

Abstract
Epidemiology and genetic studies indicate that patients with telomere length shorter than average are at higher risk of dying from heart disease or stroke. Telomeres are located at the ends of eukaryotic chromosomes which demonstrate progressive length reduction in most somatic cells during aging. The enzyme telomerase can compensate for telomere loss during cell replication. The present study is aimed to investigate the contribution of telomerase to stroke and the blood-brain barrier (BBB) dysfunction. Telomerase reverse transcriptase knock-out (TERT−/−) mice and littermate controls with normal TERT expression were subjected to a 24 h permanent middle cerebral artery occlusion (pMCAO). The stroke outcomes were assessed in terms of neurological scores and infarct volumes. In addition, we evaluated oxidative stress, permeability across the BBB, and the integrity of tight junctions in brain microvessels. Neurological testing revealed that TERT−/− mice showed enhanced deficits as compared to controls. These changes were associated with a greater infarct volume. The expression of tight junction protein ZO-1 decreased markedly in ischemic hemispheres of TERT−/− mice. The brain microvessels of TERT−/− mice also were more susceptible to oxidative stress, revealing higher superoxide and lower glutathione levels as compared to mice with normal TERT expression. Importantly, TERT deficiency potentiated the production of inflammatory mediators, such as TNF-alpha, IL-1beta and ICAM-1 in the ischemic hemispheres of mice with pMCAO. Our study suggests that TERT deficiency can predispose to the development of stroke in an experimental model of this disease.
Keywords: telomerase reverse transcriptase, stroke, blood-brain barrier, tight junction proteins, cerebral microvessels
INTRODUCTION
Telomeres are located at the ends of eukaryotic chromosomes and protect these regions from degradation. Telomerase reverse transcriptase (TERT) is the catalytic subunit of telomerase that is required for the synthesis of new telomeric DNA repeats in dividing cells (Fuster and Andres 2006). Telomerase activity is down-regulated before birth and remains at very low or undetectable levels in most human somatic tissues (Harley 2005). However, a growing body of evidence suggests that TERT expression in adult brain may be induced as the result of metabolic stress, such as hypoxia or brain ischemia, in astrocytes, microglia, vascular smooth muscles, and cortical neurons (Baek et al. 2004; Fu et al. 2002; Kang et al. 2004; Minamino et al. 2001). For example, TERT transgenic mice show a reduced myocardial infarct area after coronary artery ligation, and exogenous TERT expression in cardiac myocytes can prevent apoptosis and promote survival (Oh et al. 2001).
The blood-brain barrier (BBB) is a physical and metabolic barrier separating the microenvironment of the central nervous system (CNS) from the peripheral circulation. The BBB is mainly composed of brain endothelial cells connected by tight junctions (TJs), which are maintained by transmembrane TJ proteins, such as occludin, claudins and junctional adhesion molecules (JAMs) (Chiba et al. 2008). Moreover, cytoplasmic scaffolding proteins, such as zonula occludens (ZO) proteins, anchor the integral transmembrane proteins to actin cytoskeleton. Under ischemic stroke conditions, depletion of blood flow leads to the BBB disruption and increased paracellular permeability across cerebral vessels (Petty and Wettstein 2001). Activated brain microvascular endothelium in response to environmental stress can exhibit increased cellular oxidative stress, activation of redox-sensitive transcription factors, and production of inflammatory molecules (Frijns and Kappelle 2002). Ischemia is a potent inducer of cytokines, such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1).
The function of telomerase in the injured and ischemic brain has not been clearly defined. Therefore, the aim of our present study is to explore the influence of TERT deletion on the BBB integrity and brain inflammatory responses in the context of ischemic brain injury. Our study provides evidence that TERT deficiency potentiates ischemia-induced neurological deficits, tissue injury, and BBB dysfunction as compared to control mice with normal TERT expression.
MATERIALS AND METHODS
TERT deficient mice and the model of the permanent middle cerebral artery occlusion (pMCAO)
All experiments were performed following the protocol approved by the Institutional Animal Care and Use Committee in strict accordance with the National Institutes of Health guidelines. The focal ischemia model employed in the present study was based on a permanent occlusion of the middle cerebral artery (MCA) as described earlier (Connolly et al. 1996). Sham animals were operated upon with only ligation of the left CCA, without blocking the MCA. The severity of neurological deficit was evaluated according to the modified scale by Wauquier et al. (1989) (Table 1).
Table 1.
Locomotor scale to assess neurological deficit in mice
| points | category | |
|---|---|---|
| Ipsilateral eyelid closed | 1 | I. Head and trunk |
| Hunched back | 1 | |
| Grasping (string) upon lifting the tail | 1 | II. Limb |
| Frontpaw withdrawal upon lifting the tail | 1 | |
| Hindpaw withdrawal upon lifting the tail | 1 | |
| Circling when walking | 1 | III. Whole body activity |
| Body leaning to one side when placing on the table | 1 | |
| No spontaneous movement | 1 | |
| Total score | 8 | |
The experimental animals (4-month-old male mice) were divided into four groups: wild-type mice subjected to pMCAO, wild-type sham-operated mice; TERT−/− mice subjected to pMCAO, and TERT−/− sham-operated mice. TERT−/− mice were generated on the C57BL/6J genetic background and bred through heterozygous mating (Chiang et al. 2004). Littermate mice with normal TERT expression were used as the control group.
