

Abstract
Structures of the influenza A virus M2 proton channel have been determined by X-ray crystallography in the open conformation, and by NMR in the closed state. Whereas the X-ray structure shows a single inhibitor molecule in the middle of the channel, four inhibitor molecules bind the channel’s outer surface in the NMR structure. Although in both structures the strongest hot spots (i.e., regions which substantially contribute to the free energy of binding any potential ligand) lie inside the pore, hot spots also are found at exterior locations. By considering all available models, we propose the primary drug binding site is inside the pore, but that exterior binding also occurs under appropriate conditions.
The amantadine binding controversy
The integral membrane protein M2 of influenza virus forms an acid-activated proton conducting ion channel which functions during viral uncoating and maturation by modifying the pH in virions as well as in trans-Golgi vesicles [1, 2]. The low pH (5.5 to 6.0) of the endosome activates the M2 channel prior to hemagglutinin-mediated fusion. Proton conductance acidifies the viral interior, thereby facilitating dissociation of the matrix protein from the viral nucleoproteins—a required process for unpacking of the viral genome [1]. The channel is highly selective for protons and has low permeability for other physiological ions according to reversal potential studies [3].
M2 is a 97-residue single-pass membrane protein with 23 amino acids of the N-terminus oriented extracellularly, a single internal hydrophobic domain of 19 residues that acts as the transmembrane domain (M2TM), and a 54-residue cytoplasmic tail [4, 5]. M2TM is the target of the anti-influenza drugs amantadine and its methyl derivative rimantadine. The two drugs were recommended as the first line of defense for several years in order to reserve the only other class of anti-influenza drugs, neuraminidase inhibitors, in the event of an epidemic. Recently, however, most influenza strains have developed resistance to amantadine and rimantadine [6]. In particular, the Ser31Asn substitution renders the virus highly resistant to amantadine inhibition, and the incidence of clinical isolates bearing this mutation has jumped from a few percent to approximately 97% in recent years [6, 7]. Thus, it is important to understand how amantadine binds, why resistance to the drug develops, and whether other compounds can be found that inhibit the M2 channel.
The first structure of the M2 channel was solved for residues 22–46 by solid-state NMR in 2002 [9]. More recently Stouffer et al. [10] used X-ray crystallography to determine the structure for the same residues with and without amantadine in the detergent octyl-β-D-glucopyranoside (OG). At the same time, Schnell and Chou [11]used NMR to determine the structure of the rimantadine-bound M2 peptide of residues 18–60 solubilized in dihexanoylphosphatidylcholine (DHPC) micelles at pH 7.5. Both X-ray crystallography and NMR confirm that the channel is a pseudosymmetric tetramer with the membrane-spanning region forming a left-handed coiled coil, but the two structures disagree regarding the position of the bound inhibitor [12]. In the X-ray structure a single amantadine molecule binds in the middle of the pore, surrounded by residues Val27, Ala30, Ser31, and Gly34 (Fig. 1). By contrast, the NMR data show four rimantadine molecules bound to the lipid-exposed outer surface of the channel at the membrane boundary close to the cytoplasmic end of the helices, interacting with the residues Asp44 and Arg45 (Fig. 1).
Figure 1.

Superimposition of the X-ray and the NMR structures of the truncated M2 proton channel. The proteins are shows as cartoons, with the X-ray structure colored wheat and the NMR structure colored magenta. The amantadine bound in the middle of the channel in the X-ray structure is shown as green sticks. The four rimantadine molecules bound to the outer surface of the channel in the NMR structure are also shown in stick representation, colored magenta. The figures show the hot spot regions as transparent “clouds”, colored light blue for the X-ray structure and light green for the NMR structure. (a) Side view of the channel, with the C-terminal cytoplasmic end of the channel located on the bottom. (b) View from the C-terminal cytoplasmic end. The C-terminal helices in the NMR structure are removed to show the channel’s pore. Notice that the hot spots obtained for the two structures in the pore overlap with each other, and hence the region is shown in mixed colors. The two views also show that this hot spot is located at the amantadine binding site in the X-ray structure. For the NMR structure, the four hot spots on the outer surface overlap with the bound rimantadine molecule, and are visible on all four sides. By contrast, for the X-ray structure an external hot spot is visible only between two of the helices.
