

Drugs used in chemotherapy predominantly act by targeting the mechanisms of cell division,1 and to an extent, preferentially affect cancer cells because of their unusually high proliferation rates. Unfortunately, many healthy cells are also affected by these treatments, resulting in side effects that cause substantial discomfort for the patient. Emerging methods seek to focus the delivery of drugs on cancer tissue by targeting specific characteristics found in solid tumors.2 As a promising subset of these approaches, macromolecular drug delivery seeks to attach many small molecule drugs and targeting groups to large structures.3 Many of these delivery vehicles experience prolonged circulation time because of their increased size,4 as well as a degree of passive targeting through the enhanced permeation and retention effect, arising from unique characteristics of tumor vasculature.5 As an additional benefit, macromolecules are large enough to display multiple copies of active targeting ligands to enhance binding avidity,6 and can also provide multiple cargo attachment sites to increase the amount of payload that can be delivered.
Many macromolecules are currently being studied for use as drug carriers. Polymer therapeutics are based on a wide variety of materials, such as poly-ethylene glycol (PEG)7 or hydroxypropyl methacrylate,8 allowing carriers with a wide range of delivery behaviors to be designed. Dendritic polymers also offer these properties, with the added advantage of controlled polydispersity and globular shape.9 Liposomal10 and micellular11 systems feature high cargo loadings, but can be difficult to store and can suffer from irregular release and distribution profiles. Inorganic nanoparticles12 and carbon nanotubes13 both feature unique properties as delivery vehicles, but must undergo thorough testing to evaluate their biocompatibility. Protein based systems, such as the recent usage of serum albumin,14 are inherently biocompatible, but most current systems do not exhibit any form of active targeting. As a notable exception, thermophilic heat shock protein cages have been modified to house up to 24 doxorubicin molecules and display targeting peptide inserts on their exterior surface.15
As a flexible alternative that can house increased amounts of drug cargo, we report here the attachment of a water-soluble derivative of the chemotherapeutic taxol to a viral capsid and demonstrate the cytotoxicity of these drug-capsid conjugates in cell culture. In combination with available methods for the installation of peptide16 and aptamer-based17 targeting groups on the external surface of the capsids, this approach could provide a new series of drug carriers directed to a variety of specific tissue types.
The assembled capsid of bacteriophage MS2 provides an attractive scaffold for elaboration as a drug delivery vehicle.18 In the self assembly process, 180 copies of the MS2 protein monomer come together to form a 27 nm hollow sphere with 32 pores. These 2 nm wide pores allow the installation of small molecules on the interior surface without disassembly. The capsid protein assembles in E. coli after recombinant expression,16 yielding non-infectious genome-free particles with adjustable amino acid composition. The capsid structure is inherently monodisperse, an important advantage for the controlled biodistribution of drug delivery vehicles.
By covalently modifying specific amino acid residues on each of the MS2 protein monomers, it is possible to attach many copies of small molecules to the two surfaces of the MS2 shell. Our group has previously reported the attachment of magnetic resonance imaging contrast agents,19 positron emission tomography radioisotopes,20 and fluorescent dyes21,17 to the interior of intact capsids. The exterior surface has been modified with peptides16 and DNA aptamers17 that can target specific receptors on cancer tissue, as well as PEG chains that can protect the protein shell from antibody binding.21 Preliminary animal studies followed using PET have demonstrated the longer lifetime of capsid-bound small molecules compared to the imaging agents themselves.20 For drug delivery purposes, drug molecules would be attached to the interior surface of the capsid with the expectation that the cargo would be shielded from degradation and from non-specific interactions with healthy tissue.
