

Abstract
Reduced lung capillary expression of angiotensin I-converting enzyme (ACE), a key enzyme in cardiovascular pathophysiology, and of caveolin-1, an important regulator of endothelial cell signalling, has been demonstrated in various models of pulmonary arterial hypertension (PAH). We addressed the relationship between PAH and ACE expression in caveolin-1 knockout mice (Cav1−/−), which have moderate PAH. Tissue ACE activity was reduced by 50% in lungs from 3- month old Cav1−/− mice compared to wild type (WT). A similar reduction in lung endothelial ACE expression was observed by measuring the lung uptake of 125I-labeled monoclonal anti-ACE antibody and by quantitative immunohistochemistry. These alterations in ACE are limited to capillary segments of the pulmonary circulation. Functionally, the increase in pulmonary artery pressure (PAP) in response to ACE conversion of angiotensin I to angiotensin II in isolated, perfused mouse lungs was reduced significantly in Cav1−/− mice compared to WT. Thus, these complementary approaches demonstrate the dependence of lung microvascular endothelial cell ACE protein expression on caveolin-1 expression and underscore the vital role of caveolin-1-regulated pulmonary vascular homeostasis on endothelial ACE expression and activity. In summary, we have revealed a novel role of caveolin-1 in the regulation of ACE expression in pulmonary capillary endothelial cells. Further understanding of the mechanism by which reduced caveolin-1 expression leads altered pulmonary vascular development, PAH, and reduced ACE expression may have important clinical implications in patients with these severe lung diseases.
Keywords: pulmonary hypertension, endothelial dysfunction, anti-ACE monoclonal antibody
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a devastating illness characterized by increased pulmonary vascular resistance and right heart failure. The typical histopathologic features include luminal narrowing of muscular pulmonary arteries and plexiform lesions (Channick and Rubin, 2005). Recent studies examining plexiform lesions documented the reduced expression of caveolin-1 (Achcar et al., 2006), a multifaceted 22 kD adaptor protein localized on the inner leaflet of plasmalemma vesicles. Caveolae are 50–100 nm diameter vesicles originating from specialized sphingosine- and cholesterol-rich plasma membrane microdomains/lipid rafts (Anderson, 1998) abundant in endothelial cells and adipocytes. Caveolin-1, the major structural protein of caveolae, physically interacts with many cell signaling molecules and thus is critically involved in the regulation of cellular functions such as Ca2+ influx, cell cycle progression, trafficking of macromolecules, nitric oxide (NO) production and cholesterol transport (Anderson, 1998; Minshall et al., 2002, Minshall et al., 2003).
Caveolin-1 knockout (Cav1−/−) mice exhibit developmental abnormalities in the lung microvasculature (Drab et al., 2001, Maniatis et al, 2008, Razani et al., 2001), pulmonary arterial hypertension (PAH) (Maniatis et al., 2008, Zhao et al., 2002) and fibrosis (Drab et al., 2001, Razani et al., 2001) due to altered type I collagen deposition (Maniatis et al., 2008). The absence of caveolin-1 in endothelial cells of Cav1−/− mice leads to increased endothelial nitric oxide synthase (eNOS) activity and NO generation resulting in enhanced relaxation of preconstricted aortic rings (Drab et al., 2001) and destabilization of cell-cell junctions (Miyawaki-Schimizu et al., 2006, Schubert et al., 2002). Therefore, Cav1−/− mice are a model of lung vascular pathologies characterized by abnormal lung microvessel structure, increased pulmonary vascular resistance, thickening of alveolar septa, and PAH despite elevated plasma NO levels (Maniatis et al., 2008, Zhao et al., 2002). Moreover, administration of the caveolin-1 scaffolding domain peptide, consisting of amino acids 82–101 of caveolin-1, ameliorated experimental (monocrotaline-induced) PAH, thus implicating caveolin-1-regulated signalling events in the pathogenesis of the disease (Jasmin et al., 2006). Interestingly, Cav1−/− x eNOS−/− double knockout mice show reduced pulmonary artery pressure compared to Cav1−/− mice (Zhao et al., 2009) indicating elevated eNOS activity may play a causal role in the etiology of PAH in the absence of Cav1.
Angiotensin-Converting Enzyme (ACE) is a key enzyme in cardiovascular pathophysiology (Yang et al., 1970, Ehlers et al., 1989, Bernstein et al., 2005). ACE is critically involved in endothelial homeostasis by controlling the circulating levels of bradykinin and angiotensin II and thereby affects vascular development, tone, and permeability, particularly in the lung, the organ with the highest level of endothelial cell ACE expression. Lung and blood activity of ACE is also a sensitive marker of endothelial dysfunction, including PAH and lung vascular injury (Kay et al., 1982, Keane et al., 1982, Oparil et al., 1988, Muzykantov and Danilov, 1991, Morrell et al., 1995a, Atochina et al, 1997, Orfanos et al., 2000).
Since Cav1−/− mice have PAH in addition to alterations in pulmonary vascular development, we investigated the relationship between caveolin-1 expression, pulmonary artery pressure, and ACE expression. Endothelial ACE expression was measured by in vivo radiolabeled mAb binding (Danilov et al. 1989, Muzykantov and Danilov, 1995, Balyasnikova et al, 2006) and quantitative immunohistochemistry using a set of mAbs to denatured mouse ACE (Balyasnikova et al., 2005). The approach, based on the lung accumulation of anti-ACE monoclonal antibodies, has been proven to be an early and sensitive method to monitor endothelial dysfunction and lung injury (Danilov et al, 1989, Muzykantov and Danilov 1991, 1995) as shown in various lung injury models (Muzykantov and Danilov, 1995). In the present study, we applied this methodology in combination with classic measures to examine whether the absence of caveolin-1 affects ACE expression in different tissues. In isolated lung preparations, we measured increased pulmonary artery pressure (PAP) in response to ATI infusion as a physiological measure of ATI to ATII conversion by ACE. Using these complementary approaches, we observed significantly reduced ACE expression and activity in Cav1−/− mouse lungs and reduced ATI-mediated pulmonary artery vasoconstriction compared to WT mice.
MATERIALS and METHODS
Chemicals and Reagents
Hippuryl-L-histidyl-L-leucine (Hip-His-Leu), ATI and II and other reagents were obtained from Sigma (St. Louis, MO), or as specified. For the present studies, we used rat mAb 4B10.5, which recognizes an epitope localized on the C-terminal domain of native mouse ACE (Balyasnikova et al., 2006) and rat mAb 4G6, which recognizes both the somatic and testicular isoforms of ACE (Balyasnikova et al., 2005).