Here we review the various M2MT structures that have been published, showing that the X-ray and NMR structures represent two extremes in terms of the tilt angle of the transmembrane helices with respect to the central axis of the channel. We then use binding site analysis tools to identify the most important binding sites in the two structures. To our surprise, the results suggest that the two very different structures have fairly similar hot spots, both in the channel pore and on the outer surface. However, these hot spots occupy pockets that substantially differ in size between the two structures, resulting in the different amantadine binding modes observed experimentally. Considering the similarity of hot spots in the two structures, we propose that both binding modes are possible under appropriate conditions. Indeed, we argue that the assumption of only one binding mode leaves a number of observations unexplained.
The geometry of the M2TM channel
The X-ray structures obtained by Stouffer et al. [10] represent the open state of the channel [12], with the four helices forming a left handed, parallel bundle that resembles a truncated cone, with the narrow amino-terminal end facing the exterior of the viral envelope. The N-terminal half of the channel has nearly exact four-fold rotational symmetry; the helices are tilted by 35°±2° with respect to the central axis of the bundle. This value is in good agreement with the range of 32° – 38°, determined by various solid-state NMR (ssNMR) studies in both open and closed states [9, 13–17]. Although the NMR structure [11] also shows that MTM2 is a four-helix bundle, the channel is in closed conformation, and the tilt angle of the helices with respect to the lipid bilayer normal is only 23°, much smaller than in the X-ray structure (Fig. 1a).
Determining which of the two structures represents the biologically more relevant conformation is difficult for two main reasons. First, M2MT exhibits substantial plasticity and conformational heterogeneity, confirmed by both solid-state NMR and X-ray studies [17–22]. In particular, the transmembrane helix can be kinked around Gly34, and the kink angle can be changed by amantadine binding [17–19]. Nuclear spin relaxation data and polarization inversion spin exchange at the magic angle spectra revealed that the structure at high pH and in the absence of amantadine is more dynamic and structurally heterogeneous compared with the amantadine-bound structure [21]. This structural plasticity was suggested to be essential for channel opening, gating, and proton conduction [21]. A recent study using 1D and 2D IR spectroscopies indicates an even larger conformational change at neutral pH when the channel is closing [22]. The plasticity of the M2 channel was also confirmed by recent molecular dynamics simulations [23–25].
The second factor which makes determining the structure of the M2TM channel difficult is its dependence on the experimental conditions [26]. Indeed, the tilt angle is largely influenced by sample preparation conditions such as solvents, membrane composition, peptide concentration, and particularly bilayer thickness [27–32]. In particular, detergent micelles might cause curvature stress to membrane proteins, affecting their structure [29, 30], and this is most likely responsible for the substantial difference between the X-ray and NMR structures. In fact, Chou et al. [29, 30] noted that in the absence of the peptide, the DHPC micelle would adopt a spherical shape with the diameter of 36 Å, whereas the length of the M2 transmembrane channel is only around 30 Å [11]. Thus, the channel appears to be stretched, and this might result in a tilt angle which is substantially smaller than the one reported for the M2 channel in other biological membranes [13–20]. However, little information is available on micelle structure in the presence of a protein. For example, based on intermolecular nuclear Overhauser effects (NOEs), Fernandez et al. [32] modeled DHPC molecules around a membrane protein oriented with their hydrophobic tails perpendicular to the hydrophobic protein surface, resulting in a distorted monolayer, and thus in an environment for the protein which substantially differs from the conditions in a natural lipid bilayer
Analysis of the M2TM binding sites
The X-ray and the NMR structures represent two extremes in terms of the helical tilt angle, and hence it is interesting to determine the binding hot spots (i.e., the regions substantially contributing to the binding free energy) in both structures using computational solvent mapping (Box 1). In the X-ray structure the top ranked hot spot is located inside the channel at the amantadine binding site, interacting with amino acid residues Ser31, Val27, Ala30, and Gly34 (Fig. 1). These residues form a large pocket [38], with a volume of 210 Å3. The next three hot spots line the inside of the pore between Ile33 and His37, overlapping the water wire found in this part of the channel [33]. The fourth largest hot spot is located on the lipid-exposed outer surface of the channel (Fig. 1), at the site which binds rimantadine in the NMR structure [11]. Probe binding to the outer surface occurs only between two helices, because the 35° tilt of the truncated helices yields large openings rather than pockets on the other three sides of the four-helix bundle. However, we expect to find four pockets for a structure with slightly longer peptides.
