


Abstract
Homologous recombination (HR) plays a vital role in DNA metabolic processes including meiosis, DNA repair, DNA replication and rDNA homeostasis. HR defects can lead to pathological outcomes, including genetic diseases and cancer. Recent studies suggest that the post-translational modification by the small ubiquitin-like modifier (SUMO) protein plays an important role in mitotic and meiotic recombination. However, the precise role of SUMOylation during recombination is still unclear. Here, we characterize the effect of SUMOylation on the biochemical properties of the Saccharomyces cerevisiae recombination mediator protein Rad52. Interestingly, Rad52 SUMOylation is enhanced by single-stranded DNA, and we show that SUMOylation of Rad52 also inhibits its DNA binding and annealing activities. The biochemical effects of SUMO modification in vitro are accompanied by a shorter duration of spontaneous Rad52 foci in vivo and a shift in spontaneous mitotic recombination from single-strand annealing to gene conversion events in the SUMO-deficient Rad52 mutants. Taken together, our results highlight the importance of Rad52 SUMOylation as part of a ‘quality control’ mechanism regulating the efficiency of recombination and DNA repair.
INTRODUCTION
DNA double-strand breaks (DSBs) are one of the most toxic kinds of chromosome damage that are implicated in cancer and many other diseases. A single unrepaired break can lead to aneuploidy, genetic aberrations or cell death. DSBs are caused by a vast number of endogenous and exogenous agents including genotoxic chemicals or ionizing radiation, as well as through replication of a damaged DNA template or replication fork collapse. In the yeast Saccharomyces cerevisiae, homologous recombination (HR) is the major pathway for repair of DSBs (1).
Genes of the RAD52 epistasis group mediate HR. The members of this gene group were identified by mutants’ sensitivity to ionizing radiation or their deficiency in DSB repair, and they include RAD50, RAD51, RAD52, RAD54, RAD55, RAD57, RAD59, RDH54/TID1, MRE11 and XRS2. These genes are also needed for HR and chromosome segregation in meiosis I and are in general highly conserved among eukaryotes (2,3).
HR involves DNA homology search and DNA strand invasion to form a joint between the recombining DNA molecules. These reaction steps are mediated by the Rad51 recombinase, an orthologue of the Escherichia coli RecA recombinase (4). In the presence of ATP, Rad51 binds single-stranded DNA (ssDNA) and forms a right-handed filament called the presynaptic filament. Since the assembly of the presynaptic filament proceeds rather slowly, it is prone to interference by other competing factors such as replication protein A (RPA) (5,6). RPA is a heterotrimeric ssDNA-binding protein with a complex role in HR. In vitro, it was shown that RPA stimulates recombination by eliminating secondary structures in ssDNA and stabilizing the displaced strand of the D-loop. However, due to its very high affinity for ssDNA, it can prevent Rad51 from binding ssDNA, thus leading to the inhibition of presynaptic filament formation (7). This inhibitory effect can be overcome by the recombination mediator activity of Rad52 (1).
Rad52 multimerizes and forms a ring structure (8). The assembly of a ring structure requires sequences in the evolutionary conserved N-terminal domain of Rad52, which also possesses a DNA binding activity with a preference for ssDNA (9). Yeast Rad52 can interact with Rad51 as well as with RPA to help ensure the efficient displacement of RPA from ssDNA by Rad51 (10–12). The interaction with RPA is mediated by a domain within the middle portion of Rad52, while the carboxyl terminus harbours the Rad51 interaction domain (13,14). Moreover, the C-terminal domain also possesses a DNA binding activity and alone has recombination mediator activity (5). A robust DNA annealing activity has been described for Rad52 as well (9).
Many proteins participating in HR can be post-translationally modified by the small ubiquitin-like modifier (SUMO) protein, and this modification can potentially influence the biochemical activity and function of the target protein. Recently, Sacher et al. (15) identified the residues that serve as SUMO acceptor sites in Rad52. In addition to the SUMO-conjugating enzyme Ubc9, Rad52 modification is stimulated by the SUMO ligase Siz2 (15). SUMO was shown to protect Rad52 from proteasomal degradation and to facilitate the exclusion of Rad52 recombination foci from the nucleolus to maintain a low level of recombinational repair at the ribosomal gene locus (16).