The strategy for interior modification was based on cysteine alkylation chemistry. The wild-type MS2 coat protein contains 2 cysteine residues near the exterior surface of the capsid; however, these have previously been observed to exhibit diminished reactivity due to their poor solvent accessibility.17 Therefore, an amino acid on the interior of the capsid was mutated to a cysteine (N87C) to provide a sulfhydryl group for the attachment of cargo. The drug we chose to attach to the MS2 capsid was taxol, a potent chemotherapeutic used in current treatment of breast, lung, and ovarian cancers.22 However, taxol suffers from prohibitively low water solubility and therefore must be administered over long periods of time as an infusion containing a toxic detergent.23 Although the attachment of taxol to a water soluble protein carrier would increase the solubility of the drug during delivery, it was first necessary to solubilize it in water to allow protein bioconjugation.
With this requirement in mind, we designed a linker with 3 functionalities: an attachment site to the protein, a charged functional group to increase water solubility, and a cleavable linkage to taxol (Figure 1). Starting from mono BOC protected lysine, a maleimide group was first installed on the ε-amine using maleic anhydride, followed by DCC activation and cyclization in the presence of N-hydroxysuccinimide (NHS).24 In the same step, the carboxylic acid of the lysine core was converted to the NHS ester. Taurine was reacted with the activated acid to install a sulfonate group as a water-solubilizing agent, followed by TFA deprotection to liberate the lysine α-amino group for attachment to taxol. To install a suitable attachment group, the 2′-OH of taxol was reacted with succinic anhydride to transform the hydroxyl group into a carboxylic acid functionality (according to a previous literature procedure25), which was then attached to 4 under HATU amide bond forming conditions. After solid phase extraction with an anion exchange resin, crude 6 was purified by reversed phase HPLC and used as a 2 mM solution in 10 mM pH 7 phosphate buffer.
Figure 1.

Synthesis of taxol-MS2 conjugates. (a) Reagents and conditions: (i) maleic anhydride, DMF, 2 h, RT; (ii) NHS, DCC, DMF, 12 h, RT; (iii) taurine, DIPEA, DMF/H2O 4:1, RT; (iv) TFA, CH2Cl2, 1 h, 0 °C; (v) 5, HATU, DIPEA, DMF, 4 h, RT. DMF = N,N-dimethyl formamide, NHS = N-hydroxysuccinimide, DCC = N,N′-dicyclohexyl carbodiimide, DIPEA = N,N-diisopropylethylamine, TFA = trifluoroacetic acid, HATU = O-(7-azabenzotriazole-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate. (b) Conditions: (vi) 6, 10 mM phosphate buffer, pH 7, 1 h, RT.
Reaction of the maleimide group of 6 with the sulfhydryl group of N87C MS2 was achieved at room temperature in 10 mM phosphate buffer at pH 7. Five equivalents of 6 were added to N87C MS2 and the reaction allowed to proceed for 1 h. The use of higher pH buffers resulted in detectable degradation of the taxol component. The modified capsids were purified from small molecules via size exclusion chromatography. ESI-MS (Figure 2a) of the capsid monomers showed significant levels of single modification, while wild type MS2 showed no modification under the same reaction conditions (Supporting Information, Figure S1). Thus, the modification is most likely entirely confined to the cysteine residue introduced on the capsid interior. For analysis purposes, reversed phase HPLC (Figure 2b) was used to separate modified and unmodified MS2 monomers. Integration of the tryptophan fluorescence peaks showed up to 65% modification of the MS2 monomer proteins, which translated to ~110 molecules of taxol per capsid, or approximately 2% loading by weight.
Figure 2.