Box 1. Identification of binding hot spots by computational solvent mapping.
The binding sites of proteins generally include smaller regions called hot spots that are major contributors to the binding free energy, and hence are crucial to the binding of any ligand at that particular site [34]. In drug design applications, such hot spots can be identified by screening for the binding of fragment-sized organic molecules [34–37]. Because the binding of the small compounds is very weak, it is usually detected by Nuclear Magnetic Resonance (SAR by NMR [36]) or by X-ray crystallography [37] methods. Results show that the hot spots of proteins bind a variety of small molecules, and that the fraction of the “probe” molecules binding to a particular site predicts the potential importance of the site and can be considered a measure of druggability [34, 35]. Solvent mapping has been developed as a computational analogue of the NMR and X-ray based screening experiments [35]. The method places molecular probes - small organic molecules containing various functional groups – on a dense grid defined around the protein, finds favorable positions using empirical free energy functions, further refines the selected poses by free energy minimization, clusters the low energy conformations, and ranks the clusters on the basis of the average free energy [37]. For the identification of hot spots we generally use 16 small molecules as probes (ethanol, isopropanol, tert-butanol, acetone, acetaldehyde, dimethyl ether, cyclohexane, ethane, acetonitrile, urea, methylamine, phenol, benzaldehyde, benzene, acetamide, and N,N dimethylformamide). To determine the hot spots, we identify consensus sites, i.e., regions on the protein where clusters of different probes overlap, and rank these sites in terms of the number of overlapping probe clusters [35]. The sites with the largest number of probe clusters are considered as predictions of binding hot spots. Applications to a variety of proteins show that the probes always cluster in important subsites of the binding site and the amino acid residues that interact with many probes also bind the specific ligands of the protein [34]. For the analysis of the M2TM domain the general algorithm [35] was slightly modified to account for the membrane, which was modeled as a layer of low dielectric medium [33]. We have mapped both the unbound X-tray structure and the 15 NMR structures (PDB codes 3bkd and 2rlf) [33]. All bound ligands, ions, and water molecules were removed prior to mapping.
The mapping of the NMR structure yields interesting results. Although the NMR data [11] show four rimantadine molecules bound on the outside of the protein, the largest hot spot is within the pore near residues Ala30, Ser31, and Gly34—those that line the amantadine binding site in the X-ray structure (Fig. 1). The next four highest ranked hot spots are located on the outside of the channel at the rimantadine binding sites (Fig. 1). In this structure the four external sites (with volumes between 390 Å3 and 190 Å3) are the largest pockets, much larger than the internal site that has only a volume of 85 Å3. We note that probe binding at these sites is in part due to the favorable electrostatic interactions between the partially polar probes and the interfacial region of the lipid bilayer [33]. Indeed, amantadine concentration is much higher in this interfacial region than in the middle of the membrane [39, 40].