Here, we show that the SUMOylation of Rad52 is significantly stimulated in the presence of ssDNA in vitro. We find that enhanced SUMOylation is mediated via the C-terminal DNA binding domain of Rad52 and requires lysine K253. Moreover, we demonstrate that SUMOylated Rad52 exhibits lower affinity towards ssDNA and dsDNA, and also has reduced single-strand annealing activity. These data provide mechanistic information regarding the role of SUMOylation in the regulation of the biochemical activities of Rad52.
MATERIALS AND METHODS
Genetic methods, yeast strains and plasmids
Yeast strains were manipulated using standard genetic techniques, and media were prepared as previously described (17). All strains used in this study are RAD5 derivatives of W303 (Supplementary Table S1) (18,19). To generate the (His)6-RAD52(N + M)::pET11d plasmid, the (His)6-RAD52::pET11d plasmid was digested with BamHI. The resulting DNA product was isolated from agarose gel, ligated and sequenced. The plasmids expressing the rad52 (K43,44R), rad52 (K253R) and rad52 (K43,44,253R) mutants were constructed by site-directed mutagenesis using specific primers (Supplementary Table S2). The amino acid numbering corresponds to translation from first ATG of GenBank accession number CAA86623 for Rad52 protein.
DNA substrates
Oligonucleotides were purchased from VBC Biotech. The sequences of the oligonucleotides are shown in Supplementary Table S2. Oligo-1 and Oligo-3 were modified with fluorescein at the 5′ end. The DNA substrates were prepared by mixing an equimolar amount of the constituent oligonucleotides in the hybridization buffer H (50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 10 mM MgCl2), heated at 90°C for 3 min, and cooled slowly to room temperature to allow DNA annealing. The annealed DNA substrates were purified by fractionation in a 1-ml Mono Q column (GE Healthcare Life Sciences) with a 20-ml gradient of 50–1000 mM NaCl in 10 mM Tris–HCl, pH 7.5. Peak fractions were filtered, dialysed into 50 mM Tris–HCl, pH 7.5, containing 5 mM MgCl2 and concentrated in a Vivaspin concentrator with a 5 kDa cutoff. The concentration of the DNA substrates was determined by absorbance measurement at 260 nm.
Recombinant protein expression and purification
The various Rad52 species were expressed and purified with modifications as described (5). Rad51 and RPA proteins were purified as described (4,20).
Purification of Aos1/Uba2 complex (E1 protein)
The Escherichia coli strain BL21(DE3) transformed with plasmid for expression of E1 (Aos1/Uba2 complex) protein (a kind gift from Dr B. Schulman) containing GST-tagged Aos1 was induced with 0.5 mM IPTG for 3 h at 37°C. The cells were pelleted and stored at −80°C. The cell pellet (7 g) was resuspended in 25 ml of cell breakage buffer (CBB) (50 mM Tris–HCl, pH 7.5, 10% sucrose, 2 mM EDTA) containing 150 mM KCl, 0.01% NP40, 1 mM β-mercaptoethanol, sonicated and centrifuged (100 000g, 90 min). Clarified supernatant was loaded onto 7-ml SP-sepharose column with its outlet connected to a 7-ml Q-sepharose column (Amersham Biosciences). Both columns were pre-equilibrated with buffer K (20 mM K2HPO4, 10% glycerol, 0.5 mM EDTA) containing 100 mM KCl. The Q-sepharose column was subsequently eluted with a 70-ml gradient of 100–800 mM KCl in buffer K. Peak E1 protein fractions eluting around 330–470 mM KCl were pooled and mixed with 800 µl of GTH-sepharose (Amersham Biosciences) washed in buffer K containing 100 mM KCl for 1 h at 4°C. The GTH-sepharose with bound proteins was washed with 8 ml of 100 mM KCl in buffer K and eluted in steps with 800 µl of 10, 50, 100 or 200 mM glutathione (GTH) in buffer K containing 100 mM KCl. The peak fractions eluting within the range of 10–100 mM GTH were loaded onto a 0.5-ml Mono Q column followed by elution using 5-ml gradient of 100–700 mM KCl in buffer K. The main fractions of E1 protein eluting from the Mono Q column at ∼560 mM KCl were concentrated in a Centricon-30 to 5 µg/µl and stored in small aliquots at −80°C.