Evaluation of cerebral infarct volume
The infarct volume was assessed 24 h post induction of pMCAO using staining with 2% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma) (Lin et al. 1993) and quantified with NIH Image-J software. The infarct volume (mm3) was determined as a percentage volume of the whole brain according to the formula: Infarct volume (%) = 100×[(Vc-Vi)/2Vc]; where Vc is the total volume of the contralateral hemisphere, and Vi is the noninfarcted volume of the ipsilateral hemisphere.
Western blotting
Each mouse brain was divided into two parts: contralateral (non-ischemic) hemisphere and ipsilateral (ischemic) hemisphere. The cortical gray matter was isolated and homogenized with RIPA lysis buffer (Santa Cruz, CA). The samples were then centrifuged at 15,000 × g for 15 min at 4°C. The supernatants were collected and protein concentrations were determined using BCA protein assay kit (Pierce, Rockford, IL). Samples were separated on 4–15% Tris-HCl Ready SDS-polyacrylamide gels (Bio-Rad Laboratories, Hercules, CA), transferred onto PVDF membrane (Bio-Rad Laboratories), and incubated with the respective antibodies. Anti-ZO-1 (1:1000 dilution), anti-ZO-2 (1:500 dilution), anti-occludin (1:1000 dilution), anti-JAM-A (1:500 dilution), anti-claudin-1(1:500 dilution), and anti-claudin-5 (1:500 dilution) antibodies were purchased from Zymed (San Francisco, CA). Anti-actin antibody (1:2500 dilution) was purchased from Sigma, and all secondary antibodies were from Santa Cruz Biotechnology. For visualization of detected proteins, immunoblots were analyzed using an ECL Western blot detection kit (Amersham Biosciences, Piscataway, NJ). Quantification of immunoreactive bands was performed by scanning densitometry using UN-SCAN-IT gel image analysis software (Silk Scientific, Orem, UT).
Real-Time RT-PCR
Real-time reverse transcriptase–polymerase chain reaction (RT-PCR) was performed as described previously (Lee et al. 2004c). Briefly, cortical grey matter samples were weighed and homogenized in 1 ml of TRIZOL reagent (Invitrogen, Carlsbad, CA) per 50 mg of tissue. Total RNA was extracted according to the manufacturer’s protocol with an additional chloroform extraction and phase separation as well as an additional wash of the isolated RNA in 70% ethanol. Then, 1 µg of RNA was reverse-transcribed using the Reverse Transcription System (Promega, Madison, WI) in a total volume of 20 µL with random hexamer primers. The following conditions were employed for reverse transcription: 25°C for 10 min, 48°C for 30 min, and 95°C for 5 min. PCR amplification was performed using 3 µL of RT product, Taqman Universal PCR Master Mix (Applied Biosystems, Foster City, CA), and predeveloped primer pairs and Taqman probes (Applied Biosystems) in a total volume of 25 µL. The following thermocycling conditions were employed: 95°C for 10 min, followed by 95°C for 15 sec, and 60°C for 60 sec (for up to 40 cycles). Expression of mRNA was calculated and analyzed by the comparative CT method as described (Lee et al. 2004b). PCR amplification of mouse actin mRNA (a housekeeping gene) was performed for each sample to normalize mRNA levels of the target genes.
Measurements of glutathione (GSH) and superoxide levels
GSH content was determined by monochlorobimane (MCB; Invitrogen) staining. MCB conjugates with GSH through a reaction catalyzed by glutathione-S-transferase (GST) to form a fluorescent stable adduct (MCB-GSH) (Kamencic et al. 2000). The levels of superoxide were detected by dihydroethidium (DHE; Invitrogen) staining. DHE is cell permeable, and at the presence of superoxide, it is converted to a fluorescent product ethidium bromide (EtBR), which then is trapped by intercalating with DNA (Williams and Allen 2007). Briefly, microvessels were incubated with 1 mL MCB solution (100 µM) or 1 mL DHE solution (10 µM) at 37°C for 90 min. Fluorescence was assessed using the Olympus BX61W1 confocal microscope (Olympus Corp., Tokyo, Japan).
Statistical analysis
Data are expressed as means ± SEM. Statistical analysis was completed by using Sigma-Stat 2.03 (SPSS, Chicago, IL, USA). Two-tailed Student’s t-test or one-way ANOVA followed by Student-Newman-Keuls post hoc test was used to compare mean responses among the treatments. A statistical probability of p < 0.05 was considered significant.