Mapping results show that both the X-ray and NMR structures have the strongest hot spot located in the internal cavity and weaker hot spots on the outside of the channel (Fig. 1). Why then is the inhibitor found only inside the pore in the X-ray structure, and only on the outside in the NMR experiment? This question can be easily answered by comparing the sizes of the appropriate pockets in the two structures. In the X-ray structure the internal cavity is a large pocket which can easily accommodate an amantadine molecule. As expected, docking of amantadine to this structure places the lowest energy ligand clusters at this position [33]. We note that docked amantadine molecules can also be observed at the weaker hot spot on the lipid-exposed outer surface of the channel, at a location which binds rimantadine in the NMR structure. However, this pocket is shallow, and the average energy of amantadine binding there is much higher than the average energy in the pore [33]. By contrast, the largest pockets in the NMR structure are on the outside of the channel. The internal site with a volume of 85 Å3 is ranked ninth by its size, and it is too small to accommodate the bulky adamantane group. Indeed, the docking of amantadine to the NMR structure places ligands only at the outside binding pockets [33]. Because computational solvent mapping uses probes that are substantially smaller than amantadine, it still identifies the internal cavity as the most important hot spot.
Are two different amantadine binding modes and inhibition mechanisms possible?
The observation of the strongest hot spot in the pore for both X-ray and NMR structures indicates that amantadine first binds at this site, leading to inhibition by a pore-blocking mechanism [10, 12]. However, the existence of additional hot spots on the outside of the channel suggests that the drug can also bind on the outer surface, at least when the channel is closed. Based on the NMR structure, Schnell and Chou [11] proposed that the drug binds preferentially to, and thereby stabilizes, the closed state. They noted that amantadine action is faster at neutral pH, where the channel is mostly closed, than at low pH, where the open state is favored, in accord with closed-state stabilization but not with open-pore block [11,12]. Can the existence of both primary and secondary binding sites imply that inhibition might occur due to two different mechanisms? In fact, throughout the history of studying the M2 channel, numerous comments have suggested that one or the other mechanism leaves a number of observations unexplained. This point first was made in an early paper by Pinto and Lamb [41], who listed four electrophysiological observations that at least partially contradicted amantadine binding within the pore and suggested the possibility of an allosteric inhibition mechanism. For example, they observed that the concentration of the drug required for half-maximal inhibition of current is greater when the channel is open (low pH) than in the closed (high pH) state [41]. Second, based on solid state NMR data, Li et al [18] noted that samples that have never been exposed to amantadine display a weak signal for the amantadine-bound conformation; similarly, they noted some evidence for the amantadine-free state even in the presence of amantadine concentration sufficient to saturate more than 99% of the sites. These findings hint at the possibility that amantadine shifts a conformational equilibrium between two states that are sampled in its absence. Third, although the amantadine-resistant mutants Ser31Asn and Ala30Thr do not bind amantadine, the mutants Val27Gly, Val27Ala, Ile27Ser, and Ile27Thr are also amantadine-resistant, but bind amantadine [42]. Thus, according to these results, the virus might use two structural tactics to render a channel blocker ineffective. Fourth, the recent spectroscopic study by Manor et al. [22] reports a large scale conformational change in the channel structure when the pH is increased, suggesting that the channel pore is lined by a different set of residues in the open and closed states, and this can influence ligand binding [22].
The dual binding mechanism is compatible with available functional data that provide little information on the importance of binding at the exterior sites (figure 2). The observation that Ser31Asn interferes with both mechanisms could explain why it underlies the primary resistance mutation [43, 44]. By contrast, the Ser31Ala substitution, which does not lead to amantadine resistance, leaves both mechanisms unaffected. However, recent evidence shows that the outside binding cannot be solely responsible for inhibition. In the proposed NMR-derived external drug binding site, D44 makes a hydrogen bond with amantadine. Jing et al. [43] showed that the M2 mutant channel with the substitution Asp44Ala is a functional and amantadine-sensitive ion channel. Furthermore, the Asp44Ala substitution into the influenza A virus did affect neither growth nor sensitivity to amantadine [43].
Figure 2.
