences. 1956;242(1):192–193. [PubMed] [Google Scholar]
- 53.Kuhn R, Gauhe A. Uber Die Lacto-Difuco-Tetraose Der Frauenmilch - Ein Beitrag Zur Strukturspezifitat Der Blutgruppensubstanz Leb. Annalen Der Chemie-Justus Liebig. 1958;611(1–3):249–253. [Google Scholar]
- 54.Kuhn R, Gauhe A. Uber Ein Kristallisiertes, Lea-Aktives Hexasaccharid Aus Frauenmilch. Chemische Berichte-Recueil. 1960;93(3):647–651. [Google Scholar]
- 55.Kuhn R, Baer HH, Gauhe A. Die Konstitution Der Lacto-N-Fucopentaose Ii - Ein Beitrag Zur Spezifitat Der Blutgruppensubstanz Lea. Chemische Berichte-Recueil. 1958;91(2):364–374. [Google Scholar]
- 56.Strecker G, Montreui J. Isolation and Study on Structure of 16 Oligosaccharides Isolated from Human Urine. Comptes Rendus Hebdomadaires Des Seances De L Academie Des Sciences Serie D. 1973;277(14):1393–1396. [PubMed] [Google Scholar]
- 57.Kuhn R, Brossmer R. Uber O-Acetyllactaminsaure-Lactose Aus Kuh-Colostrum Und Ihre Spaltbarkeit Durch Influenza-Virus. Chemische Berichte-Recueil. 1956;89(9):2013–2025. [Google Scholar]
- 58.Yamashita K, Tachibana Y, Kobata A. Oligosaccharides of Human Milk.10. Structural Studies of 2 New Octasaccharides, Difucosyl Derivatives of Para-Lacto-N-Hexaose and Para-Lacto-N-Neohexaose. Journal of Biological Chemistry. 1977;252(15):5408–5411. [PubMed] [Google Scholar]
- 59.Kobata A, Ginsburg V. Oligosaccharides of Human Milk.2. Isolation and Characterization of a New Pentasaccharide, Lacto-N-Fucopentaoseiii. Journal of Biological Chemistry. 1969;244(20):5496. [PubMed] [Google Scholar]
- 60.Kuhn R, Baer HH, Gauhe A. Kristallisierte Fucosido-Lactose. Chemische Berichte-Recueil. 1956;89(11):2513–2513. [Google Scholar]
- 61.Sabharwal H, Nilsson B, Chester MA, Lindh F, Gronberg G, Sjoblad S, Lundblad A. Oligosaccharides from Feces of a Blood-Group-B, Breast-Fed Infant. Carbohydrate Research. 1988;178:145–154. doi: 10.1016/0008-6215(88)80107-1. [DOI] [PubMed] [Google Scholar]
- 62.Sabharwal H, Nilsson B, Gronberg G, Chester MA, Dakour J, Sjoblad S, Lundblad A. Oligosaccharides from Feces of Preterm Infants Fed on Breast-Milk. Archives of Biochemistry and Biophysics. 1988;265(2):390–406. doi: 10.1016/0003-9861(88)90142-7. [DOI] [PubMed] [Google Scholar]
- 63.Dua VK, Goso K, Dube VE, Bush CA. Characterization of Lacto-N-Hexaose and 2 Fucosylated Derivatives from Human-Milk by High-Performance Liquid-Chromatography and Proton Nmr-Spectroscopy. Journal of Chromatography. 1985 Jun;328:259–269. doi: 10.1016/s0021-9673(01)87396-9. [DOI] [PubMed] [Google Scholar]
- 64.Ginsburg V, Zopf DA, Yamashita K, Kobata A. Oligosaccharides of Human Milk - Isolation of a New Pentasaccharide, Lacto-N-Fucopentaosev. Archives of Biochemistry and Biophysics. 1976;175(2):565–568. doi: 10.1016/0003-9861(76)90546-4. [DOI] [PubMed] [Google Scholar]
- 65.Kuhn R, Baer HH. Die Konstitution Der Lacto-N-Tetraose. Chemische Berichte-Recueil. 1956;89(2):504–511. [Google Scholar]
- 66.Kuhn R, Gauhe A. Die Konstitution Der Lacto-N-Neotetraose. Chemische Berichte-Recueil. 1962;95(2):518. [Google Scholar]
- 67.Tachibana Y, Yamashita K, Kobata A. Oligosaccharides of Human Milk - Structural Studies of Difucosyl and Trifucosyl Derivatives of Lacto-N-Octaose and Lacto-N-Neooctaose.11. Archives of Biochemistry and Biophysics. 1978;188(1):83–89. doi: 10.1016/0003-9861(78)90359-4. [DOI] [PubMed] [Google Scholar]
- 68.Haeuwfievre S, Wieruszeski JM, Plancke Y, Michalski JC, Montreuil J, Strecker G. Primary Structure of Human-Milk Octasaccharides, Dodecasaccharides and Tridecasaccharides Determined by a Combination of H-1-Nmr Spectroscopy and Fast-Atom-Bombardment Mass-Spectrometry - Evidence for a New Core Structure, the Para-Lacto-N-Octaose. European Journal of Biochemistry. 1993;215(2):361–371. doi: 10.1111/j.1432-1033.1993.tb18042.x. [DOI] [PubMed] [Google Scholar]
- 69.Strecker G, Fievre S, Wieruszeski JM, Michalski JC, Montreuil J. Primary Structure of 4 Human-Milk Octa-Saccharides, Nona-Saccharides, and Undeca-Saccharides Established by H-1-Nuclear and C-13-Nuclear Magnetic-Resonance Spectroscopy. Carbohydrate Research. 1992;226(1):1–14. doi: 10.1016/0008-6215(92)84050-3. [DOI] [PubMed] [Google Scholar]
- 70.Kobata A, Ginsburg V. Oligosaccharides of Human Milk.3. Isolation and Characterization of a New Hexasaccharide, Lacto-N-Hexaose. Journal of Biological Chemistry. 1972;247(5):1525. [PubMed] [Google Scholar]
- 71.Kobata A, Ginsburg V. Oligosaccharides of Human Milk.4. Isolation and Characterization of a New Hexasaccharide, Lacto-N-Neohexaose. Archives of Biochemistry and Biophysics. 1972;150(1):273. doi: 10.1016/0003-9861(72)90036-7. [DOI] [PubMed] [Google Scholar]
- 72.Yamashita K, Tachibana Y, Kobata A. Oligosaccharides of Human Milk - Isolation and Characterization of 2 New Non-Asaccharides, Monofucosyllacto-N-Octaose and Monofucosyllacto-N-Neooctaose.8. Biochemistry. 1976;15(18):3950–3955. doi: 10.1021/bi00663a007. [DOI] [PubMed] [Google Scholar]
- 73.Amano J, Osanai M, Orita T, Sugahara D, Osumi K. Structural determination by negative-ion MALDI-QIT-TOF MSn after pyrene derivatization of variously fucosylated oligosaccharides with branched decaose cores from human milk. Glycobiology. 2009;19(6):601–614. doi: 10.1093/glycob/cwp026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.