

Abstract
Protracted hypoperfusion is one of the hallmarks of secondary cerebral derangement after cardiac arrest and resuscitation (CAR), and reactive oxygen species have been implicated in reperfusion abnormalities. Using transgenic (Tg) rats overexpressing copper zinc superoxide dismutase (SOD1), we investigated the role of this intrinsic antioxidant in the restoration of cerebral blood flow (CBF) after CAR. Nine Tg and 11 wild-type (WT) rats were subjected to 14-min cardiac arrest, and CBF was measured using the non-invasive arterial spin labeling magnetic resonance imaging (MRI) method before and during cardiac arrest, and 0–2 h and 1-5 days after resuscitation. The SOD1-Tg rats showed rapid normalization of CBF one day after the insult, whereas CBF in WT animals remained abnormal for at least five days, showing a progressive increase in CBF from hypo- to hyperperfusion on post-resuscitation days 1–5. The long-term outcome, as measured by survival time, change in body weight, and mapping of apparent diffusion coefficient (ADC) for ion/water homeostasis, was significantly better in the SOD1-Tg rats. Our results support the notion that reactive oxygen species are at least partially responsible for microvascular reperfusion disorders.
Introduction
Cerebral reperfusion after cardiac arrest and resuscitation (CAR) typically undergoes three critical phases: the initial hyperperfusion and reactive hyperemia upon resuscitation, the protracted hypoperfusion for hours or even days, and the delayed dissociation between perfusion pressure and microcirculation. Such secondary derangements in cerebral blood flow (CBF) are responsible for poor long-term outcome after even a brief period of circulatory arrest. The deprivation and replenishment of oxygen have been implicated as the primary cause of reperfusion injuries. Like all aerobes, mammals use molecular oxygen (O2) as a direct source of oxidants to support efficient metabolisms. This advantage does not come without a price, however, because a significant amount of O2 undergoes a one-electron reduction directly to the superoxide anion (O2•−), which in turn leads to highly cytotoxic reactive oxygen species (ROS). Most notable is the reaction of superoxide anion with hydrogen peroxide (H2O2) in the presence of transition metals to produce the extremely reactive hydroxyl radical OH•, which is responsible for most of the oxidative damage in the aerobic system.
Under normal conditions, cellular defense against the burden of O2•− and H2O2 involves superoxide dismutases (SOD), catalases, and nonspecific peroxidases.1 Under pathological conditions, however, these enzymatic defense systems are rendered insufficient or dysfunctional. After global ischemia in particular, several events can occur simultaneously to set the stage for a massive production of ROS.2 Major pathways for ROS production include: prostaglandin (PG) synthesis (i.e., the conversion of arachidonic acid to PGG2 and the subsequent peroxidized conversion to PGH2 with concomitant release of O2•−), activation of neuronal nitric oxide synthase (nNOS) leading ultimately to peroxynitrite (ONOO−), purine catabolism (i.e., O2•− release in the successive reactions of hypoxanthine to xanthine, and xanthine to urea catabolized by the xanthine oxidase, and neutrophil activation. The first two pathways are strongly dependent on cytosolic Ca2+, which is elevated during global ischemia. Upon reperfusion with a burst in O2 availability, •OH and ONOO− lead not only to protein and membrane peroxidation but also to DNA damage and apoptosis.3
ROS production through purine catabolism and neutrophil activation is highly relevant to the vascular response to ischemia. Pro-inflammatory stimulation and polymorphonuclear neutrophil infiltration, as well as endothelial cell reactions, contribute to the formation of platelet-activating factor and leukotrienes, the inactivation of anti-adhesive molecules, and the expression of endothelial cell adhesion molecules (e.g., P-selectin and intercellular adhesion molecule-1). All of these events can be triggered upon the respiratory burst of ROS production at onset of reperfusion by nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activities in neutrophils and endothelial cells, resulting in vascular dysfunction.