RESULTS
TERT deficiency increases neurological deficit and infarct volume following pMCAO
As indicated in Figure 1, TERT-deficient mice exhibited significantly higher neurologic deficits 24 h post inducing pMCAO as compared to mice with normal TERT expression. The most affected locomotor functions in TERT−/− mice were the parameters listed in category III as listed in Table 1. The functional locomotor scores were correlated with infarct volume as determined by TTC staining 24 h post the onset of pMCAO. As shown in Figure 2, the total infarct volume in the TERT−/− mice was larger by an approximately 20% as compared to control mice.
Figure 1. TERT deficiency potentiates neurological deficits induced by permanent occlusion of MCA.

Neurological deficit was evaluated 24 h post pMCAO using the eight point score system listed in Table 1; n=10. *Significantly different as compared to mice with normal TERT expression at p<0.05.
Figure 2. TERT deficiency increases infarct volume following pMCAO.
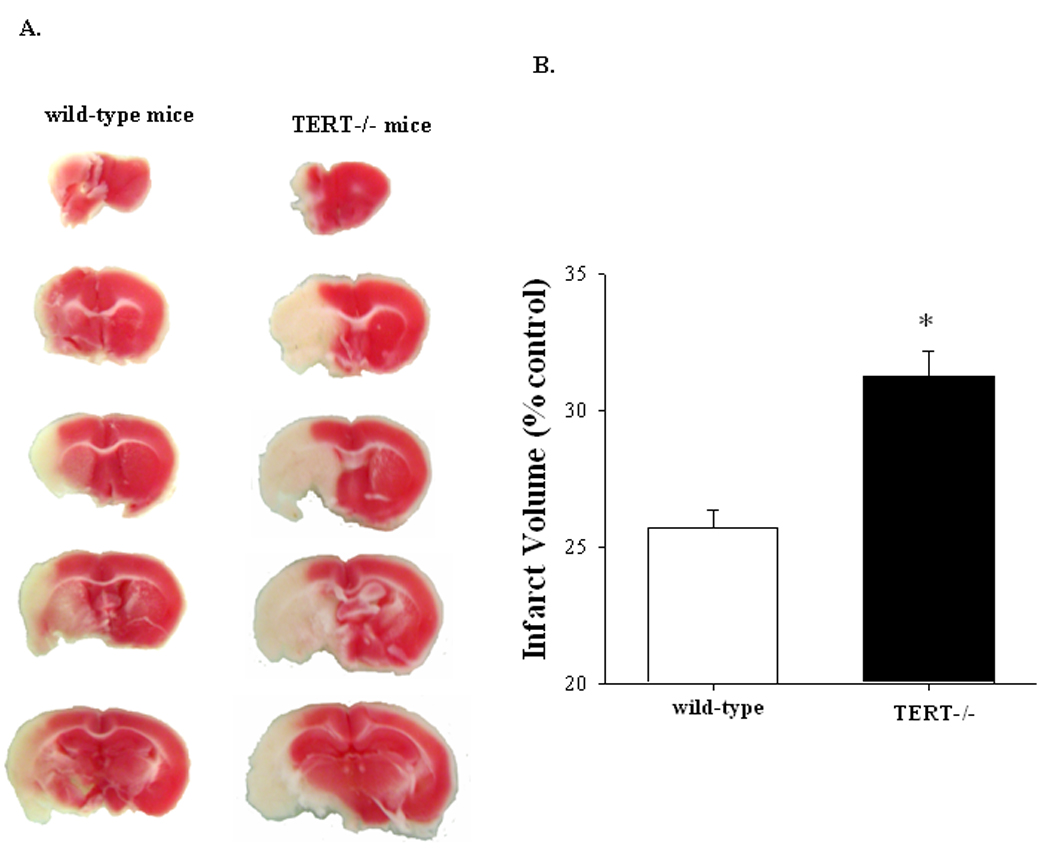
Brain sections were stained with TTC 24 h post pMCAO. White areas correspond to damaged brain tissue due to infarct (A) and quantified results are depicted in the form of bar graphs (B); n=6. *Significantly different as compared to mice with normal TERT expression at p<0.01.
TERT deficiency enhances pMCAO-induced oxidative stress in brain microvessels
In order to address potential mechanisms of increased infarct volume in TERT−/− mice, our studies focused on the involvement of the BBB and induction of oxidative stress in brain microvessels. Superoxide levels were detected in isolated brain microvessels 24 h post induction of pMCAO (Figure 3). The most pronounced increase in superoxide levels was observed in ipsilateral hemispheres of TERT−/− mice with induced pMCAO (~4.5 times of control levels). Although superoxide levels were also elevated in ipsilateral hemispheres of control mice with pMCAO, these changes were approximately 2-fold lower as compared to the corresponding values in TERT−/− animals.
Figure 3. pMCAO-induced oxidative stress in brain microvessels is enhanced in TERT deficient mice.

Twenty four hours post pMCAO or sham operation, brain microvessels were isolated and stained for superoxide levels with DHE (red color). All images were acquired using a 60× oil-immersion lens under identical instrument settings. Merged micrographs of DHE staining and phase contrast micrographs localized superoxide within the microvessels (A). The intensity of DHE fluorescence was quantified and depicted in the form of a bar graph (B). DHE fluorescence values in microvessels from ipsilateral and contralateral hemispheres of sham-operated mice were the same and the results were pooled; n=6–8. *Significantly different as compared to sham operated mice at p<0.05, * or p<0.001, ***. †Results in the TERT−/− group are statistically different from those in the corresponding hemispheres of mice with normal TERT expression at p<0.01, ††.