Purification of Ubc9 (E2 protein)
The plasmid (a kind gift from Dr Erica Johnson) expressing E2 (Ubc9) protein containing (His)6-affinity tag was introduced into E. coli strain BL21(DE3). Overnight culture grown at 37°C in 2×TY medium was diluted 100-fold into fresh 2×TY medium and incubated at 37°C. The overexpression of E2 was induced by addition of 0.1 mM IPTG followed by an incubation at 25°C overnight. To prepare cell extract, 13g of cell pellet was thawed in 20 ml of CBB containing 150 mM KCl, 0.01% NP40, 1 mM β-mercaptoethanol and protease inhibitor cocktail (pepstatin, leupeptin, aprotinin, chymostatin, benzamidine and phenylmethylsulfonyl fluoride). The cells were disrupted by sonication and the crude extract was clarified by centrifugation (100 000g, 4°C, 90 min). The supernatant was passed through a 7-ml column of Q-sepharose and the flow-through fraction was applied directly to a 7-ml SP-sepharose column that was eluted with 70-ml gradient of 100–750 mM KCl in buffer K. E2 protein eluted from SP-column at 290–370 mM KCl. The peak fractions were mixed with 750 µl of Ni–NTA agarose (Qiagen) for 75 min at 4°C. The beads were washed with 7.5 ml of buffer K containing 100 mM KCl followed by a 7.5 ml wash with buffer K containing 100 mM KCl and 10 mM imidazole. E2 protein was then eluted with buffer K containing 100 mM KCl and 50, 150 or 300 mM imidazole, respectively. The fractions eluted from Ni–NTA agarose column from 50 to 150 mM imidazole were pooled, applied onto a 1-ml Mono S column, and eluted using a 10-ml gradient of 100–750 mM KCl in buffer K. E2 protein eluted at 360–510 mM KCl was concentrated to 20 µg/µl in a Centricon-30 and stored in small aliquots at −80°C.
Purification of SUMO protein
The plasmid containing the SUMO (Smt3) gene containing both (His)6-affinity and FLAG tags (a kind gift from Dr Erica Johnson) was introduced into E. coli strain BL21(DE3). Overnight culture grown in 2×TY medium at 37°C was diluted 100-fold into fresh medium and incubated at 37°C until OD600 reached 0.8. At that time, IPTG was added to 0.5 mM, and the culture was incubated at 37°C for 3 h. The cells were harvested by centrifugation and stored at −80°C. Extract from 5 g of cell paste was prepared by sonication in 25 ml of CBB containing 150 mM KCl, 0.01% NP40, 1 mM β-mercaptoethanol and protease inhibitor cocktail. The lysate was clarified by ultracentrifugation and the resulting supernatant was passed in tandem over SP- and Q-sepharose columns (7 ml each). The Q-column was then eluted with a 70-ml gradient of 100–750 mM KCl in buffer K. The SUMO-containing fractions (370–470 mM KCl) were pooled and mixed with 600 µl Ni–NTA agarose. The bead-bound proteins were washed with 6 ml of buffer K containing 100 mM KCl, followed by 6-ml of buffer K containing 100 mM KCl and 10 mM imidazole. The bound proteins were eluted with buffer K containing 50, 150, or 300 mM imidazole and 100 mM KCl. The fractions with SUMO protein eluting from 50 to 300 mM imidazole were pooled and loaded onto a 0.5-ml Mono Q column and then eluted with a 5-ml gradient of 100–900 mM KCl in buffer K. The peak fractions (400–700 mM KCl) were pooled, concentrated in a Centricon-30 to 10 µg/µl and stored at −80°C.
In vitro SUMOylation assay
The assay was performed in a 20 µl reaction volume containing 5.6 µM of purified SUMO protein (Smt3), 400 nM E1 protein (Aos1/Uba2), 2.8 µM E2 protein (Ubc9), 2.5 mM ATP, buffer S (50 mM HEPES, 100 mM NaCl, 10 mM MgCl2, 0.1 mM DTT) and 2.7–4.1 µM of various Rad52 proteins. Reactions were incubated at 30°C for 3 h, stopped by addition of 30 µl of SDS Laemmli buffer (62.5 mM Tris–HCl, 2% SDS, 5% β-mercaptoethanol, 10% glycerol, 0.002% bromphenol blue) and analysed by SDS–PAGE and western blotting. The quantification of Rad52 SUMOylation was done using Quantity One software (Bio-Rad) and is presented as a ratio of mono-SUMOylated versus non-modified Rad52.