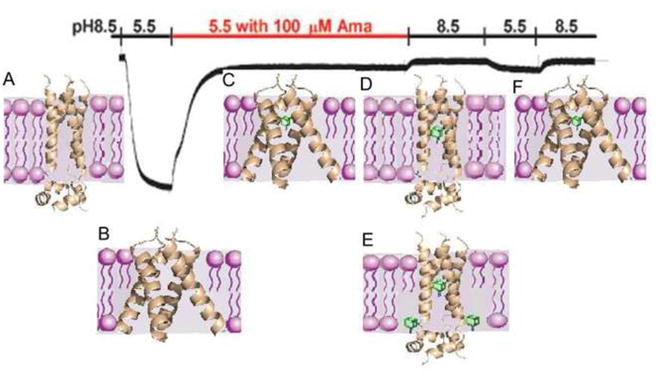
Compatibility of the dual binding mechanism with recording of surface currents through the M2 ion channel. A representative recording of surface currents through M2 channels expressed in oocytes of Xenopus laevis as the pH changes and amantadine is added, is shown; reproduced from Jing et al. [43] with permission. The experiment starts at pH 8.5. The pH is lowered to 5.5 to observe the opening of the channel, and amantadine is added in the fully open state, resulting in the observed inhibition (segment of recording shown in red). The pH is then increased to 8.5, lowered to 5.5, and increased to 5.5 again. The figure also shows the most likely channel conformations and amantadine binding modes for each segment of the experiment. (a) At pH 8.5 the channel is closed. (b) The channel opens when the pH is reduced to 5.5. (c) After amantadine is added in the open state of the channel, the drug (shown in green) binds the most favorable site in the lumen of the pore, resulting in inhibition by the pore-blocking mode. The lipid-facing sites have substantially lower affinity for amantadine than the primary site, particularly in the open state of the channel when the helices splay relatively far apart near the C terminus, and hence exterior binding is unlikely unless the amantadine/peptide ratio exceeds 1:4. Because the pore-blocking amantadine already provides inhibition, binding on the outside of the channel would not affect the current. (d) Increasing the pH closes the channel, at least to a certain degree, and most likely traps amantadine in the pore. (e) Because the external binding pockets become better defined when the channel is closed, at high enough concentration amantadine binding is likely to also occur at the lipid-facing sites. (f) The pH is reduced to 5.5, but inhibition persists due to the bound inhibitor. We believe that this segment of the experiment provides little information on the effect of the allosteric inhibition due to the amantadine bound at the secondary sites. Although Schnell and Chou [11] suggest that the outside binding would contribute to maintaining the non-conductive state when the pH is lowered, at that point the system is already saturated by amantadine, most likely bound inside the pore, and any additional contribution would be negligible.
Concluding remarks
Recently, a new solid state NMR study [45] showed that the M2 channel in its closed conformation has both a high affinity amantadine binding site inside the pore and four lower affinity sites on the C-terminal protein surface, exactly as we predicted by mapping the X-ray and NMR structures [33]. Thus, our opinion that both sites exist has been validated. The new structure also shows that at the stoichiometric amantadine:peptide ratio of 1:4, the drug binds only to the luminal site, and the exterior binding starts when it is in excess. The evidence that amantadine can bind in the closed channel [45] confirms that the NMR structure obtained by Schnell and Chou [11] is somehow distorted because its narrow pore is unable to accommodate the drug.
Although the new structure confirms that the primary binding site is in the pore, this does not exclude the potential existence of a dual inhibition mechanism. As we have discussed, electrophysiological recordings [43] neither refute nor confirm whether inhibitor binding to the secondary sites contributes to maintaining the non-conductive state when the pH is lowered (Fig. 2). Although a number of observations reviewed here support the existence of an allosteric inhibition mechanism, evidence is accumulating that in the wild type M2 channel the secondary sites have limited pharmacological significance [43]. However, assuming that the allosteric inhibition mechanism is confirmed, the secondary sites might become potentially more important in drug resistant mutants which are now found in over 97% of clinical isolates. Because neither the pore blocking nor the allosteric inhibition mechanisms work in these mutants, the binding site in the pore does not necessarily have higher affinity any more, and hence both the internal and external sites are potential targets for the development of novel inhibitors that would evade drug resistance.