Overexpression of SOD has long been suggested as an effective way to enhance the cellular defense against ROS toxicity and reperfusion injuries.4–6 Three types of SOD isoenzymes exist in mammals with different prosthetic metal ions and cellular locations: SOD1 (or Cu/Zn-SOD) is present in the cytoplasm and binds to copper and zinc in a dimer form; SOD2 (or Mn-SOD) is an inducible mitochondrial enzyme and binds to manganese; and SOD3 also uses copper and zinc as a prosthetic ions but is present only in the extracellular space in a tetrameric form. About 90% of the total SODs in mammals are SOD1.7 In this study, we combined the use of transgenic (Tg) rats overexpressing SOD1 and non-invasive magnetic resonance imaging (MRI) techniques to investigate the recovery of cerebral perfusion and ion-water homeostasis after a nominal 14-min cardiac arrest, using a clinically relevant outcome model of CAR in rats. Our results demonstrated the profound protective effect of SOD1 overexpression on the rapid normalization of cerebral perfusion in Tg rats compared to wild-type (WT) animals, showing a significantly better long-term outcome in the Tg animals.
Materials and Methods
The experimental protocols were approved by the Institutional Animal Care and Use Committees of the University of Pittsburgh and Stanford University. Sprague-Dawley rats (Harlan Sprague Dawley, Inc., Indianapolis, IN), transgenically modified to overexpress SOD1, were produced as detailed previously.5 Nine male Tg rats and 11 male WT controls were used in the present study.
The CAR procedures were essentially the same as described previously.8–10 Briefly, rats were surgically prepared by cannulating both femoral arteries and one femoral vein under 1–1.25% isoflurane general anesthesia. Mechanical ventilation with an approximate 1:1 mixture of oxygen and air was controlled and carefully adjusted to maintain physiologically normal arterial blood gases before CAR. Arterial blood gases were measured frequently using a Corning 278 pH/blood gas analyzer (Chiron Diagnostics, East Walpole, MA, USA). Once surgically prepared, the animals were placed snugly in the cradle of a custom-designed birdcage imaging probe and positioned inside the 9.4-T magnet of a Varian CMXW-400SLI imaging spectrometer (Varian Inc., Palo Alto, CA) for MRI scans. Cardiac arrest was induced remotely by intravenous (iv) injection of the short-acting beta-1 adrenergic blocker esmolol combined with apnea, which was achieved by stoppage of mechanical ventilation under complete muscle relaxation with pancuronium bromide (2.0 mg/kg/hr iv) and vecuronium bromide (1.0 mg/kg iv given 5 min before apnea). Resuscitation began with restoration of mechanical ventilation and retrograde intra-aortic infusion of donor arterial blood mixed with epinephrine (8 μg/ml), sodium bicarbonate (0.05 mEq/ml), and heparin (5 U/ml) until the first sign of return of spontaneous circulation (ROSC) was observed in the arterial blood pressure recording. MRI scans were collected continuously before and during cardiac arrest and after resuscitation. At least two hours after resuscitation, rats were taken out of the magnet, catheters were removed, and wounds were sutured. On post-resuscitation days 1–5, rats were again anesthetized, intubated, and placed inside the magnet for repeated evaluations with MRI. To assess long-term outcome, the total survival time censored at 120 hours post-resuscitation, body weight changes, and the neurological deficit score (NDS) were evaluated on the fifth day after the CAR episode. The NDS is a numeric system based on the empirical evaluation of the neurological appearance of the animals,9, 11 with 500 being neurologically normal and 0 being brain dead. For histology evaluation, hematoxylin and eosin (H&E) staining was performed, and the histology outcome was quantified by counting healthy neurons, normalized against the area, in a predetermined segment in the CA1 region of the left hippocampus.
The non-invasive MRI evaluations included CBF measurements using the arterial spin labeling technique12 and water apparent diffusion coefficient (ADC) mapping. The established methods, as detailed previously,9, 13–16 were used with the following parameters: imaging plane at the dorsal hippocampal level (plane 29 to 32 in Sawnson’s rat brain atlas17), plane offset for the arterial spin tagging = ± 1.4 cm, duration of tagging pulse = 0.8 s, recycle delay (TR) = 1 s, field-of-view (FOV) = 44 × 44 mm2, data matrix = 128 × 64, and acquisition time = 5 min for each pair of perfusion images. ADC maps were derived from pixel-by-pixel exponential fitting of five spin-echo images acquired with a pair of 5-ms Stejskal-Tanner18 diffusion-weighting gradients, applied in the readout direction on both sides of the 180° refocusing pulse to produce diffusion-weighting b factors of 0, 20, 81, 182, and 324 s/mm2. Other acquisition parameters for ADC mapping were the same as those used for the perfusion measurements. ADC maps were acquired before cardiac arrest and 80 min and 5 d after resuscitation. Temporal resolution was 12.5 min per ADC map. The average perfusion and ADC in the brain regions were calculated using LabVIEW software (National Instruments, Austin, TX) by scaling and fitting individual images to the reference anatomical masks.9 Statistical analysis was performed using SPSS software (SPSS Inc., Chicago, IL).