Circular dichroism spectroscopy
Circular dichroism (CD) spectra were recorded at 10°C using a Jasco J-810 spectrometer (Jasco, Tokyo, Japan) coupled with Peltier temperature controller. Data were collected from 195 to 340 nm, at 100 nm/min, 1 s response time and 2 nm bandwidth using a 0.1 cm quartz cuvette containing studied protein/DNA in 20 mM phosphate buffer and 50 mM KCl (pH 7.5). CD difference spectrum was calculated by subtracting spectrum of DNA from that of protein–DNA complex. Each spectrum represents an average of 10 individual scans and is corrected for absorbance caused by the buffer. CD data were expressed in millidegrees (instrument units of CD).
Electrophoretic mobility shift assay
Indicated amounts of various forms of Rad52 protein were incubated with fluorescently-labelled DNA substrates (0.44 µM nucleotides) at 37°C in 10 µl of buffer B (50 mM Tris–HCl, pH 7.8, 5 mM MgCl2 and 1 mM DTT) for 10 min. After the addition of gel loading buffer (60% glycerol, 10 mM Tris–HCl, pH 7.4 and 60 mM EDTA), the reaction mixtures were resolved in 7.5% native polyacrylamide gels in TBE buffer (40 mM Tris–HCl, 20 mM boric acid, 2 mM EDTA, pH 7.5) at 4°C, and the DNA species were quantified using Quantity One software (Bio-Rad).
Single-strand annealing assay
The assay was essentially done as described previously (14). Oligo-2 and fluorescently labelled Oligo-1 (0.26 µM nucleotides) were incubated in separate tubes at 37°C for 3 min in the absence or presence of RPA (20 nM) in 20 µl of buffer D (40 mM Tris–HCl, pH 7.5, 50 mM KCl, 1 mM DTT and 100 µg/ml bovine serum albumin (BSA)). Varying amounts of Rad52 or Smt3-Rad52 were added to the tube containing Oligo-2 and then mixed with Oligo-1. The completed reactions were incubated at 37°C for 8 min. At the indicated times, 9 µl of the annealing reactions was removed and treated with 0.5% SDS, and 500 µg/ml proteinase K at 37°C for 10 min in a total volume of 10 µl. The individual samples were resolved in 12% native polyacrylamide gels run in TBE buffer. DNA annealing was quantified as the portion of the fluorescently-labelled Oligo-1 that had been converted into the double-stranded form.
Binding of Rad52 to Rad51 and RPA immobilized on Affi-gels
Affi-gel 15 beads containing either Rad51 (Affi-Rad51; 300 ng/µl) or RPA (Affi-RPA; 350 ng/µl) were prepared as described previously (21). Purified Rad52 or Smt3-Rad52 were mixed with 30 µl of Affi-Rad51 or Affi-RPA in 25 µl of buffer T (20 mM Tris–HCl, pH 7.5, 150 mM KCl, 1 mM DTT, 0.5 mM EDTA and 0.01% NP40) for 30 min at 4°C. The beads were washed twice with 100 µl of buffer T before being treated with 30 µl of SDS Laemmli buffer to elute bound proteins. The supernatant that contained unbound Rad52 or Smt3-Rad52 protein, and the SDS eluate (10 µl each) were analysed by SDS–PAGE in 10% gel.
Gel filtration
A Sephacryl S400 column (25 ml total) was used to monitor the oligomeric status of Rad52 and SUMOylated Rad52 proteins. The proteins were filtered through the sizing column at 0.1 ml/min in buffer K containing 200 mM KCl, collecting 0.5 ml fractions. The indicated column fractions were separated by SDS–PAGE electrophoresis to determine contents of the Rad52, or SUMOylated Rad52 proteins, respectively.