Acknowledgments
This work has been supported by grant GM064700 from the National Institutes of Health
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Helenius A. Unpacking the incoming influenza-virus. Cell. 1992;69:577–578. doi: 10.1016/0092-8674(92)90219-3. [DOI] [PubMed] [Google Scholar]
- 2.Sugrue RJ, Hay AJ. Structural characteristics of the M2 protein of influenza A viruses: evidence that it forms a tetrameric channel. Virology. 1991;180:617–624. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chizhmakov IV, et al. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J Physiol. 1996;494:329–336. doi: 10.1113/jphysiol.1996.sp021495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moffat JC, et al. Proton transport through influenza A virus M2 protein reconstituted in vesicles. Biophys J. 2008;94(2):434–45. doi: 10.1529/biophysj.107.109082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang Y, et al. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J Biol Chem. 2002;277:39880–39886. doi: 10.1074/jbc.M206582200. [DOI] [PubMed] [Google Scholar]
- 6.Deyde VM, et al. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis. 2007;196:249–257. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 7.Bright RA, et al. Adamantine resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. J Am Med Assoc. 2006;295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 8.Hayden FG. Antiviral resistance in influenza viruses—implications for management and pandemic response. N Engl J Med. 2006;354:785–788. doi: 10.1056/NEJMp068030. [DOI] [PubMed] [Google Scholar]
- 9.Nishimura K, et al. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry. 2002;41:13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 10.Stouffer AL, et al. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller C. Ion channels: Coughing up flu’s proton channels. Nature. 2008;451:532–533. doi: 10.1038/451532a. [DOI] [PubMed] [Google Scholar]
- 13.Kovacs FA, Cross TA. Transmembrane four-helix bundle of influenza A M2 protein channel: structural implications from helix tilt and orientation. Biophys J. 1997;73:2511–2517. doi: 10.1016/S0006-3495(97)78279-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song Z, et al. Transmembrane domain of M2 protein from influenza A virus studied by solid-state 15N polarization inversion spin exchange at magic angle NMR. Biophys J. 2000;79:767–775. doi: 10.1016/S0006-3495(00)76334-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kovacs FA, et al. Helix tilt of the M2 transmembrane peptide from influenza A virus: an intrinsic property. J Mol Biol. 2000;295:117–125. doi: 10.1006/jmbi.1999.3322. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, et al. Structure of the transmembrane region of the M2 protein H+ channel. Protein Sci. 2001;10:2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu J, et al. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C, et al. Solid-state NMR characterization of conformational plasticity within the transmembrane domain of the influenza A M2 proton channel. Biochim Biophys Acta. 2007;1768:3162–3170. doi: 10.1016/j.bbamem.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cady SD, et al. Structure of amantadine-bound M2 transmembrane peptide of influenza a in lipid bilayers from magic-angle-spinning solid-state NMR: The role of Ser31 in amantadine binding. J Mol Biol. 2009;385:1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cady SD, Hong M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proc Natl Acad Sci USA. 2008;105:1483–1488. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu J, et al. The chemical and dynamical influence of the anti-viral drug amantadine on the M2 proton channel transmembrane domain. Biophys J. 2007;93:276–283. doi: 10.1529/biophysj.106.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manor J, et al. Gating mechanism of the influenza A M2 channel revealed by 1D and 2D IR spectroscopies. Structure. 2009;17:247–254. doi: 10.1016/j.str.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yi M, et al. Conformational heterogeneity of the M2 proton channel: astructural model for channel activation. Proc Natl Acad Sci USA. 2009;106:13311–13316. doi: 10.1073/pnas.0906553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leonov H, Arkin IT. Structure and dynamics of the influenza A M2 Channel: a comparison of three structures. J Mol Model. 