Results
All animals were subjected to 14 min of apnea, beginning with the stoppage of ventilation and bolus esmolol injection to the onset of the retrograde infusion of oxygenated blood and restart of mechanical ventilation. Resuscitation was highly reproducible, with an average resuscitation time of 1.72 ± 0.49 min among all animals. Table 1 summarizes the physiological parameters related to CAR experiments for the control and Tg animal groups. Total arrest time was calculated from the point when the mean and pulse arterial pressures dropped below 10 and 5 mmHg, respectively, to the point when the first sign of ROSC was observed. The mean arterial blood pressure (MABP) and heart rate (HR) before, during, and after the in-magnet CAR are depicted in Figure 1. No statistical difference was found in MABP, HR, initial body weight, PaCO2, total arrest time, and resuscitation time (time to ROSC) between groups. Although the averaged PaO2 values showed a significant difference, all PaO2 values were above 180 mmHg, corresponding to arterial oxygen saturation over 99.5% before cardiac arrest in all animals.
Table 1.
Physiological parameters of cardiac arrest and resuscitation*
| Group | N | Weight, g | PaO2, mmHg† | PaCO2, mmHg† | Total Arrest Time, min | Time to ROSC, min |
|---|---|---|---|---|---|---|
| SOD-Tg | 9 | 200 ± 31 | 284 ± 12 | 39.1 ± 4.7 | 15.07 ± 0.18 | 1.48 ± 0.16 |
| Wild-Type | 11 | 197 ± 22 | 246 ± 34 | 40.0 ± 3.5 | 15.51 ± 0.59 | 1.92 ± 0.59 |
Data are presented as Mean ± SD
Blood gas values before the induction of cardiac arrest
Figure 1.

Comparison of (A) mean arterial blood pressure (MABP) and (B) heart rate (HR) before and during a nominal 15-min cardiac arrest and the first 2 h after resuscitation between Cu/Zn superoxide dismutase-overexpressed rats (SOD) and the wild-type controls (CON). Repeated measures analysis of variance showed no significant difference in MABP and HR between groups.
Changes in cerebral perfusion as a function of time, before and during cardiac arrest and immediately after resuscitation, exhibited the same pattern in the Tg rats as in the WT controls. Figure 2 shows arrays of representative CBF maps from a typical rat in each group. The regional CBF in the unit of “ml of blood per gram of brain tissue per min” is color-coded using the color scale shown in the figure. Hyperperfusion occurred within a few minutes of ROSC, followed by protracted hypoperfusion until the end of MRI measurements on the day of CAR experiments. Major differences in cerebral reperfusion between the two groups became apparent 1–5 days after the resuscitation. Whereas animals in the WT control group still showed hypoperfusion 24 h after resuscitation and demonstrated gradual increases in CBF from hypoperfusion to hyperperfusion in the next 4 days, CBF in the Tg rats seemed to have returned to the normal level within one day. The brain-averaged perfusion values as a function of time are plotted for the early CAR period in Figure 3 and for 1–5 days after resuscitation in Figure 4. The region-averaged CBF in the cortex, hippocampus, thalamus, and amygdala followed similar patterns. Before and during cardiac arrest and within 120 min after resuscitation, CBF showed no significant difference between groups except for the first ~5 min immediately after ROSC (brain-averaged PCA-5min = 0.037 by repeated measures ANOVA). This initial perfusion difference is particularly profound in the hippocampus (PCA-5min = 0.026) and thalamus (PCA-5min = 0.004). It is interesting to note that in all brain regions, Tg rats showed slightly less hyperperfusion and reactive hyperemia immediately after ROSC.
Figure 2.

Representative cerebral perfusion images measured by the arterial spin labeling MRI techniques for a typical Cu/Zn superoxide dismutase-overexpressed rat (SOD) and a wild-type control (CON) before and during 15-min cardiac arrest and immediately and 1–5 days after resuscitation. The perfusion values are color-coded using the scale bar shown above the images.
Figure 3.