Animals
All animal procedures were approved by the University of Illinois Institutional Animal Care and Use Committee. Mice were housed in groups of maximally five mice/cage and were maintained at a 12-hr light-dark cycle, species-appropriate relative humidity and temperature and ad libitum access to food and water. Mice used in this study were 12–16 week-old Cav1−/− mice (Jackson Laboratory, Bar Harbor, ME) and strain-matched Black Swiss control mice. [Note: the background strain of Cav1−/− mice has subsequently changed to B6/129SJ2, which continue to show reduced ACE expression (unpublished observation)]. For organ harvesting, isoflurane-anesthetized mice underwent laparotomy followed by blood sampling from the inferior vena cava and exsanguination by severing the abdominal aorta. Blood was flushed by injecting 10 ml of PBS into the right ventricle before organ collection.
ACE activity assay
ACE activity was measured as described (Friedland and Silverstein, 1976). Briefly, 20 μl of plasma or tissue homogenate (1/10, w/v) diluted in PBS-BSA (0.1 mg/ml) were added to 200 μl of ACE substrate (5 mM Hip-His-Leu) and incubated 30 min at 37°C. The reaction was terminated with 0.28 M NaOH, and the His-Leu product was estimated by incubation with O-phthaldialdehyde. All samples were centrifuged for 3 min at 3000 g before measurement of the fluorescence using 365 nm excitation and 500 nm emission wavelengths.
Antibody radiolabeling and in vivo biodistribution
Radio-iodination of antibodies with 125I was performed in Iodo-Gen precoated tubes (Pierce, Rockford, USA). Briefly, 100 μg of antibody was incubated for 5 min on ice with 100 μCi of Na125I. Excess iodine was removed by gel-filtration on a PD-10 (Sephadex G-25) mini-column (Pharmacia, Uppsala, Sweden). Mice were injected with antibodies (0.5 μCi) via a jugular vein PE-10 catheter under isoflurane anesthesia. After 1 hr, the animals were sacrificed and tissue radioactivity was determined by gamma scintillation counter. Results are expressed as radioactivity per g of wet tissue (cpm/g) as well as organ/blood radioactivity ratio (Muzykantov and Danilov 1991, 1995).
Immunohistochemistry
To determine possible changes in local ACE expression in lungs and other organs of Cav1−/− mice versus WT, we performed a systematic study of ACE expression using mAb 4G6 to denatured mouse ACE, suitable for immunohistochemistry (IHC) on paraffin-embedded tissues (Balyasnikova et al., 2005). Routinely processed formalin-fixed and paraffin-embedded and native mouse tissues of from 6 mice (3 wild-type Black Swiss mice and 3 Cav 1 KO mice) were obtained from normal parts of several organs with known and characteristic expression sites of ACE: lung, heart, vessels, liver, kidney, spleen, and testis.
Formalin-fixed and paraffin-embedded 2 to 4 μm tissue sections were generated from lung, heart, liver, spleen and kidney. The paraffin-embedded material was mounted on super-plan slides and dried overnight at 37°C. Microwave treatment was performed in citrate target retrieval solution (pH 6.0, Dako, Hamburg, Germany). Immunohistochemistry was performed using the rat alkaline-phosphatase-anti-alkaline-phosphatase (APAAP) technique as described (Balyasnikova et al., 2005). Sections were incubated with the primary mAb followed by the secondary rabbit anti-rat immunoglobulin (1:40, Dako, Hamburg, Germany) supplemented with reconstituted lyophilized mouse serum (1:600, Dianova, Hamburg, Germany) and the rat APAAP complex (1:50, Dako Hamburg, Germany). Each step lasted 30 min at RT. Alkaline phosphatase substrate reaction with fuchsin (100 μg/ml) and levamisole (400 μg/ml) was performed for 20 min at RT. Sections were counterstained with hematoxylin and mounted in gelatin.
Semi-quantitative analysis of ACE immunostaining
In order to estimate the level of ACE expression, semi-quantitative analysis of immunostaining was performed visually and also by computer-assisted pixel analysis (CAPA; DatInf® GmbH, Tübingen, Germany). The visual analysis of endothelial ACE was estimated using a four scale system of no (--), weak (+), moderate (++), or strong expression (+++) which we have previously used to quantify changes in local ACE expression in rat models pulmonary hypertension (Morrell et al., 1995) and human non-neoplastic kidney diseases (Metzger et al., 1999). From each slide, 3 representative pictures were taken (Zeiss, Axio-Imager A1; camera: Zeiss Ic3; magnification: x10), analysed by CAPA (DatInf® GmbH, Tübingen, Germany), and averaged (MS Excel). All together, more than 600 slides of all organs from 6 mice were analyzed.
Isolated mouse lung preparation
Ketamine (100 mg/Kg ip)/Xylazine (10 mg ip)-anesthetized mice were placed on mechanical ventilation via tracheostomy, and following sternotomy, a PE10 catheter was placed in the pulmonary artery (PA) and the left atrium. The lungs were perfused via the PA catheter with HEPES-buffered RPMI medium and the PAP was recorded continuously via a force displacement transducer (Maniatis et al., 2006). Drugs were infused through a side port at 10x concentration and diluted to desired concentration in the main perfusion line.
Statistical analysis
All data are presented as mean ± SD. Statistical comparisons were made using Student’s t-test or one-way analysis of variance (ANOVA). P values < 0.05 were considered significant.
RESULTS
Tissue ACE activity in Cav1−/− mice
We undertook an initial survey of ACE activity in various organs from WT and Cav1−/− mice, including lungs, kidneys, brain, spleen, small intestine, heart, striated muscle, liver, testes and plasma. Whole organ ACE activity of tissue homogenates was taken as the amount of His-Leu produced by hydrolysis of ACE substrate Hip-His-Leu. In Cav1−/− mice, we observed a significant reduction in ACE activity in various organs compared to WT mice (Fig. 1A, B). The most striking differences were found in the lungs, where ACE activity was reduced by 44% in Cav1−/− lungs (11,522 ± 908 vs 6452 ± 709 mU/g tissue; n=4, p<0.05). ACE activity was also significantly reduced in the kidneys (6164 ± 535 vs 5227 ± 418 mU/g tissue; n=4, p<0.05), brain (125 ± 17 vs 96 ± 5 mU/g tissue; n=4, p<0.05) and spleen (88 ± 5 vs 62 ± 8 mU/g tissue; n=4, p<0.05) of Cav1−/− mice. No differences in ACE activity between WT and Cav1−/−mice were observed in the intestine, heart, striated muscle, liver, testes or plasma (Fig. 1A, B).