Determination of mitotic recombination rates
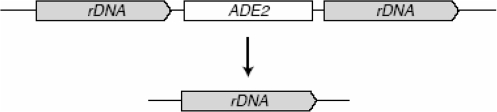
Interchromosomal and direct-repeat recombination between non-functional leu2 heteroalleles was measured as described (22), except that the single colonies were inoculated into liquid SC medium containing 100 µg/ml adenine. The plating efficiency and the number of recombinants were determined by plating an appropriate number of cells on SC and SC-Leu medium. After 3 days at 30°C, the number of colonies was counted. The Leu+ colonies were replica-plated to SC-Leu-Ura in order to distinguish between gene conversion (Ura+) and single-strand annealing (Ura−) events in the direct-repeat assay. For each strain, 15–19 independent trials were carried out. The corresponding recombination rates and their standard deviations were calculated according to the median method (23). The rate of ADE2 marker loss at the rDNA was determined as described (24). In brief, single colonies were grown overnight in liquid YPD medium at 30°C. Then, cultures grown to exponential phase were diluted before plating an appropriate number of cells on YPD. After 3 days at 30°C, the number of half-sectored red–white colonies was counted as a direct measure of the rDNA recombination rate.
Analysis of meiotic recombination
Sporulation was induced by replica-plating of the fresh patch of cells from solid YPD to SPO medium and followed by incubation at 30°C for 3 days. Next, cells were resuspended in H2O and inspected microscopically to determine the sporulation efficiency. For each strain, 18 four-spore tetrads were dissected to determine the germination efficiency. To determine the meiotic recombination frequency and relative viability, OD600 was measured and cells were sonicated before plating an appropriate number of cells on SC and SC-Leu medium.
Analysis of inverted-repeat substrate recombination and break-induced replication
Recombination between inverted repeats was assayed using the ade2-5′Δ::TRP1::ade2-n system (provided by L. Symington, Columbia University, New York, NY, USA). The experiments were essentially performed as described (23). A chromosome fragmentation assay was used to measure the efficiency of break-induced replication (BIR) in Rad52 SUMOylation-defective mutant. The experiments were performed as previously described (25).
Microscopy
Cells were processed for fluorescence microscopy as described previously (26). Yellow fluorescent protein (YFP) was visualized using a band-pass YFP (Cat. no. 41028) filter set from Chroma (Brattleboro, VT, USA). Images were acquired with a 10% neutral density filter in place to reduce photobleaching and phototoxicity. Fluorescence intensities were measured in arbitrary units (AU) using Volocity software (Improvision).
Results
Enhancement of Rad52 SUMOylation by ssDNA
Previous studies have shown that Rad52 protein is SUMOylated at lysines K43, K44 and K253 in response to DNA damage [Figure 1A, (15)]. To define the biochemical requirements of this reaction, we have established an in vitro SUMOylation assay using purified yeast Ubc9, Aos1, Uba2 and Smt3 (SUMO in yeast) proteins (Figure 1B and Supplementary Figure S3). Addition of Rad52 protein to this reaction resulted in its modification (Figure 2A). We have also confirmed that lysines K43, 44 and 253 are the major in vitro SUMO conjugation sites as observed in vivo [(15), Supplementary Table S3)]. Since Rad52 is known to bind both ssDNA and dsDNA, we decided to assess the effect of DNA on Rad52 SUMOylation. Importantly, when the SUMOylation of Rad52 was performed in the presence of ssDNA cofactor, we noticed a significant stimulation of the SUMO modification (∼3-fold, Figure 2A). That Rad52 was modified by SUMO conjugation was confirmed by immunoblotting with anti–Smt3 and anti–Rad52 antibodies (Figure 2B and C). In contrast, the addition of dsDNA did not enhance the level of SUMOylated Rad52 species (Figure 2A), suggesting that only ssDNA binding confers this stimulation. Interestingly, experiments with titration of ssDNA showed that stimulation of Rad52 SUMOylation was optimal when the ratio of Rad52 to nucleotides reached approximate ratio 1:37 (Figure 2D and E). This is in good correlation with previously published data indicating that Rad52 protein binds ∼36 nt of ssDNA (27). Maximum stimulation corresponded up to 25% of modified Rad52 protein. Altogether these data show that: (i) Rad52 can be SUMOylated in vitro, (ii) the pattern of SUMOylation is identical to that observed in vivo and (iii) Rad52 binding to ssDNA stimulates its SUMOylation.