Induction of pMCAO resulted in decreased glutathione levels in ipsilateral hemispheres in both control and TERT−/− mice. However, this effect was significantly enhanced in TERT-deficient animals, indicating severely diminished antioxidant protection in this group of mice. In contrast to control mice, glutathione levels were also diminished in contralateral hemispheres in TERT−/− animals (Figure 4).
Figure 4. TERT deficiency potentiates pMCAO-induced glutathione depletion in brain microvessels.

Twenty four hours post pMCAO or sham operation, brain microvessels were isolated and stained for glutathione levels with MCB (blue color). Images were acquired as in Figure 3. Merged micrographs of glutathione staining and phase contrast micrographs localized glutathione within the microvessels (A). The intensity of glutathione staining was quantified and depicted in the form of a bar graph (B); n=6–8. Fluorescence intensity in microvessels from ipsilateral and contralateral hemispheres of sham-operated mice were the same and the results were pooled. *Significantly different as compared to sham operated mice at p<0.05. †Results in the TERT−/− group are statistically different from those in the corresponding hemispheres of mice with normal TERT expression at p<0.05, † or p<0.01, ††.
TERT deficiency differentially influences pMCAO-induced changes in TJ protein expression
TJs are the main structural elements of cerebral microvessels that regulate the integrity of the BBB. Therefore, we evaluated the effects of TERT deficiency on the levels of TJ proteins in mice with pMCAO. As illustrated in Figure 5, occludin and ZO-1 exhibited different expression patterns following MCA occlusion. Occludin expression was significantly increased in the ipsilateral hemisphere of control mice with pMCAO; however, the level of this TJ protein was not affected in the brains of TERT−/− mice with pMCAO as compared to sham controls. In contrast, expression of ZO-1 decreased to almost negligible levels in the ipsilateral hemisphere of TERT deficient mice with pMCAO. Similar tendency was observed in mice with normal TERT expression; however, these changes were significantly less pronounced. Expression of other TJ proteins, such as ZO-2, JAM-A, claudin-5, and claudin-1, was not affected 24 h post pMCAO in either TERT−/− or control mice (data not shown).
Figure 5. TERT deficiency differentially influences pMCAO-induced changes in TJ protein expression.
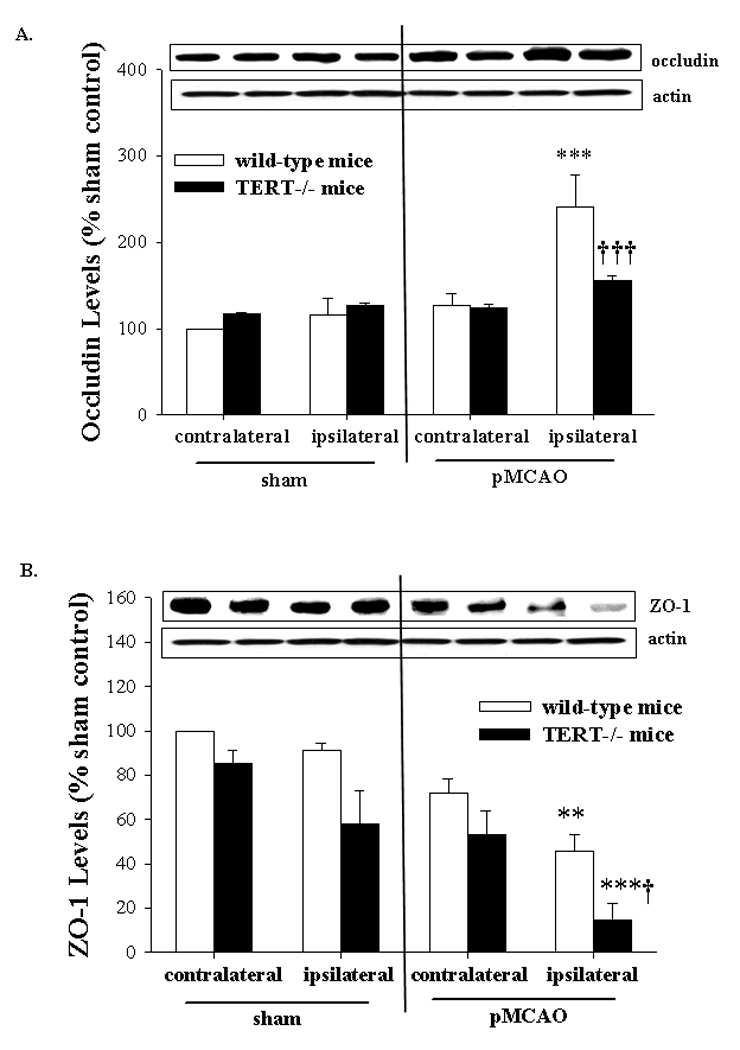
Expression of TJ proteins occludin (A) and ZO-1 (B) was analyzed by Western blotting 24 h post pMCAO or sham operation separately in ipsilateral (ischemic) and contralateral (non-ischemic) hemispheres. The blots are representative images from four mice per group and the quantified results are depicted in the form of bar graphs; n=4. *Significantly different as compared to the respective hemispheres of sham operated mice at p<0.01, ** or p<0.001, ***. †Results in the TERT−/− group are statistically different from those in the corresponding hemispheres of mice with normal TERT expression at p<0.05, † or p<0.001, †††.