Figure 1. Tissue ACE activity.

Mice were sacrificed, blood and tissues weighed, and homogenates (1:10 w/v ratio) prepared as described in Methods. ACE activity was measured fluorimetrically with Hip-Hs-Leu as substrate. A. ACE activity of blood (mU/ml) and tissues (mU/g of tissue) from WT mice. (B) ACE activity in Cav1−/− mice expressed as percentage of wild-type mice. Data are mean ± SD, N =7; * p< 0.05.
Endothelial ACE expression in Cav1−/− mice
To assess ACE expression specifically in endothelial cells, we measured the accumulation of intravenously injected 125I-labeled mAb 4G10.5 directed against native, catalytically active mouse ACE (Balyasnikova et al., 2006). This approach has been used previously to quantify endothelial-bound ACE in various experimental rat injury models, using mAb 9B9 recognizing rat ACE (Muzykantov and Danilov, 1991, 1995). Consistent with the notion that the pulmonary capillary endothelium is the main site of ACE expression, the majority of intravenously administered 125I-labeled mAb 4G10.5 was retained in lung tissue. In WT mice, 77% of the tracer accumulated in the lung, resulting in a 6-fold enrichment in 125I-labeled mAb 4G10.5 compared to plasma counts of 125I-labeled mAb 4G10.5 (Fig. 2A).
Figure 2. Endothelial ACE expression.

Accumulation of intravenously injected 125I-labeled mAb 4B10.5 was used to assess intravascular endothelial cell surface ACE expression. Radiolabeled mAb injected intravenously into WT and Cav1−/− mice was measured in blood and tissues after circulating for 1 hr. A, B. 125I-mAb 4B10.5 accumulation is expressed as cpm/ml of plasma or cpm/gram of tissue (A) and as the organ/blood ratio (B). C. Accumulation of mAb 4B10.5 in organs of Cav1−/− mice is expressed as a % of WT animals. Data are mean ±SD, n=7; * p< 0.05.
In Cav1−/− lungs, however, we observed only a 4-fold increase in anti-ACE mAb accumulation relative to plasma (Fig. 2B). All other organs tested had much lower endothelial ACE expression and thus anti-ACE mAb binding was well below the plasma level of radiolabeled anti-ACE mAb (Fig. 2A, B). Non-specific accumulation of 125I-labeled rat non-immune IgG into the studied organs (negative control) did not exceed 10 % of the injected dose per gram of tissue (in contrast to over 500 % of the injected dose per gram of lung with specific 125I-mAb to ACE (4B10.5).
Thus, we found a significant (50%) decrease in anti-ACE mAb accumulation in the lungs, which likely reflects a decrease in endothelial ACE expression (Fig 2C, p<0.05), whereas no differences were observed between Cav1−/− and WT mouse ACE expression in the plasma, heart, liver, kidney, spleen, striated muscle and testes (Fig. 2C). These results reflect the difference between high-level ACE expression of pulmonary ECs and low-level ACE expression seen in endothelia of the systemic circulation (Franke et al., 1997, Danilov et al., 2001), including the near absence of ACE in renal endothelia (Metzger et al., 1999)
Histology and immunohistochemistry of tissue ACE
The morphology of Cav1−/− tissues was evaluated in paraffin sections by hematoxylin and eosin staining. Compared to WT lungs, Cav1−/− lungs showed collapsed alveolae and septal thickening (Fig. 3A and 3B) as reported previously (Maniatis et al., 2008, Zhao et al., 2002). Immunohistochemical staining profiles in both Cav1−/− and WT mice were consistent with known ACE expression sites. In WT and Cav1−/− lungs, we observed homogeneous and robust ACE staining in all large vessels, including the aorta, large and small arteries, arterioles, veins, and the pulmonary microcirculation. In most tissues (other than the kidney and lung), ACE expression in the microcirculation (capillaries and small veins) was detected in approximately 10% of endothelial cells, in agreement with our previous report (Balyasnikova et al., 2005) , whereas WT and Cav1−/− lungs showed uniform ACE-labelling in the endothelium of the entire microcirculation.
Figure 3. Tissue histology and immunohistochemistry of lung ACE.
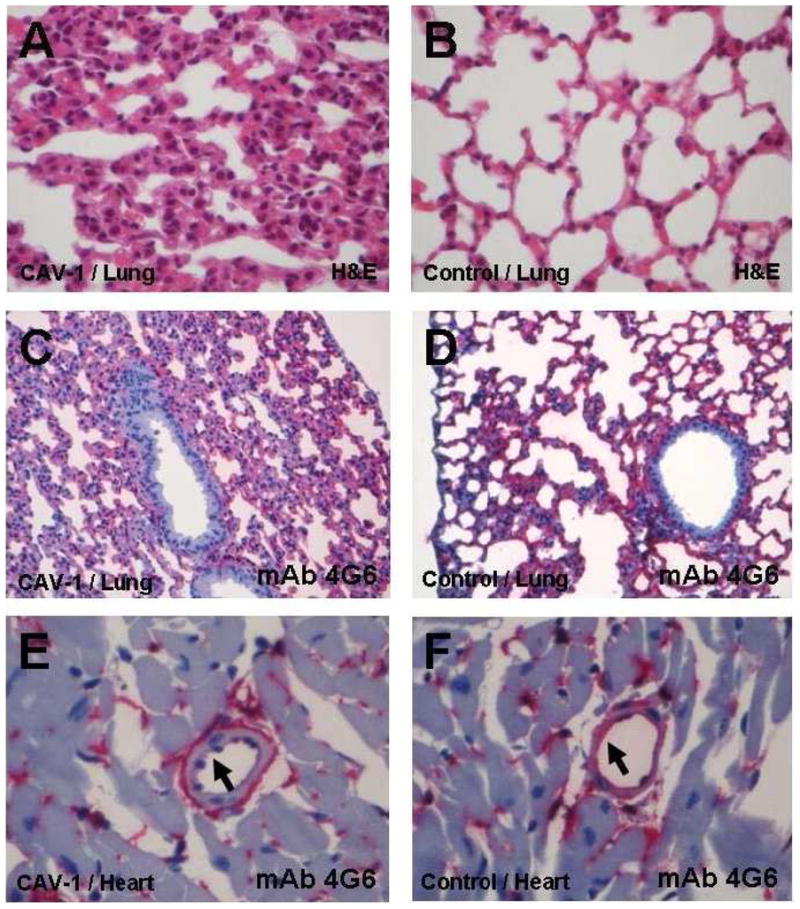
Representative paraffin sections from Cav1−/− (A, C, and E) and WT mice (B, D, and F). Hematoxylin-eosin staining of lung tissue sections shows a substantial thickening of the alveolar wall and collapsed alveolae apparent in Cav1−/− mice (A) compared with the WT in B. Original magnification, x400, H&E. ACE immunostaining shows uniform ACE labelling in the endothelium of the entire alveolar microcirculation of Cav1−/− mice (C) and WT (D). Quantitative immunostaining for ACE revealed a 40% decrease in ACE expression in Cav1−/− lungs compared to WT, as quantified by computer-assisted pixel analysis, based on at least 10 lung sections from 3 animals from each strain (WT and Cav1−/−) and at least 5 views from each section. C/D mouse lung: mAb 4G6, original magnification × 200, rat APAAP
In all organs the percentage of ACE-positive EC in Cav1−/− mice (E) and WT (F) is equal, but the intensity of staining per EC is significantly reduced in Cav1−/− mice. E and F show heart muscle with a small muscular artery. Visual quantification revealed weak expression in EC of Cav1−/− mice (arrow in E) and strong expression in EC of WT (arrow in F). E/F mouse heart: mAb 4G6, original magnification x400, rat APAAP.