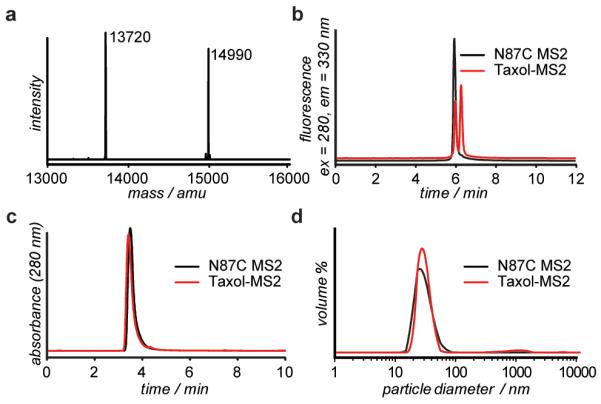
Analysis of taxol-MS2 conjugates. (a) ESI-MS reconstruction of N87C MS2 and the taxol-MS2 conjugate. Expected mass of N87C MS2 m/z [M + H+] = 13719 amu. Expected mass of taxol-MS2 m/z [M + H+] =14987 amu. (b) Reversed-phase HPLC was used to quantify the modification level based on the increased retention caused by the hydrophobic taxol group. (c) Size exclusion chromatography and (d) dynamic light scattering analysis of N87C MS2 and Taxol-MS2 confirm that the capsids remained assembled after modification.
The attachment of so many hydrophobic molecules to the capsid could potentially disturb the stability of the protein assembly. However, size exclusion chromatography (Figure 2c) indicated that the protein remained as intact capsids. TEM imaging (Supporting Information, Figure S2) confirmed the presence of assembled capsids, and dynamic light scattering (DLS) measurements (Figure 2d) showed that the modified MS2 capsids were of comparable diameter to wild-type MS2. Thermal denaturation experiments (also followed by DLS) indicated that the capsids were stable up to 64 °C (Supporting Information Figure S4). The modified capsids remained soluble in phosphate buffer at 200 μM (based on MS2 monomer).
Antibody binding experiments were performed to confirm the presence of taxol on the interior of N87C MS2 capsids. Western blot analysis (Figure 3a) showed the presence of taxol only on the N87C MS2 mutants. Wild-type capsids that had been exposed to maleimide 6 gave no signal, providing further evidence that the native cysteines in the MS2 coat protein were unreactive. The internal location of the taxol molecules was confirmed by performing an immuno dot blot (Figure 3b) on intact versus GnHCl-denatured MS2 capsids. α-MS2 antibodies were used to assay for the presence of MS2 protein with similar results, while α-taxol antibodies showed significantly different signal intensities for intact and denatured capsids. Presumably, the taxol molecules on the interior of the capsid are blocked from interaction with the antibodies, which are not able to fit through the capsid pores to access the interior surface. These molecules are only allowed to interact with the antibodies upon protein denaturation and capsid disassembly.
Figure 3.
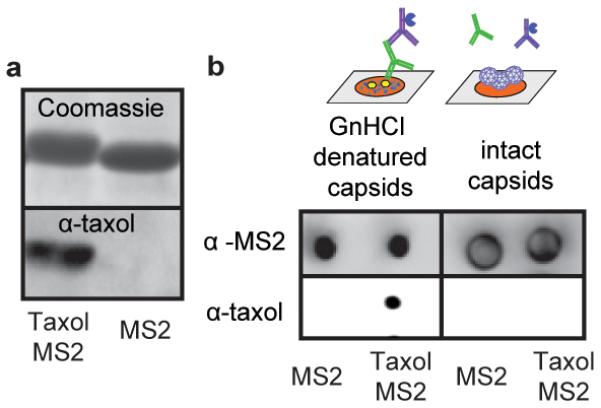
Antibody analysis of taxol-MS2 conjugates (a) Western blot analysis indicated the presence of taxol on MS2 capsid monomers. (b) Immuno dot blot analysis showed different responses to taxol molecules attached to denatured monomers (left) and inside assembled capsids (right).
To determine capsid stability under physiological conditions, samples of MS2 internally labelled with Oregon Green 488 maleimide (to facilitate detection) were incubated for 6 d at 37 °C in PBS, 10% FBS, and cell culture media. In each case, SEC analysis revealed that >80% of the initial protein sample remained as assembled capsids (Supporting Information, Figure S5a). Thus, little disassembly is to be expected in the absence of cellular targets.