TERT deficiency potentiates inflammatory responses following pMCAO
We also examined brain tissues for mRNA expression of TNF-α, IL-1β, and ICAM-1 24 h post induction of pMCAO. The choice of these inflammatory mediators includes cytokines (TNF-α and IL-1β), and an adhesion molecule (ICAM-1). These agents are critical in the induction of inflammatory reactions in the vascular endothelium by stimulating leukocyte recruitment, adhesion, and transendothelial migration. pMCAO resulted in a significant elevation of mRNA levels of all studied inflammatory mediators in ipsilateral hemispheres of TERT−/− and control mice (Figure 6A–C). Importantly, these levels were more advanced in TERT deficient mice. Specifically, mRNA levels of TNF-α, IL-1β and ICAM-1 in the ischemic hemisphere of TERT−/− mice increased 3, 2.6, and 2.4-fold, respectively, as compared to the corresponding hemisphere of control mice with pMCAO. TNF-α and ICAM-1 mRNA expression was also elevated in contralateral hemispheres of TERT−/− mice.
Figure 6. TERT deficiency potentiates pMCAO-induced inflammatory responses.

Experiments were performed as described in Figure 5. mRNA levels of TNF-α (A), IL-1β (B), and ICAM-1 (C) were determined by real-time PCR; n=4. *Significantly different as compared to the corresponding hemispheres of sham operated animals at p<0.05, * or p<0.001, ***. †Results in the TERT−/− group are statistically different from those in the corresponding hemispheres of mice with normal TERT expression at p<0.05, †; p<0.01, ††; or p<0.001, †††.
DISCUSSION
The BBB is a dynamic interface between the peripheral circulation and the CNS. In the present study, we provide in vivo evidence for the role of TERT deficiency in the BBB dysfunction and brain injury during ischemic stroke. Specifically, TERT−/− mice showed severe neurological deficits and increased infarct volume as compared to animals with normal TERT expression 24 h post induction of pMCAO. These alterations were closely associated with increased oxidative stress in brain capillaries, alterations of TJ protein expression and increased inflammatory responses.
The presence of telomerase activity in the brain during development characterizes the role of this enzyme in neuronal differentiation and survival (Greenberg et al. 1998). Both telomerase and TERT levels are generally decreased during embryonic and early postnatal development in somatic cells. Particularly, telomerase activity is reduced as proliferation of neuroblasts is decreased, whereas TERT levels are reduced in conjunction with neuronal differentiation and natural cell death (Klapper et al. 2001). This dissociation between telomerase activity and TERT levels suggests that TERT may function to promote cell survival through a mechanism other than maintaining telomere length (Mattson and Klapper 2001). In addition, recent reports from epidemiological and genetic studies indicate that humans with shorter telomeres than average are at higher risk of heart disease, stroke and degenerative diseases (Harley 2005).
Our study indicated that TERT−/− mice are more susceptible to ischemic brain injury. To support these observations, it was recently reported that TERT transgenic mice were characterized by significantly reduced infarct volume after MCAO and protective effects against NMDA-induced neurotoxicity (Kang et al. 2004). In addition, TERT transgenic mice exhibited a reduced myocardial infarct area after coronary artery ligation, and exogenous TERT expression in cardiac myocytes promoted survival by protection against apoptosis (Oh et al. 2001).
Impairment of TJs and the accompanied dysfunction of the BBB are typical events during cerebral ischemia (Ballabh et al. 2004). Indeed, our studies indicated a marked loss of ZO-1 in the ipsilateral hemisphere of the mice with pMCAO. Importantly, this effect was potentiated in TERT−/− mice as compared to control animals. ZO-1 is essential for TJ assembly (Kniesel and Wolburg 2000) as it forms a scaffold complex with other cytosolic accessory proteins, such as ZO-2 and AF6, to anchor integral membrane proteins to the actin cytoskeleton. Disruption of ZO-1 at TJs has been observed in response to cytokines (Blum et al. 1997) and hypoxia (Fischer et al. 2002), and has been correlated with increased transendothelial permeability. In the context of stroke, an ex vivo study of cerebral ischemia identified translocation and decreased expression of occludin and ZO-1 in brain capillaries after microsphere-induced cerebral embolism in rats (Kago et al. 2006). In contrast, we observed increased occludin levels in control mice following pMCAO but no apparent changes in TERT−/− mice. The influence of alterations of occludin expression on the BBB integrity is not fully understood. For example, increased occludin levels frequently correspond with decreased permeability across the BBB (Balda et al. 2000); however, others reported also the opposite phenomenon (Lee et al. 2004a). It appears that interactions of occludin with other TJ proteins, including ZO-1, may have more prominent consequences on the BBB integrity than changes in levels of this TJ protein.