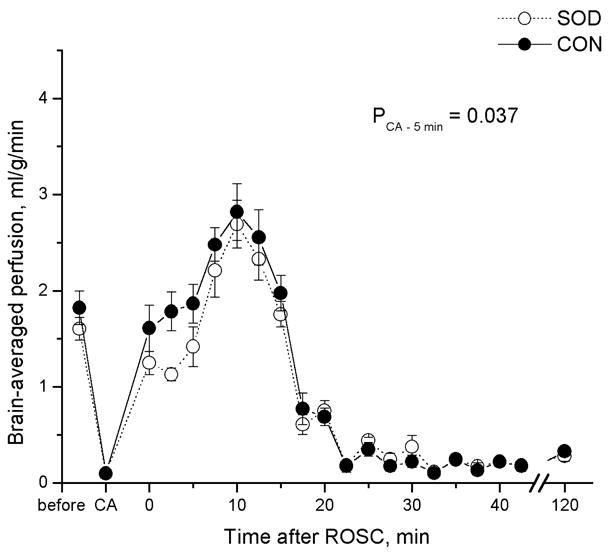
Comparison of brain-averaged cerebral perfusion changes between Cu/Zn superoxide dismutase-overexpressed rats (SOD) and the wild-type controls (CON) before and during 15-min cardiac arrest and the first 2 h after resuscitation. PCA-5min is the p value for the first 5 min of reperfusion between the two groups. No perfusion difference was measureable in the remaining 2-h period.
Figure 4.
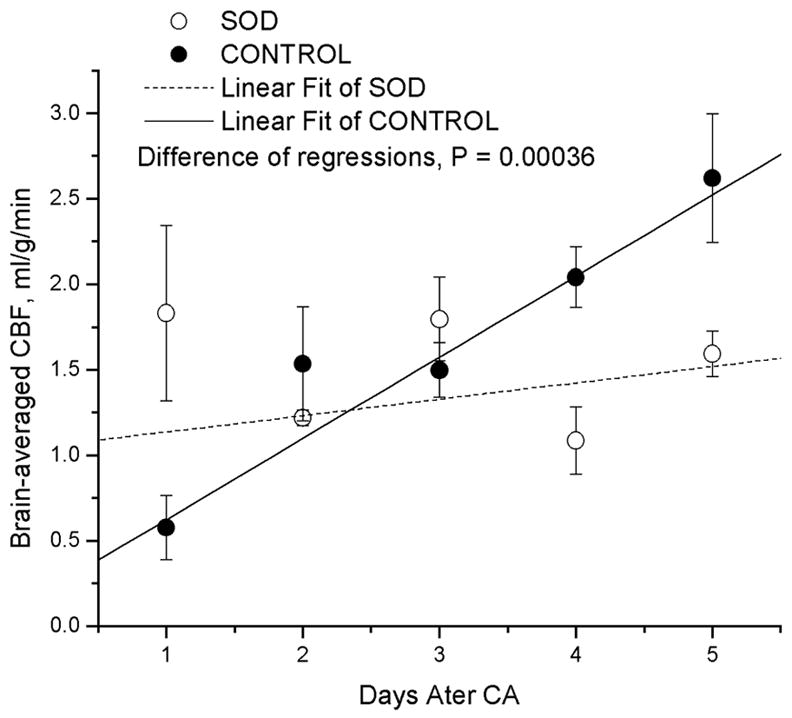
Comparison of brain-averaged cerebral perfusion changes between Cu/Zn superoxide dismutase-overexpressed rats (SOD) and the wild-type controls (CON) 1–5 days after resuscitation. Lines are error-weighted linear regression to the data. Notice the quick normalization of CBF in the SOD1 transgenetic rats and the protracted abnormality from hypo-to hyperperfusion in the wild-type animals.
Recovery of cerebral perfusion was significantly faster in the Tg rats than in the WT controls. After one day, CBF in Tg rats returned to and remained near the normal level. The solid and dashed lines in Figure 4 are error-weighted linear least squares fitting to the brain-averaged CBF among all animals in the control and Tg groups, respectively. The slopes between the two groups are significantly different (P<0.0004). Notice that the CBF in the control animals changed from hypoperfusion to profound hyperperfusion over the 5-day recovery period, indicating a long period (up to 5 days) of abnormality in CBF.
Changes in the ion and water homeostasis after cardiac arrest and resuscitation can be inferred from MRI diffusion mapping. Indeed, the apparent diffusion coefficients are significantly depressed in the lateral cortex, thalamus, and hypothalamus 80 min after CAR (Figure 5), implicating disturbances to the distribution and compartmentation of tissue water. Five days after the CAR episode, ADC maps appeared normal in all Tg rats but showed scattered abnormalities in the WT animals (see Figure 5).