A taxol release profile was also determined at pH 7.4. Samples of MS2-taxol conjugates were incubated in 10% FBS for 5 d. At 24 h intervals, samples were taken and the remaining capsids were isolated via selective precipitation and SEC. RP-HPLC analysis of the resulting material allowed quantification of the amount of taxol that had been released (Supporting Information, Figure S5b). 26 These experiments indicated a 1.9 d half life for the linker under these conditions.
A cell viability assay was used to test the effect of MS2 and taxol-MS2 on cancer cells in vitro (Figure 4 and Supporting Information, Figure S3). The release of taxol was expected to occur via hydrolysis of the ester bond at the 2′-OH position.28 MCF-7 breast cancer cells were assayed using Alamar Blue dye, a fluorescent indicator of cellular metabolic processes.29 Unmodified N87C MS2 was found to have no noticeable effect on MCF-7 viability. However, taxol-containing MS2 caused a significant lowering of cell viability, which was comparable to that occurring through the administration of equivalent amounts of taxol solubilized with DMSO. Virtually identical results were obtained after 3 d and 5 d incubation times. While it is not known how efficiently the untargeted capsids are uptaken via pinocytosis, the drug release experiments suggest that a reduced amount of taxol would be available through hydrolysis alone over the 3 d time period. However, the equivalent toxicity to that of free taxol suggests that the cells may be playing an active role in the cleavage process. Whether or not this is the case, the addition of targeting groups that bind the drug conjugates to specific cell types, would be expected to focus this activity only on the relevant cancer tissue.
Figure 4.
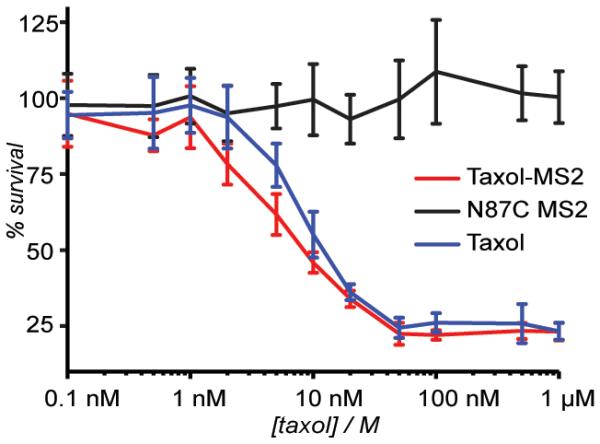
Cell viability assay using an MCF-7 cell line. Five days after the initial treatment, the cell survival was calculated relative to an untreated control by measuring the fluorescence resulting from treatment with Alamar Blue. Taxol-MS2 conjugates were delivered as a solution in 10 mM phosphate buffer, and free taxol was applied as a <1% solution of dimethyl sulfoxide in phosphate buffer. Error bars represent the standard deviation resulting from six replicate experiments.
In conclusion, a water-soluble derivative of the chemotherapeutic drug taxol containing bioconjugation functionality was synthesized and attached to MS2 viral capsids. The modified capsids remained in their capsid form and released taxol when incubated with MCF-7 cells, resulting in cell viability levels similar to that of free taxol in solution. We are presently investigating the use of linkers that undergo cell-specific cleavage between cargo and capsid, as well as the generation of MS2 capsids with both drug cargo and active targeting groups.
Supplementary Material
Acknowledgement
This work was generously supported by the NIH (R01 GM072700). The Bertozzi group is gratefully acknowledged for cell culture assistance.