The loss of blood supply to the brain associated with the onset of pMCAO results in a cascade of events including inflammation and induction of oxidative stress around the infarcted region. Inflammatory responses include upregulation of cytokines (e.g. TNF-α and IL-1β) (Tarkowski et al. 1997) which have been shown to precede BBB permeability and promote leukocyte infiltration into the brain tissue through induction of adhesion molecules on the surface of endothelial cells (Wong et al. 2007). In addition, activated microglia can release a variety of inflammatory mediators upon brain ischemia, while blood-borne leukocytes can migrate into the brain and subsequently contribute to production of inflammatory substances, increased permeability of the BBB and secondary injury to the brain tissue (Chamorro and Hallenbeck 2006; Wang et al. 2007). Our novel findings illustrate that pMCAO resulted in more pronounced expression of TNF-α, IL-1β, and ICAM-1 in TERT deficient mice as compared to mice with normal TERT expression. These results are in agreement with the report that TNF-α-mediated increase in monocyte adhesion in senescent human endothelial cells can be attenuated in TERT transfected cells (Matsushita et al. 2001), indicating a relation between telomerase activity and endothelial dysfunction.
Another novel observation of the present study indicates that superoxide production and decreased GSH levels in brain capillaries are more pronounced in TERT−/− mice as compared to controls with normal TERT expression upon induction of pMCAO. Increased oxidative stress is known to stimulate redox-regulated signaling pathways that dysregulate TJ protein expression and affect the BBB integrity (Pun et al. 2009). Persistent oxidative stress can lead to diminished antioxidant protection (Balaban et al. 2005; Sohal et al. 1994), as observed in the present study by decreased GSH levels in brain microvessels. GSH is a critical antioxidant in vascular endothelial cells and its depletion is strongly associated with BBB dysfunction (Agarwal and Shukla 1999). In agreement with our study, recent evidence emphasizes the role of telomerase in the protection against induction of oxidative stress. For example, overexpression of TERT increased oxidative stress resistance, improved antioxidant defense, and differentiation capacity in mouse embryonic stem cells (Armstrong et al. 2005). Overexpression of TERT also resulted in improved mitochondrial functions, protecting against production of superoxide radicals and increased cellular ROS levels (Ahmed et al. 2008). To further demonstrate the interrelationship between oxidative stress and telomerase activity, it was demonstrated that cellular oxidation can impair telomerase activity and accelerate the telomere shortening in endothelial (Furumoto et al. 1998; Kurz et al. 2004) and vascular smooth muscle cells (Matthews et al. 2006), as well as result in telomerase exclusion from the nucleus to the mitochondria (Haendeler et al. 2003; Haendeler et al. 2004).
Increased oxidative stress and diminished antioxidative protection in brain microvessels of TERT−/− animals may further contribute to the disruption of the BBB integrity as observed in the present study. In fact, it was demonstrated that oxidative stress can alter TJs at the BBB level by alterations of phosphorylation and expression of TJ proteins (Fischer et al. 2005; Lee et al. 2004a; Haorah et al. 2005; Rao et al. 2002). Increased levels of ROS can also stimulate NF-κB activation, leading to increased production of inflammatory cytokines and adhesion molecules (Kim et al. 2008), such as TNF-α, IL-1β, and ICAM-1 evaluated in the present manuscript. In the present study, elevated superoxide levels were mostly limited to the ipsilateral hemisphere following the pMCAO-based experimental stroke model. In agreement with these results, increased expression of inflammatory genes and disruption of TJ proteins were also mostly affected in ipsilateral hemisphere.
In conclusion, the results of the present study indicate that TERT deficiency potentiates the BBB dysfunction, inflammatory responses, and oxidative stress associated with experimental stroke induced by the permanent occlusion of MCA. Importantly, these alterations were associated with increased neurological deficit and infarct volume. The present results emphasize the importance of TERT in the protection of brain injury against ischemic stroke.
Acknowledgments
Grant information: This study was supported in part by grants from the NIH: P42 ES 07380, MH63022, MH072567, and NS39254, the American Heart Association Postdoctoral Scholarship, and the University of Kentucky Department of Surgery Research Grant Program.