Semi-quantitative measurements of ACE expression in the lungs and different organs of WT and Cav1−/− mice revealed significant differences. In Figure 3, representative images of the lung histology and anti-mouse ACE mAb 4G6 immunostaining of lung and heart are shown. Whereas the percentage of ACE-positive capillaries in Cav1−/− mice and WT was equal, the intensity of staining was significantly reduced in Cav1−/− mice lungs (Fig. 3C–F). Quantification by computer-assisted pixel analysis showed a 40% decrease in ACE expression in Cav1−/− lungs compared to WT, predominantly in the alveolar microvessels. The value of 40% reduction in ACE expression in the lung of CAV-1 mice was based on the analysis of at least 10 lung sections from each animal and at least 5 views from each section. Therefore, this value was based on the analysis of more than 300 views from lungs of 6 mice.
Pulmonary artery presser response to angiotensin I (ATI) and angiotensin II (ATII)
PAP was measured in isolated perfused mouse lungs during infusion of ATI or ATII. Because the perfusate flow and venous outflow pressure were held constant, changes in PAP in response to pharmacologic interventions reflect changes in pulmonary vascular resistance. Baseline PAP during a perfusate flow of 2.0 ml/min was elevated in Cav1−/− lungs compared to WT (6.9 ± 0.14 cmH2O vs. 6.0 ± 0.27 cmH2O; p<0.05, n=3–4). Infusion of 1 μM ATI increased PAP by 29.3 ± 3.9% in WT lungs and 17.2 ± 3.0% in Cav1−/− lungs (p<0.05, n=3–4) and 10 μM AT I raised PAP by 31 ± 11.0% in WT lungs and 19.4 ± 1.5% in Cav1−/− lungs (p<0.05, n=3–4). Similarly, infusion of 10 μM AT II raised PAP by 36.3 ± 16.0% in WT lungs and 13.6 ± 0.3% in Cav1−/− lungs (p<0.05, n=3–4) (Fig. 4), indicating pulmonary arteries in the Cav1−/− mouse have reduced reactivity to AT I and II.
Figure 4. Effect of AI Infusion on Pulmonary Artery Pressure in the Isolated Perfused Mouse lung.

Pulmonary artery pressure (PAP) was measured in isolated buffer-perfused and ventilated mouse lung preparations (flow rate = 2.0 ml/min) at baseline and following infusion of angiotensin I (ATI) and angiotensin II (ATII). Data presented in panel A are absolute PAP values in cmH2O and in panel B are as percent change from baseline following infusion of ATI and ATII. PAP at baseline was significantly increased (by 15%) in Cav1−/− lungs compared to WT (A). Infusion of 1 μM and 10 μM ATI increased PAP in both groups (A, B), but the response was attenuated in Cav1−/− lungs (A, B). Infusion of 10μM ATII also raised PAP to a greater extent in WT lungs compared to Cav1−/− lungs (A, B) (*p<0.05, n=3–4).
Wunderlich et al. (Wunderlich et al., 2008) recently demonstrated that increased plasma NO2 and NO3 levels reflect the generation of reactive oxygen species (ROS) rather than purely increased NO production. To test whether significant increase in plasma ROS in Cav1−/− mice at 3 and 5 months age (181% and 177%, respectively) reduces plasma ACE activity via probably oxidative inactivation of ACE denaturation, we determined the ratio of Hip-His-Leu to Z-Phe-His-Leu hydrolysis by plasma ACE from Cav1−/− and WT mice. Similar to our recent report indicating that this ratio is sensitive to even small denaturation/inactivation in the N and C domain active centers in somatic ACE (Danilov et al., 2008), the ZPHL/HHL ratio was weakly albeit statistically higher in Cav1−/− mice (0.91+0.026 versus 0.86+0.025, p=0.026).
DISCUSSION
Caveolae, the primary vesicular carriers and important signalling hubs in endothelial cells, are required for proper endothelial function (Minshall et al., 2002; Minshall et al., 2003). Mice lacking caveolae have severe phenotypic defects due to endothelial impairment resulting in altered microvessel development (Razani et al, 2001) and barrier integrity (Schubert et al., 2002), and PAH and right ventricular remodelling despite the paradoxically elevated level of plasma NO (Zhao et al., 2002).
Previously, using specific monoclonal antibodies to defined epitopes on ACE, we identified the lung microvascular endothelium as the primary site of lung ACE expression in the rat, human and mice (Franke et al., 1997, Morrell et al., 1995a, Danilov et al. 2001, Balyasnikova et al. 2006). ACE distribution pattern is altered in hypoxia-induced PAH, predominantly due to a reduction in capillary ACE expression, whereas in small muscularized arteries, ACE is up-regulated (Morrell et al., 1995a). Differences in ACE expression in the lung vasculature provide a unifying explanation for the paradoxical result that ACE inhibitors attenuated pulmonary hypertension (Morrell et al., 1995b; Niazova et al., 1996) despite a reduction in total lung ACE activity (Morrell et al., 1995a).