References
- 1.Chabner BA, Roberts TG. Nat. Rev. Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 2.a) Allen TM. Nat. Rev. Cancer. 2002;2:750–763. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]; b) Kratz F, Müller IA, Ryppa C, Warnecke A. ChemMedChem. 2008;3:20–53. doi: 10.1002/cmdc.200700159. [DOI] [PubMed] [Google Scholar]
- 3.a) Haag R, Kratz F. Angew. Chem. 2006;118:1218–1237. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]; Haag R, Kratz F. Angew. Chem., Int. Ed. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]; b) Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat. Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 4.a) Gabizon AA. Adv. Drug Delivery Rev. 1995;16:285–294. [Google Scholar]; b) Harris JM, Chess RB. Nat. Rev. Drug Discovery. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 5.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. J. Controlled Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 6.a) Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker JR, Banaszak Holl MM. Chem. Biol. 2007;14:107–115. doi: 10.1016/j.chembiol.2006.11.015. [DOI] [PubMed] [Google Scholar]; b) Joshi A, Vance D, Rai P, Thiyagarajan A, Kane RS. Chem. Eur. J. 2008;14:7738–7747. doi: 10.1002/chem.200800278. [DOI] [PubMed] [Google Scholar]
- 7.a) Zalipsky S, Gilon C, Zilkha A. Eur. Polym. J. 1983;19:1177–1183. [Google Scholar]; b) Greenwald R. Adv. Drug Delivery Rev. 2003;55:217–250. doi: 10.1016/s0169-409x(02)00180-1. [DOI] [PubMed] [Google Scholar]
- 8.a) Duncan R, Kopeckova-Rejmanova P, Strohalm J, Hume I, Cable HC, Pohl J, Lloyd JB, Kopecek J. Br. J. Cancer. 1987;55:165–174. doi: 10.1038/bjc.1987.33. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Duncan R, Seymour LW, Ulbrich K, Kopecek J. J. Bioact. Compat. Pol. 1988;3:4–15. [Google Scholar]
- 9.a) Ihre HR, Padilla De Jesús OL, Szoka Francis C., Fréchet JMJ. Bioconjug. Chem. 2002;13:443–452. doi: 10.1021/bc010102u. [DOI] [PubMed] [Google Scholar]; b) Padilla De Jesús OL, Ihre HR, Gagne L, Fréchet JMJ, Szoka FC. Bioconjug. Chem. 2002;13:453–461. doi: 10.1021/bc010103m. [DOI] [PubMed] [Google Scholar]; c) Gillies ER, Fréchet JMJ. J. Am. Chem. Soc. 2002;124:14137–14146. doi: 10.1021/ja028100n. [DOI] [PubMed] [Google Scholar]; d) Wolinsky JB, Grinstaff MW. Adv. Drug Delivery Rev. 2008;60:1037–1055. doi: 10.1016/j.addr.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 10.Torchilin VP. Nat.Rev. Drug discovery. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 11.a) Masayuki Y, Mizue M, Noriko Y, Teruo O, Yasuhisa S, Kazunori K, Shohei I. J. Controlled Release. 1990;11:269–278. [Google Scholar]; b) Torchilin VP. Pharm. Res. 2007;24:1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 12.Gibson JD, Khanal BP, Zubarev ER. J. Am. Chem. Soc. 2007;129:11653–11661. doi: 10.1021/ja075181k. [DOI] [PubMed] [Google Scholar]