Figure 5.
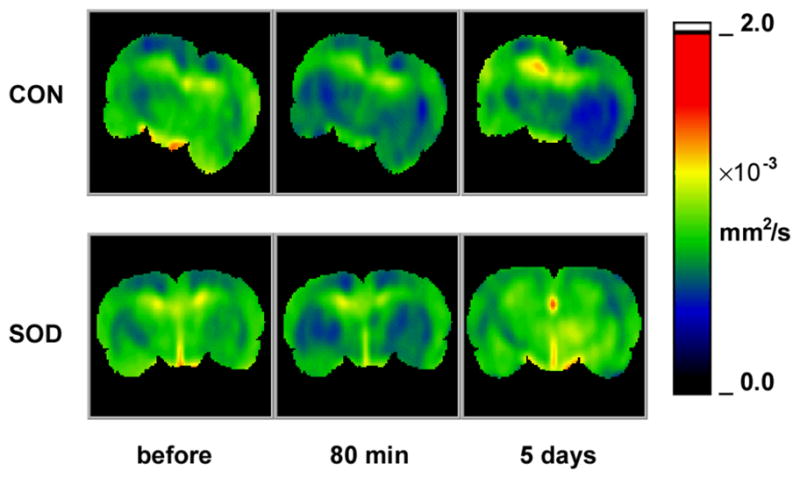
Comparison of typical apparent-diffusion coefficient (ADC) maps between Cu/Zn superoxide dismutase-overexpressed rats (SOD) and the wild-type controls (CON) before cardiac arrest and 80 min and 5 d after resuscitation. ADC values, color-coded using the scale bar to the right of the images, reflect changes in ion-water homeostasis. Notice the scattered ADC abnormalities in the control rats and rapid recovery in the SOD rats.
Consistent with the degree of CBF and ADC recovery, the long-term outcome, as measured by mean survival time censored at 120 hours (i.e., the end of the observation period), body weight change after CAR, and NDS, was significantly better in the Tg group. Table 2 summarizes the long-term outcome measures. All Tg animals were alive at the end of the observation period, gained on average 9 g in 5 days, and had a median NDS of 460. In contrast, 36% of the animals in the control group died before the end of the 120-h observation period (one survived for ~100 hours and died at the beginning of the fifth day). The remaining WT rats lost ~30 g of body weight, and the average NDS was significantly lower than in the Tg group. It is interesting to note that the histology score by neuron counting in the hippocampal CA1 region was an insensitive measure of long-term outcome. The number of healthy neurons, normalized within a predetermined area, was not significantly different between the two groups, being 162±100 and 103±62 for the control and Tg group, respectively (p=0.18, t-test). This is in agreement with a previous study,19 which also showed that the histology damage in the hippocampal CA1 region was not significantly different between SOD1 Tg mice and WT mice after KCl-induced cardiac arrest under hyperthermia.
Table 2.
Parameters of outcome evaluation after cardiac arrest and resuscitation
| Group | 120-h survival, % | Mean survival time, h* | Weight gain, g¶ | NDS§ |
|---|---|---|---|---|
| SOD-Tg | 100 (9/9) | > 120† | +9 ± 4# | 460 (433, 500)& |
| Wild-Type | 64 (7/11) | 94 ± 12† | −30 ± 9# | 400 (0, 450)& |
Survival time was calculated by Kaplan-Weier survival analysis and log-rank test, with censored values set to 120 hours. Data are presented as Mean ± SD
Data are presented as Mean ± SEM
Data are presented as Median (25th percentile, 75th percentile).