Because mice lacking caveolin-1 also demonstrated PAH (Drab et al., 2001; Maniatis et al., 2008; Zhao et al., 2002), we assessed ACE expression in these mice. Using complementary techniques to discern endothelial from non-endothelial ACE expression, we demonstrated a significant reduction in endothelial ACE activity and expression level in the lung of Cav1−/− mice. Whole organ ACE activity was diminished by 50% in lung homogenates, whereas smaller, albeit significant decreases were also found in the kidney, brain and spleen of Cav1−/− mice. Because endothelial cell ACE expression in these organs is very low to undetectable, the observed differences likely reflect changes in non-endothelial ACE.
The tissue distribution of radiolabeled anti-mouse ACE mAb in mice (Balyasnikova et al., 2006) correlates with ACE expression on the surface of endothelial cells as shown previously for mAbs against rat and human ACE (Danilov et al., 1989). In our studies, 77% percent of intravenously injected ACE mAb was retained in the lung, consistent with the high level of ACE expression in pulmonary microvessel endothelial cells (Danilov et al., 2001; Franke et al., 1997). The level of anti-ACE mAb binding in the lung exceeded the plasma concentration by a factor of 6, whereas anti-ACE mAb binding in the remaining organs was far below the plasma levels, reflecting lower level ACE expression in endothelial cells in the systemic circulation. Using this method, we showed a 50% decrease in anti-ACE mAb accumulation in the lung of Cav1−/− mice compared to WT mouse lungs, which likely reflects decreased endothelial cell ACE expression. This decrease is the same magnitude of difference observed when total lung ACE activity is measured. ACE expression in other organs was much lower relative to the lung, and no differences were observed between Cav1−/− and WT mice in these other organs. This result reflects a fundamental difference between the two methods of ACE quantification. The determination of ACE activity in a whole-organ homogenates reflects total ACE in all cells of the organ, whereas intravenous injection of radiolabeled antibodies to ACE detects enzyme expression specifically on the surface of endothelial cells. As shown previously by other methods (Maniatis et al., 2008), the capillary density in Cav1−/− mice is not decreased and our findings cannot be explained by diminished microvascular surface area. Also, because plasma ACE levels did not change, the observed effect is probably not attributed to shedding of ACE into the circulation. Due to the low-level endothelial ACE expression in extrapulmonary tissues, no differences in antibody uptake were observed between Cav1−/− and WT mice.
In order to determine the anatomic distribution of ACE expressed in the lungs of Cav1−/− mice, we performed immunohistochemical staining of lung sections using our mouse-specific mAbs and quantified the intensity of staining. ACE was distributed almost exclusively in pulmonary capillaries in WT and Cav1−/− mouse lungs and was not detected in larger pulmonary blood vessels. However, using this technique, we observed a 40% reduction in ACE expression in Cav1−/− lungs compared to WT, which was approximately the same difference observed with the two other methods.
In addition to showing a significant effect of the absence of caveolin-1 on tissue ACE expression, the present study indicates that evaluation of ACE expression may be an approach to assess lung endothelial injury in patients with lung abnormalities. Previously, an increase in serum ACE activity was thought to indicate lung endothelial injury (Kay et al. 1982, Muzykantov and Danilov, 1991; Orfanos et al., 2000). However, the extent of the increase in blood ACE activity depends not only on the rate of ACE shedding from lung endothelial cells, but also on the hepatic clearance rate of ACE from the circulation (Orfanos et al., 2000). The ability of the liver to metabolize circulating proteins might be also compromised in pathological conditions and therefore measurement of lung ACE may be a more useful approach.
How can lung ACE be measured in patients? There are at least three approaches. One approach utilizes injection of labeled ACE substrates and estimation of the rate of the hydrolysis of these in the blood during passage through the lung (Orfanos et al, 2000, 2001). Another approach utilizes injection of a labeled ACE inhibitor and determination of the dose required to specifically block pulmonary ACE activity, which is proportional to the mass of ACE in the lung, using positron emission tomography (Qing et al., 2000). Both techniques demonstrated reliable sensitivity in the detection of lung ACE content and changes induced by pathological conditions.
Our approach utilizes selective vascular endothelial cell binding of monoclonal antibodies to ACE, which indicate the amount of ACE in the lung. We found that lung accumulation of mAb 9B9 is a sensitive and early marker to detect acute lung injury before pulmonary edema formation (Muzykantov and Danilov, 1995; Muzykantov et al., 1991). These prior studies reproducibly demonstrate that lung accumulation of anti-ACE mAb 9B9 is significantly reduced in the endotoxin-induced model of septic shock in rats. Even in non-edematous endotoxemia, accumulation of mAb 9B9 was 30% less than control (Muzykantov et al., 1991). This suggests that the measurement of anti-ACE mAb accumulation in patients with septic shock may be useful for estimating the degree of lung injury. For example, gamma-scintigraphy of radiolabeled anti-ACE mAb (Muzykantov and Danilov, 1995) in septic patients could be used clinically for early detection of potentially fatal pulmonary microvascular injury.
The detection of lung endothelial ACE by monoclonal antibodies could be a feasible approach to surveying endothelial involvement in various pathological conditions in human lungs as well as other vascular beds. The advantage of this approach compared to currently existing methods would be the minimal invasiveness, since it would obviate the need for arterial and central venous catheterization and cardiac output measurement. Thus, it would be possible for many investigators to widely use the technique in order to assess its validity as a diagnostic tool and as a means to monitor treatment responses by obtaining serial measurements.
We confirmed the presence of increased PAP in isolated perfused Cav1−/− lungs compared to WT mouse lungs. Because the left atrial pressure was held constant, an increase in PAP reflects increased pulmonary vascular resistance as recently described (Maniatis et al, 2008). PAH in Cav-1−/− mice is related to a reduction in the density of perfused precapillary vessels due to distortion and compression of capillaries (Maniatis et al., 2008). Infusion of ATI led to vasoconstriction of pulmonary arteries and increased PAP in WT lungs upon conversion to ATII by ACE. This effect was attenuated in Cav1−/− lungs, in line with reduced ACE levels. However, we also observed a blunted response to direct ATII infusion indicating the Cav1−/− vascular muscle cells may be less reactive to vasoconstrictors in general, perhaps due to the higher NO levels, disruption of ATII signalling (Zuo et al., 2005), or reduced vascular muscle cell Ca2+ influx as observed in caveolin-1 null endothelial cells (Kwiatek et al., 2006). Therefore, the diminished presser effect of ATI may be only partially due to reduced ACE expression.