REFERENCES
- Agarwal R, Shukla GS. Potential role of cerebral glutathione in the maintenance of blood-brain barrier integrity in rat. Neurochem Res. 1999;24(12):1507–1514. doi: 10.1023/a:1021191729865. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci. 2008;121(Pt 7):1046–1053. doi: 10.1242/jcs.019372. [DOI] [PubMed] [Google Scholar]
- Armstrong L, Saretzki G, Peters H, Wappler I, Evans J, Hole N, von Zglinicki T, Lako M. Overexpression of telomerase confers growth advantage, stress resistance, and enhanced differentiation of ESCs toward the hematopoietic lineage. Stem Cells. 2005;23(4):516–529. doi: 10.1634/stemcells.2004-0269. [DOI] [PubMed] [Google Scholar]
- Baek S, Bu Y, Kim H. Telomerase induction in astrocytes of Sprague-Dawley rat after ischemic brain injury. Neurosci Lett. 2004;363(1):94–96. doi: 10.1016/j.neulet.2004.03.059. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Balda MS, Flores-Maldonado C, Cereijido M, Matter K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J Cell Biochem. 2000;78(1):85–96. [PubMed] [Google Scholar]
- Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16(1):1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Blum MS, Toninelli E, Anderson JM, Balda MS, Zhou J, O'Donnell L, Pardi R, Bender JR. Cytoskeletal rearrangement mediates human microvascular endothelial tight junction modulation by cytokines. Am J Physiol. 1997;273(1 Pt 2):H286–H294. doi: 10.1152/ajpheart.1997.273.1.H286. [DOI] [PubMed] [Google Scholar]
- Chamorro A, Hallenbeck J. The harms and benefits of inflammatory and immune responses in vascular disease. Stroke. 2006;37(2):291–293. doi: 10.1161/01.STR.0000200561.69611.f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang YJ, Hemann MT, Hathcock KS, Tessarollo L, Feigenbaum L, Hahn WC, Hodes RJ. Expression of telomerase RNA template, but not telomerase reverse transcriptase, is limiting for telomere length maintenance in vivo. Mol Cell Biol. 2004;24(16):7024–7031. doi: 10.1128/MCB.24.16.7024-7031.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba H, Osanai M, Murata M, Kojima T, Sawada N. Transmembrane proteins of tight junctions. Biochim Biophys Acta. 2008;1778(3):588–600. doi: 10.1016/j.bbamem.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Connolly ES, Jr, Winfree CJ, Stern DM, Solomon RA, Pinsky DJ. Procedural and strain-related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery. 1996;38(3):523–531. doi: 10.1097/00006123-199603000-00021. discussion 532. [DOI] [PubMed] [Google Scholar]
- Fischer S, Wiesnet M, Renz D, Schaper W. H2O2 induces paracellular permeability of porcine brain-derived microvascular endothelial cells by activation of the p44/42 MAP kinase pathway. Eur J Cell Biol. 2005;84(7):687–697. doi: 10.1016/j.ejcb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Fischer S, Wobben M, Marti HH, Renz D, Schaper W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res. 2002;63(1):70–80. doi: 10.1006/mvre.2001.2367. [DOI] [PubMed] [Google Scholar]
- Frijns CJ, Kappelle LJ. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke. 2002;33(8):2115–2122. doi: 10.1161/01.str.0000021902.33129.69. [DOI] [PubMed] [Google Scholar]
- Fu W, Lee J, Guo Z, Mattson MP. Seizures and tissue injury induce telomerase in hippocampal microglial cells. Exp Neurol. 2002;178(2):294–300. doi: 10.1006/exnr.2002.8030. [DOI] [PubMed] [Google Scholar]
- Furumoto K, Inoue E, Nagao N, Hiyama E, Miwa N. Age-dependent telomere shortening is slowed down by enrichment of intracellular vitamin C via suppression of oxidative stress. Life Sci. 1998;63(11):935–948. doi: 10.1016/s0024-3205(98)00351-8. [DOI] [PubMed] [Google Scholar]
- Fuster JJ, Andres V. Telomere biology and cardiovascular disease. Circ Res. 2006;99(11):1167–1180. doi: 10.1161/01.RES.0000251281.00845.18. [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Allsopp RC, Chin L, Morin GB, DePinho RA. Expression of mouse telomerase reverse transcriptase during development, differentiation and proliferation. Oncogene. 1998;16(13):1723–1730. doi: 10.1038/sj.onc.1201933. [DOI] [PubMed] [Google Scholar]
- Haendeler J, Hoffmann J, Brandes RP, Zeiher AM, Dimmeler S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol Cell Biol. 2003;23(13):4598–4610. doi: 10.1128/MCB.23.13.4598-4610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haendeler J, Hoffmann J, Diehl JF, Vasa M, Spyridopoulos I, Zeiher AM, Dimmeler S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ Res. 2004;94(6):768–775. doi: 10.1161/01.RES.0000121104.05977.F3. [DOI] [PubMed] [Google Scholar]
- Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol. 2005;78(6):1223–1232. doi: 10.1189/jlb.0605340. [DOI] [PubMed] [Google Scholar]
- Harley CB. Telomerase therapeutics for degenerative diseases. Curr Mol Med. 2005;5(2):205–211. doi: 10.2174/1566524053586671. [DOI] [PubMed] [Google Scholar]
- Kago T, Takagi N, Date I, Takenaga Y, Takagi K, Takeo S. Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun. 2006;339(4):1197–1203. doi: 10.1016/j.bbrc.2005.11.133. [DOI] [PubMed] [Google Scholar]