- 13.a) Bianco A, Kostarelos K, Prato M. Curr. Opin. Chem. Biol. 2005;9:674–679. doi: 10.1016/j.cbpa.2005.10.005. [DOI] [PubMed] [Google Scholar]; b) Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H. Cancer Res. 2008;68:6652–6660. doi: 10.1158/0008-5472.CAN-08-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Kratz F. J. Controlled Release. 2008;132:171–183. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]; b) Hawkins MJ, Soon-Shiong P, Desai N. Adv. Drug Delivery Rev. 2008;60:876–885. doi: 10.1016/j.addr.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 15.a) Flenniken ML, Willits DA, Harmsen AL, Liepold LO, Harmsen AG, Young MJ, Douglas T. Chem. Biol. 2006;13:161–170. doi: 10.1016/j.chembiol.2005.11.007. [DOI] [PubMed] [Google Scholar]; b) Flenniken ML, Liepold LO, Crowley BE, Willits DA, Young MJ, Douglas T. Chem. Comm. 2005;2:447. doi: 10.1039/b413435d. [DOI] [PubMed] [Google Scholar]
- 16.Carrico ZM, Romanini DW, Mehl RA, Francis MB. Chem. Commun. 2008:1205–1207. doi: 10.1039/b717826c. [DOI] [PubMed] [Google Scholar]
- 17.Tong GJ, Hsiao SC, Carrico ZM, Francis MB. J. Am. Chem. Soc. 2009;131 doi: 10.1021/ja903857f. ASAP article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valegård K, Liljas L, Fridborg K, Unge T. Nature. 1990;345:36–41. doi: 10.1038/345036a0. [DOI] [PubMed] [Google Scholar]
- 19.a) Hooker JM, Datta A, Botta M, Raymond KN, Francis MB. Nano Lett. 2007;7:2207–2210. doi: 10.1021/nl070512c. [DOI] [PubMed] [Google Scholar]; b) Datta A, Hooker JM, Botta M, Francis MB, Aime S, Raymond KN. J. Am. Chem. Soc. 2008;130:2546–2552. doi: 10.1021/ja0765363. [DOI] [PubMed] [Google Scholar]
- 20.Hooker JM, O'Neil JP, Romanini DW, Taylor SE, Francis MB. Mol. Imaging Biol. 2008;10:182–191. doi: 10.1007/s11307-008-0136-5. [DOI] [PubMed] [Google Scholar]
- 21.Kovacs EW, Hooker JM, Romanini DW, Holder PG, Berry KE, Francis MB. Bioconjugate Chem. 2007;18:1140–1147. doi: 10.1021/bc070006e. [DOI] [PubMed] [Google Scholar]
- 22.a) Khayat D, Antoine E-C, Coeffic D. Cancer Invest. 2000;18:242–260. doi: 10.3109/07357900009031828. [DOI] [PubMed] [Google Scholar]; b) Saijo N. Cancer Sci. 2006;97:448–452. doi: 10.1111/j.1349-7006.2006.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Sparreboom A, Van Tellingen O, Nooijen WJ, Beijnen JH. Cancer Res. 1996;56:2112–2115. [PubMed] [Google Scholar]; b) Singla A, Garg A, Aggarwal D. Int. J. Pharm. 2002;235:179–192. doi: 10.1016/s0378-5173(01)00986-3. [DOI] [PubMed] [Google Scholar]
- 24.Ede NJ, Tregear GW, Haralambidis J. Bioconjugate Chem. 1994;5:373–378. doi: 10.1021/bc00028a016. [DOI] [PubMed] [Google Scholar]
- 25.Deutsch HM, Glinski JA, Hernandez M, Haugwitz RD, Narayanan VL, Suffness M, Zalkow LH. J. Med. Chem. 1989;32:788–792. doi: 10.1021/jm00124a011. [DOI] [PubMed] [Google Scholar]
- 26.Direct detection of the released taxol using RP-HPLC yielded data that were consistent with these findings. However, the lack of a distinct chromophore and the large number of small molecules in FBS limited the accuracy of this method.
- 28.a) Mathew AE, Mejillano MR, Nath JP, Himes RH, Stella VJ. J. Med. Chem. 1992;35:145–151. doi: 10.1021/jm00079a019. [DOI] [PubMed] [Google Scholar]; b) Chen X, Plasencia C, Hou Y, Neamati N. J. Med. Chem. 2005;48:1098–1106. doi: 10.1021/jm049165z. [DOI] [PubMed] [Google Scholar]; c) Lavis LD. ACS Chem. Biol. 2008;3:203–206. doi: 10.1021/cb800065s. [DOI] [PubMed] [Google Scholar]
- 29.Nakayama GR, Caton MC, Nova MP, Parandoosh Z. J. Immunol. Methods. 1997;204:205–208. doi: 10.1016/s0022-1759(97)00043-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.