Using a variety of techniques, we show a significant and highly reproducible reduction in ACE levels in the Cav1−/− mouse lung, suggesting a role for caveolin-1 in regulating ACE expression. Possible mechanisms may include alterations of endothelial cell phenotype with failure of endothelial cells and microvessels to mature, perhaps due in part to elevated levels of NO (Fernandez-Alfonso and Gonzàlez, 1999). Recent data by Wunderlich et al., 2008 demonstrated that increased plasma NO2 and NO3 levels may in fact reflect increased ROS generation rather than simply enhanced NO production. Our results showing decreased ACE activity in the lung and increased blood levels of NO3 and NO2 in Cav1−/− mice (Maniatis et al., 2008) may indicate partial denaturation of lung ACE by ROS and thus a decrease in ACE activity, immunostaining, and surface expression in Cav1−/− mice. We and others previously demonstrated oxidative inactivation of ACE (Sakharov et al., 1991; Michel et al., 2001). Bearing in mind that the C domain of ACE is more sensitive (susceptible) to denaturation (Marcic et al., 2000; Voronov et al., 2002), the slight decrease in the hydrolysis ratio of ACE substrates ZPHL and HHL by plasma ACE and overall lung ACE activity in Cav1−/− mice perhaps can be explained, at least in part, by accumulation of reactive oxygen species in 3 and 5 month old Cav1−/− mice (Wunderlich et al., 2008).
An alternative explanation for decreased lung capillary ACE in Cav1−/− mice might be due to the increase in pressure per se, i.e., the “pressure hypothesis”. We previously made the observation that capillary endothelial ACE expression appears to respond to changes in systemic and pulmonary arterial pressure. Thus, only 10–15 % of capillary endothelial cells in the systemic circulation (having a pressure of approximately 25–30 mmHg) express ACE. The important exception to this general pattern of endothelial ACE expression was found in the lung where all endothelial cells (100%) of pulmonary capillaries strongly expressed ACE (Morrell et al., 1995a; Franke et al., 1997; Danilov et al., 2001), and where the pressure in healthy adults does not exceed 15–18 mmHg. The opposite was shown for kidney endothelial cells which do not express ACE and are exposed to pressures of more than 50 mm Hg (Morrell et al., 1995a; Orfanos et al., 2001). Additional confirmation of the validity of this hypothesis comes from a study of hypoxia-induced pulmonary hypertension in rats (Morrell et al., 1995a). Exposure of rats to hypoxic conditions lead to a marked increase in pulmonary pressure (from 17.8 mmHg to 27–31 mmHg mean PAP) and to a significant (50%) decrease in ACE expression in lung capillaries (Morrell et al., 1995a). The inverse relationship between pulmonary capillary pressure and ACE expression was also noted in rats with pulmonary hypertension induced by monocrotaline (Kay et al., 1982). Finally, Quing et al., 2000, using PET scanning with fluorocaptopril, demonstrated that lungs of patients with PPH have 30% of the ACE expression of healthy controls. It is therefore plausible to suggest that reduction in lung ACE expression in Cav1−/− mice may be due to an affect of the hypertension per se.
Our finding may also be relevant to the pathogenesis of pulmonary hypertension. Previous studies have shown a reduction in caveolin-1 expression in plexiform lesions from patients with PAH (Achcar et al., 2006). Furthermore, ACE expression is also reduced in patients with scleroderma (Matucci-Cerinic et al., 1990; Orfanos et al., 2001), a disease associated with PAH. In addition, caveolin-1 was shown to control collagen production in lung fibroblasts from scleroderma patients (Tourkina et al., 2005). Therefore, it appears worthwhile to investigate the function of caveolin-1 in patients with these disorders and to look for caveolin-1 gene polymorphisms in patients with PAH or scleroderma with lung involvement. In summary, we have revealed a novel role of caveolin-1 in the regulation of ACE expression in pulmonary capillary endothelial cells. Further understanding of the mechanism by which reduced caveolin-1 expression leads altered pulmonary vascular development, PAH, and reduced ACE expression may have important clinical implications in patients with these severe lung diseases.
Acknowledgments
This research was supported by American Lung Association Career Investigator Award CI21610 (SMD) and NIH NHLBI grants HL60678 and HL71626 (RDM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achcar RO, Demura Y, Rai PR, Taraseviciene Stewart L, Kasper M, Voelkel NF, Cool CD. Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest. 2006;129:696–705. doi: 10.1378/chest.129.3.696. [DOI] [PubMed] [Google Scholar]
- Anderson RG. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- Atochina E, Muzykantov V, Al Mehdi A, Danilov S, Fisher A. Lung ischemia/reperfusion releases angiotensin converting enzyme from the pulmonary endothelium. Am J Resp Crit Care Med. 1997;156:1114–1119. doi: 10.1164/ajrccm.156.4.96-12116. [DOI] [PubMed] [Google Scholar]
- Balyasnikova IV, Metzger R, Sun ZL, Berestetskaya YV, Albrecht RF, Danilov SM. Development and characterization of rat monoclonal antibodies to denatured mouse angiotensin-converting enzyme. Tissue Antigens. 2005;65:24–51. doi: 10.1111/j.1399-0039.2005.00364.x. [DOI] [PubMed] [Google Scholar]
- Balyasnikova I, Sun Z, Metzger R, Taylor P, Vicini E, Muciaccia B, Visintine D, Berestetskaya Y, McDonald T, Danilov S. Monoclonal antibodies to native mouse angiotensin converting enzyme (CD143): ACE expression quantification, lung endothelial cell targeting and gene delivery. Tissue Antigens. 2006;67:10–29. doi: 10.1111/j.1399-0039.2005.00516.x. [DOI] [PubMed] [Google Scholar]