Figure 1.

Purification of Rad52 species and SUMO machinery proteins. (A) Schematic representation of functional domain within S. cerevisiae Rad52 protein, including the positions of known SUMO acceptor lysines. The RPA interaction domain (black box, residues 308–311) and Rad51 interaction domain (grey box, residues 409–412) are indicated. (B) Purified SUMO machinery proteins: GST-tagged Aos1/Uba2 heterodimer (1 μg), (His)6-tagged Ubc9 and Smt3 proteins (1 μg each) were run on a 15% SDS–PAGE and stained with Coomassie blue. (C) Purified (His)6-tagged Rad52 species (1 μg each): Rad52, Rad52 (K43,44R), Rad52 (K253R) and Rad52 (K43,44,253R) were run on a 10% SDS–PAGE and stained with Coomassie blue.
Figure 2.

In vitro SUMOylation of Rad52 is stimulated by ssDNA. (A) The effect of ssDNA and dsDNA on Rad52 SUMOylation. In vitro SUMOylation assays were performed with recombinant Aos1/Uba2 (400 nM), Ubc9 (2.8 μM) and Smt3 (5.6 μM) in the presence or absence of ATP (2.5 mM), Rad52 (2.7 μM), 83-mer ss-DNA (100 μM nucleotides) or dsDNA as indicated. The reaction mixtures were incubated for 3 h at 30°C, stopped by adding 30 μl of SDS Laemmli buffer and analysed on 7% SDS–PAGE followed by silver staining. The hash symbol indicates high molecular poly-SUMOylated species of Rad52 and E1. The number at the bottom of the gel represents the ratio of modified and unmodified Rad52. SUMOylated Rad52 protein was confirmed by western blotting using anti-Smt3 (B) or anti-Rad52 (C) antibodies. (D) In vitro SUMOylation assay on Rad52 protein carried out without DNA (lanes 1 and 2) or with increasing amount of 83-mer ssDNA (15, 50, 100, 150, 300 μM nucleotides; lanes 3–7). The hash symbol indicates high molecular weight SUMOylated species. (E) Graphical representation of the gel in panel D, as a ratio of mono-SUMOylated versus non-modified Rad52. (F) The CD spectrum of 83-mer ssDNA (black), Rad52 (red) and the Rad52-ssDNA complex (white). The difference spectrum (green) represents values for Rad52 in the complex with DNA from which the spectrum of DNA has been subtracted. To form the complex, protein and DNA were incubated together for 10 min at 37°C prior to the analysis. The concentration of the protein was 3.87 μM and the concentration of DNA was 2.24 μM. The samples were measured at 10°C in 20 mM phosphate buffer containing 50 mM KCl, pH 7.5.
Binding of ssDNA causes a conformational change in Rad52
Stimulation of Rad52 SUMOylation by ssDNA prompted us to test whether ssDNA binding could cause conformational changes in the Rad52 protein. Therefore, we measured and compared CD spectrum of Rad52 alone and difference spectrum of Rad52 in the presence of DNA (Figure 2F; red and green squares, respectively). The results provided evidence that interaction of Rad52 protein with ssDNA induced a conformational change in the protein. This conformational change takes place without any alteration in the structure of the DNA, since the DNA spectrum did not change above 240 nm. We attribute the increased magnitude of the negative bands to an increase in the α-helicity of Rad52 (28). In summary, binding of ssDNA results in a conformational change within Rad52 equivalent to an increase in the α-helicity.