2009 Apr 18; doi: 10.1007/s00894-009-0493-2. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 25.Khurana E, et al. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. Proc Natl Acad Sci U S A. 2009;106(4):1069–74. doi: 10.1073/pnas.0811720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duong-Ly KC, et al. The conformation of the pore region of the M2 proton channel depends on lipid bilayer environment. Protein Sci. 2005;14:856–861. doi: 10.1110/ps.041185805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinto LH, Lamb RA. Controlling influenza virus replication by inhibiting its proton channel. Mol Biosyst. 2007;3:18–23. doi: 10.1039/b611613m. [DOI] [PubMed] [Google Scholar]
- 28.Im W, et al. An implicit membrane generalized Born theory for the study of structure, stability, and interactions of membrane proteins. Biophys J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou JJ, et al. Micelle-induced curvature in a water-insoluble HIV-1 Env peptide revealed by NMR dipolar coupling measurement in stretched polyacrylamide gel. J Am Chem Soc. 2002;124:2450–2451. doi: 10.1021/ja017875d. [DOI] [PubMed] [Google Scholar]
- 30.Chou JJ, et al. Characterization of phospholipid mixed micelles by translational diffusion. J Biomol NMR. 2004;29:299–308. doi: 10.1023/B:JNMR.0000032560.43738.6a. [DOI] [PubMed] [Google Scholar]
- 31.Poget SF, Girvin ME. Solution NMR of membrane proteins in bilayer mimics: Small is beautiful, but sometimes bigger is better. Biochim Biophys Acta. 2007;1768:3098–3106. doi: 10.1016/j.bbamem.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernández C. Lipid-protein interactions in DHPC micelles containing the integral membrane protein OmpX investigated by NMR spectroscopy. Proc Natl Acad Sci U S A. 2002;99:13533–13537. doi: 10.1073/pnas.212515099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chuang G-Y, et al. Binding hot spots and amantadine orientation in the influenza A virus M2 proton channel. Biophys J. 2009;97:2846–2853. doi: 10.1016/j.bpj.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landon MR, et al. Identification of hot spots within druggable binding sites of proteins by computational solvent mapping. J Med Chem. 2007;50:1231–1240. doi: 10.1021/jm061134b. [DOI] [PubMed] [Google Scholar]
- 35.Brenke R, et al. Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25:621–627. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hajduk PJ, et al. Druggability indices for protein targets derived from NMR-based screening data. J Med Chem. 2005;48:2518–2525. doi: 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]
- 37.Mattos C, Ringe D. Locating and characterizing binding sites on proteins. Nat Biotech. 1996;14:595–599. doi: 10.1038/nbt0596-595. [DOI] [PubMed] [Google Scholar]
- 38.Laurie ATR, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein–ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 39.Chew CF. Distribution and dynamics of adamantanes in a lipid bilayer. Biophys J. 2008;95:5627–5636. doi: 10.1529/biophysj.108.139477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C, et al. Solid-state NMR and MD simulations of the antiviral drug amantadine solubilized in DMPC bilayers. Biophys J. 2008;94:1295–1302. doi: 10.1529/biophysj.107.112482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pinto LH, Lamb RA. Understanding the mechanism of action of the anti-influenza virus drug amantadine. Trends Microbiol. 1995;3:271. doi: 10.1016/s0966-842x(00)88942-8. [DOI] [PubMed] [Google Scholar]
- 42.Astrahan P, et al. A novel method of resistance for influenza against a channel-blocking antiviral drug. Proteins. 2004;55:251–257. doi: 10.1002/prot.20018. [DOI] [PubMed] [Google Scholar]
- 43.Jing X, et al. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc Natl Acad Sci U S A. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pielak RM, et al. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci U S A. 2009;106:7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cady SD, et al. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]