- Bernstein KE, Xiao HD, Frenzel K, Li P, Shen XZ, Adams JW, Fuchs S. Six truisms concerning ACE and the renin-angiotensin system from the genetic analysis of mice. Circ Res. 2005;96:1135–1144. doi: 10.1161/01.RES.0000169536.73576.66. [DOI] [PubMed] [Google Scholar]
- Chandrashekhar Y, Narula J. Exposing ACE up the sleeve. J Nucl Med. 2007;48:173–174. [PubMed] [Google Scholar]
- Channick RN, Rubin LJ. Pulmonary Vasculitis and Primary Pulmonary Hypertension. In: Mason, editor. Murray & Nadel’s Textbook of Respiratory Medicine. 4. Saunders; 2005. [Google Scholar]
- Danilov SM, Balyasnikova IV, Albrecht RF, 2nd, Kost OA. Simultaneous determination of ACE activity with 2 substrates provides information on the status of somatic ACE and allows detection of inhibitors in human blood. J Cardiovasc Pharmacol. 2008;52:90–103. doi: 10.1097/FJC.0b013e31817fd3bc. [DOI] [PubMed] [Google Scholar]
- Danilov SM, Gavriljuk VD, Franke FE, Harshaw W, Granger DN, Miletich DJ, Muzykantov VR. Antibody-mediated lung endothelium targeting: surface endothelial antigens as targets for drug/gene delivery to the pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1335–L1347. doi: 10.1152/ajplung.2001.280.6.L1335. [DOI] [PubMed] [Google Scholar]
- Danilov S, Sakharov I, Martynov A, Faerman A, Muzykantov V, Klibanov A, Trakht I. Monoclonal antibody to angiotensin converting enzyme: a powerful tool for lung and vessel studies. J Mol Cell Cardiol. 1989;21(suppl 1):165–170. doi: 10.1016/0022-2828(89)90853-5. [DOI] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Ehlers MRW, Riordan JF. Angiotensin-converting enzyme: new concepts concerning its biological role. Biochemistry. 1989;28:5311–5318. doi: 10.1021/bi00439a001. [DOI] [PubMed] [Google Scholar]
- Fernández Alfonso MS, González C. Nitric oxide and the renin-angiotensin system. Is there a physiological interplay between the systems? J Hypertens. 1999;17:1355–61. doi: 10.1097/00004872-199917100-00001. [DOI] [PubMed] [Google Scholar]
- Franke FE, Metzger R, Bohle RM, Kerkman L, Alhenc Gelas F, Danilov SM. Angiotensin I-Converting Enzyme (CD 143) on endothelial cells in normal and in pathological conditions. In: Kishimoto T, et al., editors. Leucocyte Typing VI: White Cell Differentiation Antigens. Garland Publishing Inc; New York: 1997. pp. 749–751. [Google Scholar]
- Friedland J, Silverstein EA. A sensitive fluorometric assay for serum angiotensin-converting enzyme. Am J Clin Path. 1976;66:416–424. doi: 10.1093/ajcp/66.2.416. [DOI] [PubMed] [Google Scholar]
- Jasmin J, Mercier I, Dupuis J, Tanowitz H, Lisanti M. Short-term administration of a cell-permeable caveolin-1 peptide prevents the development of monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Ciiculation. 2006;114:912–920. doi: 10.1161/CIRCULATIONAHA.106.634709. [DOI] [PubMed] [Google Scholar]
- Kay JM, Keane PM, Suyama KL, Gauthier D. Angiotensin converting enzyme activity and evolution of pulmonary vascular disease in rats with monocrotaline pulmonary hypertension. Thorax. 1982;37:88–96. doi: 10.1136/thx.37.2.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane PM, Kay JM, Suyama KL, Gauthier D, Andrew K. Lung angiotensin converting enzyme activity in rats with pulmonary hypertension. Thorax. 1982:198–204. 198–204. doi: 10.1136/thx.37.3.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatek AM, Minshall RD, Cool DR, Skidgel RA, Malik AB, Tiruppathi C. Caveolin-1 regulates store-operated Ca2+ influx by binding of its scaffolding domain to TRPC1 in endothelial cells. Mol Pharmacol. 2006;70:1174–1183. doi: 10.1124/mol.105.021741. [DOI] [PubMed] [Google Scholar]
- Maniatis NA, Brovkovych V, Allen SE, John TA, Shajahan AN, Tiruppathi C, Vogel SM, Skidgel R, Malik AB, Minshall RD. Novel mechanism of endothelial nitric oxide synthase activation mediated by caveolae internalization in endothelial cells. Circ Res. 2006;99:870–877. doi: 10.1161/01.RES.0000245187.08026.47. [DOI] [PubMed] [Google Scholar]
- Maniatis NA, Shinin V, Schraufnagel DE, Okada S, Vogel SM, Malik AB, Minshall RD. Increased Pulmonary Vascular Resistance and Defective Pulmonary Artery Filling in Caveolin-1−/− Mice. Am J Physiol-Lung Cell Mol Physiol. 2008;294:L865–L873. doi: 10.1152/ajplung.00079.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcic B, Deddish PA, Jackman HL, Erdös EG, Tan F. Effects of the N-terminal sequence of ACE on the properties of its C-domain. Hypertension. 2000;36:116–121. doi: 10.1161/01.hyp.36.1.116-a. [DOI] [PubMed] [Google Scholar]
- Matucci-Cerinic M, Pignone A, Iannone F, Lotti T, Pesciullesi E, Spillantini G, Falcini F, Cagnoni M. Clinical correlations of plasma angiotensin converting enzyme (ACE) activity in systemic sclerosis: a longitudinal study of plasma ACE level, endothelial injury and lung involvement. Respir Med. 1990;84:283–287. doi: 10.1016/s0954-6111(08)80054-6. [DOI] [PubMed] [Google Scholar]
- Metzger R, Bohle RM, Kerkman L, Eichner G, Alhenc Gelas F, Danilov SM, Franke FE. Distribution of angiotensin I-converting enzyme (CD 143) in the normal human kidney and in non-neoplastic kidney diseases. Kidney Int. 1999;56:1442–1454. doi: 10.1046/j.1523-1755.1999.00660.x. [DOI] [PubMed] [Google Scholar]
- Michel B, Grima M, Nirina LB, Ingert C, Coquard, Barthelmebs M, Imbs JL. Inhibitory effect of reactive oxygen species on angiotensin I-converting enzyme (kininase II) Clin Exp Pharmacol Physiol. 2001;28:212–218. doi: 10.1046/j.1440-1681.2001.03419.x. [DOI] [PubMed] [Google Scholar]