ssDNA bound by RPA but not by Rad51 can stimulate SUMOylation of Rad52
The ssDNA generated during DSB repair is often protected by interaction with other ssDNA binding proteins, namely RPA and Rad51. Therefore, we tested if RPA- or Rad51-bound ssDNA is still accessible for interaction with Rad52 and whether it enhances Rad52 SUMOylation. We pre-incubated ssDNA with increasing amounts of RPA protein and examined the effect of RPA on Rad52 SUMOylation in vitro. Under the experimental conditions, RPA bound efficiently to ssDNA (Supplementary Figure S4) with stoichiometry of one RPA trimer per up to 30 nt. As shown in Figure 3A and B, the level of stimulation was similar to that without RPA, suggesting that ssDNA coated with RPA can enhance Rad52 SUMOylation. Even with a 2-fold molar excess of RPA over the ssDNA (Figure 3A, lane 8), stimulation of Rad52 SUMOylation is comparable to that observed with ssDNA (Figure 3A, lane 3). Next, we pre-incubated ssDNA with increasing amounts of Rad51 protein to form presynaptic filaments. Notably, this DNA was no longer accessible for Rad52 and resulted in decreased Rad52 SUMOylation to the basal level observed in the absence of ssDNA (Figure 3C and D). Specifically, the maximum inhibition of Rad52 SUMOylation occurred at 3 nt of ssDNA per Rad51 monomer, corresponding to the ssDNA binding site size of Rad51 and ensuring fully coated DNA (20). Further increasing the Rad51 amount did not have any additional effect on Rad52 SUMOylation (Figure 3C, lane 10 and data not shown). Taken together, these data indicate that ssDNA bound by RPA is fully accessible for binding by Rad52, whereas the DNA in a Rad51 nucleoprotein filament is not.
Figure 3.

Effect of accessibility of RPA- or Rad51-coated ssDNA on Rad52 SUMOylation. (A) RPA bound to ssDNA does not affect Rad52 SUMOylation. Increasing amounts of RPA protein (1.2, 3.5, 4.6, 5.9 and 8.3 μM) were pre-incubated with 83-mer ssDNA (100 μM nucleotides) for 10 min at 37°C and then mixed with E1 (400 nM), E2 (2.8 μM), Smt3 (5.6 μM), Rad52 (2.7 μM) and 2.5 mM ATP. After 3 h incubation at 30°C, the reactions were analysed by 7.5% SDS–PAGE and silver-stained. (B) The ratio of mono-SUMOylated versus non-modified Rad52 is presented by quantifying the corresponding bands in (A). (C) Rad51-coated ssDNA does not stimulate Rad52 SUMOylation. The reactions were carried out as in (A) except increasing amounts of Rad51 were used (3, 7, 14, 21, 27, 34, 41 μM). The reaction mixtures were stopped and analysed by 10% SDS–PAGE followed by silver staining. (D) Quantification of Rad52-Smt3 conjugate from C, as a ratio of mono-SUMOylated versus non-modified Rad52. The arrow indicates amount of Rad51 that fully coats ssDNA. The hash symbol indicates high molecular poly-SUMO chains of Rad52 and E1 proteins.
The enhancement of Rad52 SUMOylation at residue K253 is mediated via the C-terminal DNA binding domain
Rad52 SUMOylation occurs at three lysine residues: K43, K44 and K253 (15), so we constructed and purified Rad52 mutant proteins lacking one or more of these SUMO-conjugation sites for the in vitro studies (Figure 1C). Interestingly, the Rad52 (K43,44R) mutant showed a pattern of SUMOylation similar to wild-type Rad52 both in the absence and presence of ssDNA (Figure 4A), suggesting that ssDNA-enhanced SUMOylation can occur at K253 alone. The other two Rad52 mutant proteins containing the K253R mutation [Rad52 (K253R) and Rad52 (K43,44,253R)] are strongly impaired for Rad52 SUMOylation (Figure 4A), indicating that residues K43 and K44 are modified as a consequence of SUMOylation at lysine 253 (Figure 4A). This is different to previously published data which could reflect the presence of E3 ligase in the in vivo situation (15). To help eliminate the possibility that the individual mutations could affect the protein folding, we tested all our mutant proteins for DNA binding and DNA strand annealing, and found no difference between wild-type and mutant proteins (Supplementary Figure S1).
Figure 4.

Binding of Rad52 to ssDNA via C-terminal domain enhances the SUMOylation of lysine K253. (A) SUMOylation at lysine K253 is stimulated by ssDNA. The standard in vitro SUMOylation reaction was done with wild-type Rad52 (2.7 μM) or SUMO-deficient acceptor lysine Rad52 mutants: Rad52 (K43,44R), Rad52 (K253R) and Rad52 (K43,44,253R). (B) The C-terminal DNA binding domain of Rad52 is responsible for stimulation of SUMOylation. Rad52 protein and its fragments: Rad52 (N + M) (4.13 μM), Rad52 (M) containing GST-tag (3.52 μM) and Rad52 (M + C) (4.03 μM) were SUMOylated in vitro in the presence or absence of 83-mer ssDNA (100 μM nucleotides) and analysed. The asterisks indicate main SUMOylated Rad52 species. The hash symbol indicates high molecular poly-SUMO chains of Rad52 and E1 proteins.