Significant differences between groups (†, P = 0.05, log-rank test; #, P = 0.001, t-test; &, P = 0.029, non-parametric Mann-Whitney U test)
Discussion
The availability of SOD1-overexpressed rats and a long-term outcome model of CAR allowed us to study the role of SOD1 in mitigating cerebral reperfusion impairment after a clinically relevant episode of global ischemia. The combination of non-invasive MRI techniques with the transgenic approach provided a unique experimental paradigm to test the hypothesis that enhancement of cellular defenses against oxidative damage can greatly improve overall long-term outcome as judged by the speed of physiological and behavioral recovery. In this study, we found that SOD1 Tg rats recovered from a severe ischemic insult significantly better than WT rats. This finding is consistent with results from previous studies using the same Tg rats with a transient two-vessel-occlusion model.5 The previous studies focused on cellular events in selectively vulnerable neurons and found that SOD1 overexpression confers protection by blocking the mitochondrial pathway of caspase activation.6 Involvement of caspase-independent apoptotic pathways was also suggested, particularly the translocation of apoptosis-induced factor and endonuclease G from mitochondria to nuclei.20 More recent studies seem to also suggest that after a severe ischemic and hypoxic insult, very few cells can reach the stage of caspase-3 activation to complete the apoptosis process.21 These findings underscore the complexity of cellular pathways of cell death after global ischemia and reperfusion.
In complement with the previous cellular level investigations, the present study focused on macroscopic recovery on a global scale. Based on the initial vascular response to resuscitation, we found that SOD1 overexpression had little effect on CBF during the first two hours of reperfusion (except for the initial 5 min after ROSC). This is in accordance with the notion that overproduction of SOD1 after global ischemia and reperfusion takes several hours.22 The only detectable difference in initial reperfusion between the two groups was in the first 5 min after ROSC, during which the WT animals showed slightly higher levels of reactive hyperemia. Our previous report concluded that after a long period of circulatory arrest, the first 15 to 30 min of hyperperfusion is controlled by the resuscitation efficacy and is crucial for the long-term outcome.14 For both the Tg and WT groups in the present study, the resuscitations were highly efficacious and the initial reperfusion was more than adequate to account for the better than usual 5-day survival rate after ~15 min of complete circulatory arrest. However, the overall neurological deficit score for the WT group was significantly worse than that of the SOD1 Tg group, suggesting that events beyond the first few hours of reperfusion might have accounted for the difference between the two groups. Therefore, it can be concluded that the therapeutic window to improve long-term outcome after a prolonged cardiac arrest can possibly be extended to several hours or even days. It should be noted, however, that transgenic approach represents the maximum therapeutic potentials achievable by the action of superoxide dismutase.
The time course of superoxide radical production was compared previously between the SOD1 Tg rats and WT rats using a somewhat invasive technique (2-vessel occlusion plus hypotension).6 In the WT rats, an early, sharp increase in superoxide radicals in hippocampal CA1 neurons was found one hour after ischemia and reperfusion. The increased level was elevated even after one day, and then returned to the basal level after three days. In contrast, the increase in superoxide radicals was significantly reduced one hour post-ischemia in the SOD1 Tg rats, and quickly returned to the basal level after one day (see Figure 2 in Sugawara et al.6). Interestingly, the complete return of the superoxide radical level to the basal level in the SOD1 Tg rats within one day in the transient global cerebral ischemia model correlates well with the rapid normalization of CBF within one day after the cardiac arrest and resuscitation insult in this model.
Although the WT animals showed reasonable survival rates due to effective resuscitation, disturbance to cerebral perfusion after resuscitation lasted significantly longer in these animals than in their Tg counterparts. The well-documented protracted hypoperfusion after cardiac arrest apparently lasted for more than 24 h in the WT rats, as judged by both the CBF maps (Figure 2) and the brain-averaged perfusion values (Figure 4). In the following days, until the end of the observation period, the CBF steadily increased to a state of severe hyperperfusion. We first documented this kind of delayed CBF reversal in focal ischemia using the middle cerebral arterial occlusion model,15 and the present study is perhaps the first report of the similar CBF overshoot after global ischemia. In our earlier studies with shorter cardiac arrest times, the overshoot was less apparent in the averaged perfusion values.9 While it is generally believed that the hypoperfusion is a consequence of vasospasms in arterioles and capillaries and activation of endothelial cell adhesive molecules, the mechanisms of subsequent hyperperfusion are not entirely clear. We speculated in the previous focal ischemia study that the CBF overshoot was a sign of vascular injuries caused by ROS. Indeed, the SOD1-overexpressed Tg rats in the present study showed no sign of delayed perfusion reversal and overshoot. Moreover, after an identical global ischemic insult, the Tg rats exhibited much faster normalization in perfusion.