- Kamencic H, Lyon A, Paterson PG, Juurlink BH. Monochlorobimane fluorometric method to measure tissue glutathione. Anal Biochem. 2000;286(1):35–37. doi: 10.1006/abio.2000.4765. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Choi YS, Hong SB, Kim KW, Woo RS, Won SJ, Kim EJ, Jeon HK, Jo SY, Kim TK, Bachoo R, Reynolds IJ, Gwag BJ, Lee HW. Ectopic expression of the catalytic subunit of telomerase protects against brain injury resulting from ischemia and NMDA-induced neurotoxicity. J Neurosci. 2004;24(6):1280–1287. doi: 10.1523/JNEUROSCI.4082-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Bae YH, Bae SK, Choi KS, Yoon KH, Koo TH, Jang HO, Yun I, Kim KW, Kwon YG, Yoo MA, Bae MK. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim Biophys Acta. 2008;1783(5):886–895. doi: 10.1016/j.bbamcr.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Klapper W, Shin T, Mattson MP. Differential regulation of telomerase activity and TERT expression during brain development in mice. J Neurosci Res. 2001;64(3):252–260. doi: 10.1002/jnr.1073. [DOI] [PubMed] [Google Scholar]
- Kniesel U, Wolburg H. Tight junctions of the blood-brain barrier. Cell Mol Neurobiol. 2000;20(1):57–76. doi: 10.1023/A:1006995910836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz DJ, Decary S, Hong Y, Trivier E, Akhmedov A, Erusalimsky JD. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci. 2004;117(Pt 11):2417–2426. doi: 10.1242/jcs.01097. [DOI] [PubMed] [Google Scholar]
- Lee HS, Namkoong K, Kim DH, Kim KJ, Cheong YH, Kim SS, Lee WB, Kim KY. Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvasc Res. 2004a;68(3):231–238. doi: 10.1016/j.mvr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Lee YW, Eum SY, Chen KC, Hennig B, Toborek M. Gene expression profile in interleukin-4-stimulated human vascular endothelial cells. Mol Med. 2004b;10(1–6):19–27. doi: 10.2119/2004-00024.lee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YW, Eum SY, Nath A, Toborek M. Estrogen-mediated protection against HIV Tat protein-induced inflammatory pathways in human vascular endothelial cells. Cardiovasc Res. 2004c;63(1):139–148. doi: 10.1016/j.cardiores.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24(1):117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao PS. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res. 2001;89(9):793–798. doi: 10.1161/hh2101.098443. [DOI] [PubMed] [Google Scholar]
- Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, Bennett M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99(2):156–164. doi: 10.1161/01.RES.0000233315.38086.bc. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Klapper W. Emerging roles for telomerase in neuronal development and apoptosis. J Neurosci Res. 2001;63(1):1–9. doi: 10.1002/1097-4547(20010101)63:1<1::AID-JNR1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Minamino T, Mitsialis SA, Kourembanas S. Hypoxia extends the life span of vascular smooth muscle cells through telomerase activation. Mol Cell Biol. 2001;21(10):3336–3342. doi: 10.1128/MCB.21.10.3336-3342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Taffet GE, Youker KA, Entman ML, Overbeek PA, Michael LH, Schneider MD. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc Natl Acad Sci U S A. 2001;98(18):10308–10313. doi: 10.1073/pnas.191169098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty MA, Wettstein JG. Elements of cerebral microvascular ischaemia. Brain Res Brain Res Rev. 2001;36(1):23–34. doi: 10.1016/s0165-0173(01)00062-5. [DOI] [PubMed] [Google Scholar]
- Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43(4):348–364. doi: 10.1080/10715760902751902. [DOI] [PubMed] [Google Scholar]
- Rao RK, Basuroy S, Rao VU, Karnaky KJ, Jr, Gupta A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J. 2002;368(Pt 2):471–481. doi: 10.1042/BJ20011804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev. 1994;74(1–2):121–133. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Rosengren L, Blomstrand C, Wikkelso C, Jensen C, Ekholm S, Tarkowski A. Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin Exp Immunol. 1997;110(3):492–499. doi: 10.1046/j.1365-2249.1997.4621483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184(1–2):53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauquier A, Melis W, Janssen PA. Long-term neurological assessment of the post-resuscitative effects of flunarizine, verapamil and nimodipine in a new model of global complete ischaemia. Neuropharmacology. 1989;28(8):837–846. doi: 10.1016/0028-3908(89)90176-7. [DOI] [PubMed] [Google Scholar]
- Williams IA, Allen DG. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2007;293(3):H1969–H1977. doi: 10.1152/ajpheart.00489.2007. [DOI] [PubMed] [Google Scholar]
- Wong D, Prameya R, Dorovini-Zis K. Adhesion and migration of polymorphonuclear leukocytes across human brain microvessel endothelial cells are differentially regulated by endothelial cell adhesion molecules and modulate monolayer permeability. J Neuroimmunol. 2007;184(1–2):136–148. doi: 10.1016/j.jneuroim.2006.12.003. [DOI] [PubMed] [Google Scholar]