- Minshall RD, Sessa WC, Stan RV, Anderson RG, Malik AB. Caveolin regulation of endothelial function. Am J Physiol Lung Cell Mol Physiol. 2003;285:1179–83. doi: 10.1152/ajplung.00242.2003. [DOI] [PubMed] [Google Scholar]
- Minshall RD, Tiruppathi C, Vogel SM, Malik AB. Vesicle formation and trafficking in endothelial cells and regulation of endothelial barrier function. Histochem Cell Biol. 2002;117:105–112. doi: 10.1007/s00418-001-0367-x. [DOI] [PubMed] [Google Scholar]
- Miyawaki-Shimizu K, Predescu D, Shimizu J, Broman M, Predescu S, Malik AB. siRNA-induced caveolin-1 knockdown in mice increases lung vascular permeability via the junctional pathway. Am J Physiol Lung Cell Mol Physiol. 2006;290:405–413. doi: 10.1152/ajplung.00292.2005. [DOI] [PubMed] [Google Scholar]
- Morrell NW, Atochina EN, Morris KG, Danilov SM, Stenmark KR. Angiotensin converting enzyme expression is increased in small pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. J Clin Invest. 1995;96:1823–1833. doi: 10.1172/JCI118228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell NW, Morris KG, Stenmark KR. Role of angiotensin-converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am J Physiol. 1995;269:1186–1194. doi: 10.1152/ajpheart.1995.269.4.H1186. [DOI] [PubMed] [Google Scholar]
- Muzykantov VR, Danilov SM. New Approaches to investigations of oxidative injury to the pulmonary endothelium: a minireview. Biomed Sci. 1991;2:11–22. [PubMed] [Google Scholar]
- Muzykantov VR, Danilov SM. Targeting of radiolabelled monoclonal antibody against angiotensin-converting enzyme to the pulmonary vasculature. In: Torchilin VP, editor. Handbook of Targeting Delivery of Imaging Agents. CRC; Boca Raton, FL: 1995. pp. 465–486. [Google Scholar]
- Muzykantov VR, Puchnina EA, Atochina EN, Slinkin M, Hiemish H, Meertsuk F, Danilov SM. Endotoxin reduces specific pulmonary uptake of radiolabeled monoclonal antibody to angiotensin-converting enzyme. J Nucl Med. 1991;3:2453–2460. [PubMed] [Google Scholar]
- Niazova ZA, Batyraliev TA, Aikimbaev KS, Kudaiberdieva GZ, Akgul F, Soodanbekova YK, Birand A. High-altitude pulmonary hypertension: effects of captopril on pulmonary and systemic arterial pressures. J Hum Hypertens. 1996;10:S141–S142. [PubMed] [Google Scholar]
- Oparil S, Narkates AJ, Jackson RM, Ann HS. Altered angiotensin-converting enzyme in lung and extrapulmonary tissues of hypoxia-adapted rats. J Appl Physiol. 1988;65:218–227. doi: 10.1152/jappl.1988.65.1.218. [DOI] [PubMed] [Google Scholar]
- Orfanos SE, Armaganidis A, Glynos C, Psevdi E, Kaltsas P, Sarafidou P, Catravas JD, Dafni UG, Langleben D, Roussos C. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in acute lung injury. Circulation. 2000;102:2011–2018. doi: 10.1161/01.cir.102.16.2011. [DOI] [PubMed] [Google Scholar]
- Orfanos SE, Psevdi E, Stratigis N, Langleben D, Catravas JD, Kyriakidis M, Moutsopoulos HM, Roussos C, Vlachoyiannopoulos PG. Pulmonary capillary endothelial dysfunction in early systemic sclerosis. Arthritis Rheum. 2001;44:902–911. doi: 10.1002/1529-0131(200104)44:4<902::AID-ANR147>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Qing F, McCarthy TJ, Markham J, Schuster DP. Pulmonary angiotensin-converting enzyme (ACE) binding and inhibition in humans. A positron emission tomography study. Am J Respir Crit Care Med. 2000;161:2019–2025. doi: 10.1164/ajrccm.161.6.9907036. [DOI] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- Sakharov IY, Dukhanina EA, Puchnina EA, Danilov SM, Muzykantov VR. Oxidative inactivation of angiotensin-converting enzyme. Biokhimiia. 1991;56:55–62. [PubMed] [Google Scholar]
- Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW, Lisanti MP. Microvascular hyperpermeability in Caveolin-1−/− knockout mice. J Biol Chem. 2002;277:40091–40098. doi: 10.1074/jbc.M205948200. [DOI] [PubMed] [Google Scholar]
- Tourkina E, Gooz P, Pannu J, Bonner M, Scholz D, Hacker S, Silver RM, Trojanowska M, Hoffman S. Opposing effects of protein kinase Calpha and protein kinase Cepsilon on collagen expression by human lung fibroblasts are mediated via MEK/ERK and caveolin-1 signaling. J Biol Chem. 2005;280:13879–13887. doi: 10.1074/jbc.M412551200. [DOI] [PubMed] [Google Scholar]
- Voronov S, Zueva N, Orlov V, Arutyunyan A, Kost O. Temperature-induced selective death of the C-domain within angiotensin-converting enzyme molecule. FEBS Lett. 2002;522:77–82. doi: 10.1016/s0014-5793(02)02888-0. [DOI] [PubMed] [Google Scholar]
- Wunderlich C, Schober K, Schmeisser A, Heerwagen C, Tausche AK, Steinbronn N, Brandt A, Kasper M, Schwencke C, Braun-Dullaeus RC, Strasser RH. The adverse cardiopulmonary phenotype of caveolin-1 deficient mice is mediated by a dysfunctional endothelium. J Mol Cell Cardiol. 2008;44:938–947. doi: 10.1016/j.yjmcc.2008.02.275. [DOI] [PubMed] [Google Scholar]
- Yang HY, Erdös EG, Levin Y. A dipeptidyl carboxypeptidase that converts angiotensin I and inactivates bradykinin. Biochim Biophys Acta. 1970;214:374–376. doi: 10.1016/0005-2795(70)90017-6. [DOI] [PubMed] [Google Scholar]
- Zhao YY, Liu Y, Stan RV, Fan L, Gu Y, Dalton N, Chu PH, Peterson K, Ross J, Jr, Chien KR. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A. 2002;99:11375–11380. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, Brovkovych V, Yuan JX, Wharton J, Malik AB. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. 2009;119:2009–2018. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L, Ushio-Fukai M, Ikeda S, Hilenski L, Patrushev N, Alexander RW. cAbl tyrosine kinase mediates reactive oxygen species- and caveolin-dependent AT1 receptor signaling in vascular smooth muscle: role in vascular hypertrophy. Circ Res. 2005;97:829–836. doi: 10.1161/01.RES.0000185322.46009.F5. [DOI] [PubMed] [Google Scholar]