Thus, in addition to the devastating neuronal events in the selectively vulnerable regions, vascular events also likely determine the overall long-term outcome. Several factors can contribute to the delayed but prolonged hyperperfusion in the WT animals. The pro-inflammatory reactions stimulate and mobilize neutrophils and quickly up-regulate cytokines and chemokines in response to ischemia. These processes are known to activate nitric oxide synthases and NADH/NADPH oxidase in the endothelia and neutrophils upon re-oxygenation. Following the initial hemodynamic response to the activation of platelets and the inactivation of anti-adhesive molecules, which cause hypoperfusion, the over-production of O2•− and H2O2, both potent vasodilators, can lead to uncontrollable vasodilation. Although arteriole dilatation is possibly an over-correction reaction to ischemia, when the production of ROS becomes uncontrolled, the result is the loss of blood vessel’s ability to respond to changes in perfusion pressure, thus leading to autoregulatory dysfunction. Indeed, microarray and protein expression analyses of the temporal profiles of various genes after ischemia and reperfusion23 showed that proinflammatory gene expression was greatly reduced in Tg animals overexpressing SOD1, and the protein expression of monocyte chemoattractant protein 1 and macrophage inflammatory protein-1 alpha was significantly lower in Tg animals than in the WT controls.23 Gene transfer experiments to overexpress SOD1 have shown amelioration to CBF autoregulatory dysfunction after ischemia and brain injuries.24
In summary, using transgenic technology combined with non-invasive MRI for cerebral perfusion and diffusion measurements, we demonstrated that rats overexpressing Cu/Zn superoxide dismutase show much faster CBF recovery after a severe global ischemia and reperfusion insult. The SOD1 effects on long-term recovery and outcome seem to be associated with the prevention and amelioration of ROS-induced vascular deterioration. While WT rats exhibited at least 5 days of perfusion abnormalities, ranging from hypoperfusion to prolonged hyperperfusion, CBF and ion-water homeostasis, as measured by ADC maps, quickly returned to a normal level after 24 h of reperfusion in SOD1-Tg rats.
Acknowledgments
The authors would like to thank Mr. Bernard Calagui for technical assistance in making the transgenic animals. This work was supported in part from grants from the National Institute of Neurological Disorder and Stroke, National Institutes of Health (R01NS036124, P50NS014543, R01NS025372, R01NS036147, and R01NS038653).
References
- 1.Maier CM, Chan PH. Role of superoxide dismutases in oxidative damage and neurodegenerative disorders. Neuroscientist. 2002;8:323–334. doi: 10.1177/107385840200800408. [DOI] [PubMed] [Google Scholar]
- 2.Saito A, Maier CM, Narasimhan P, Nishi T, Song YS, Yu F, Liu J, Lee YS, Nito C, Kamada H, Dodd RL, Hsieh LB, Hassid B, Kim EE, Gonzalez M, Chan PH. Oxidative stress and neuronal death/survival signaling in cerebral ischemia. Mol Neurobiol. 2005;31:105–116. doi: 10.1385/MN:31:1-3:105. [DOI] [PubMed] [Google Scholar]
- 3.Chan PH, Kinouchi H, Epstein CJ, Carlson E, Chen SF, Imaizumi S, Yang GY. Role of superoxide dismutase in ischemic brain injury: Reduction of edema and infarction in transgenic mice following focal cerebral ischemia. Prog Brain Res. 1993;96:97–104. doi: 10.1016/s0079-6123(08)63260-4. [DOI] [PubMed] [Google Scholar]
- 4.Chan PH, Epstein CJ, Kinouchi H, Kamii H, Chen SF, Carlson E, Gafni J, Yang G, Reola L. Neuroprotective role of cuzn-superoxide dismutase in ischemic brain damage. Adv Neurol. 1996;71:271–280. [PubMed] [Google Scholar]
- 5.Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ. Overexpression of sod1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugawara T, Noshita N, Lewen A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH. Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci. 2002;22:209–217. doi: 10.1523/JNEUROSCI.22-01-00209.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noor R, Mittal S, Iqbal J. Superoxide dismutase--applications and relevance to human diseases. Med Sci Monit. 2002;8:RA210–215. [PubMed] [Google Scholar]
- 8.Liachenko S, Tang P, Hamilton RL, Xu Y. A reproducible model of circulatory arrest and remote resuscitation in rats for nmr investigation. Stroke. 1998;29:1229–1238. doi: 10.1161/01.str.29.6.1229. discussion 1238–1229. [DOI] [PubMed] [Google Scholar]
- 9.Liachenko S, Tang P, Hamilton RL, Xu Y. Regional dependence of cerebral reperfusion after circulatory arrest in rats. J Cereb Blood Flow Metab. 2001;21:1320–1329. doi: 10.1097/00004647-200111000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Jomura S, Uy M, Mitchell K, Dallasen R, Bode CJ, Xu Y. Potential treatment of cerebral global ischemia with oct-4+ umbilical cord matrix cells. Stem Cells. 2007;25:98–106. doi: 10.1634/stemcells.2006-0055. [DOI] [PubMed] [Google Scholar]
- 11.Neumar RW, Bircher NG, Sim KM, Xiao F, Zadach KS, Radovsky A, Katz L, Ebmeyer E, Safar P. Epinephrine and sodium bicarbonate during cpr following asphyxial cardiac arrest in rats. Resuscitation. 1995;29:249–263. doi: 10.1016/0300-9572(94)00827-3. [DOI] [PubMed] [Google Scholar]
- 12.Williams DS, Detre JA, Leigh JS, Koretsky AP. Magnetic resonance imaging of perfusion using spin inversion of arterial water. Proc Natl Acad Sci U S A. 1992;89:212–216. doi: 10.1073/pnas.89.1.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liachenko S, Tang P, Xu Y. Deferoxamine improves early postresuscitation reperfusion after prolonged cardiac arrest in rats. J Cereb Blood Flow Metab. 2003;23:574–581. doi: 10.1097/01.WCB.0000057742.00152.3F. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, Liachenko S, Tang P. Dependence of early cerebral reperfusion and long-term outcome on resuscitation efficiency after cardiac arrest in rats. Stroke. 2002;33:837–843. doi: 10.1161/hs0302.104198. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Yushmanov VE, Liachenko SM, Tang P, Hamilton RL, Xu Y. Late reversal of cerebral perfusion and water diffusion after transient focal ischemia in rats. J Cereb Blood Flow Metab. 2002;22:253–261. doi: 10.1097/00004647-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Yushmanov VE, Wang L, Liachenko S, Tang P, Xu Y. Adc characterization of region-specific response to cerebral perfusion deficit in rats by mri at 9.4 t. Magn Reson Med. 2002;47:562–570. doi: 10.1002/mrm.10103. [DOI] [PubMed] [Google Scholar]
- 17.Swanson LQ. Brain maps: Structure of the rat brain. Amsterdam: Elsevier; 1998. [Google Scholar]
- 18.Stejskal EO, Tanner JE. Spin diffusion measurements: Spin echoes in the presence of a time-dependent field gradient. J Chem Phys. 1965;42:288–292. [Google Scholar]
- 19.Kofler J, Hurn PD, Traystman RJ. Sod1 overexpression and female sex exhibit region-specific neuroprotection after global cerebral ischemia due to cardiac arrest. J Cereb Blood Flow Metab. 2005;25:1130–1137. doi: 10.1038/sj.jcbfm.9600119. [DOI] [PubMed] [Google Scholar]
- 20.Yu F, Sugawara T, Nishi T, Liu J, Chan PH. Overexpression of sod1 in transgenic rats attenuates nuclear translocation of endonuclease g and apoptosis after spinal cord injury. J Neurotrauma. 2006;23:595–603. doi: 10.1089/neu.2006.23.595. [DOI] [PubMed] [Google Scholar]
- 21.Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, Davis RJ, Mizushima N, Rakic P, Dardzinski BJ, Holland SK, Sharp FR, Kuan CY. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato H, Kogure K, Araki T, Liu XH, Kato K, Itoyama Y. Immunohistochemical localization of superoxide dismutase in the hippocampus following ischemia in a gerbil model of ischemic tolerance. J Cereb Blood Flow Metab. 1995;15:60–70. doi: 10.1038/jcbfm.1995.7. [DOI] [PubMed] [Google Scholar]
- 23.Nishi T, Maier CM, Hayashi T, Saito A, Chan PH. Superoxide dismutase 1 overexpression reduces mcp-1 and mip-1 alpha expression after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:1312–1324. doi: 10.1038/sj.jcbfm.9600124. [DOI] [PubMed] [Google Scholar]
- 24.Shin HK, Hong KW. Importance of calcitonin gene-related peptide, adenosine and reactive oxygen species in cerebral autoregulation under normal and diseased conditions. Clin Exp Pharmacol Physiol. 2004;31:1–7. doi: 10.1111/j.1440-1681.2004.03943.x. [DOI] [PubMed] [Google Scholar]