Rad52 was shown to harbour two DNA binding domains, one at the amino-terminus and the other at its carboxyl-terminus [Figure 1A, (5)]. We wished to examine which of these domains are responsible for enhancement of Rad52 SUMOylation. Since this stimulation is mediated via K253, located at the middle region of Rad52, we checked the SUMOylation status and its enhancement after ssDNA binding using three Rad52 fragments that harbour either the amino-terminus and the middle portion of the protein (N + M), the middle portion only (M) or both the middle portion and carboxyl-terminus (M + C). Even though all three fragments can be SUMOylated in vitro, only the reaction involving Rad52 (M + C) was stimulated by ssDNA (Figure 4B), suggesting that the C-terminal binding region is responsible for the SUMOylation enhancement.
SUMOylation attenuates DNA binding and strand annealing
The effect of ssDNA on SUMOylation efficiency prompted us to test the biochemical properties of SUMOylated Rad52 protein. The oligomeric status of Rad52 prevents purification of the SUMOylated fraction from the in vitro SUMOylation reaction. Therefore, the SUMOylation reaction was performed in the presence or absence of ATP to ensure the same quantity of Rad52 for individual experiments, leading to up to 10% of modified Rad52. First, we tested both free and SUMOylated Rad52 for its ability to bind DNA using a well-established electrophoretic mobility shift assay. The SUMOylated Rad52 showed a significant decrease in DNA binding efficiency towards both ssDNA and dsDNA (Figure 5A and B, and data not shown). Next, we addressed whether the lower affinity towards DNA can also affect the annealing activity of Rad52 protein. Both unmodified and SUMOylated Rad52 were incubated with complementary ssDNA strands, and their annealing ability was monitored. Importantly, SUMOylated Rad52 showed a lower activity in the annealing reaction (Supplementary Figure S2). This has been further confirmed in the reaction when ssDNA is coated by RPA to reflect the in vivo conditions and to prevent the effect of possible DNA self-annealing (Figure 5C and D). No effect of components of the SUMOylation reaction or ATP alone on DNA binding or single-strand annealing activity was observed (data not shown). In summary, SUMOylation of as little as 10% of the total Rad52 protein is sufficient to significantly decrease Rad52 affinity towards both ssDNA and dsDNA and decrease single-strand annealing activity.
Figure 5.

Rad52 SUMOylation affects its biochemical activities. (A) SUMOylation of Rad52 inhibits its binding to DNA. Increasing amounts of Rad52 or Smt3-Rad52 (20, 40, 80, 200 nM) were incubated with fluorescently labelled 49-mer ssDNA (0.49 µM nucleotides) at 37°C for 10 min. The reaction mixtures were resolved in 7.5% native polyacrylamide gels, and the DNA species were quantified using Quantity One software (Bio-Rad). (B) The results from (A) plotted. (C) SUMOylation of Rad52 inhibits its strand annealing activity. Labelled Oligo-1 (0.25 µM nucleotides) and Oligo-2 (0.25 µM nucleotides) were incubated separately with RPA (20 nM) for 3 min at 37°C. The annealing reactions were initiated by mixing RPA-coated oligonucleotides and Rad52 or Smt3-Rad52 proteins (0.7, 5, 10, 20 nM) and incubated at 37°C. After 8 min of incubation, 9 µl of the annealing reactions was removed and treated with 0.5% SDS, and 500 µg/ml proteinase K at 37°C for 10 min. The reaction mixtures were resolved in 12% native polyacrylamide gels. (D) The averaged values of results from three independent experiments are plotted.
SUMOylation of Rad52 does not affect its interactions with Rad51 and RPA
Rad52 is known to self-associate (8) and interact with other HR proteins including Rad51 (10,11) and RPA proteins (12,29,30). We have tested the effect of Rad52 SUMOylation on these interactions. Since Smt3 protein in this experiment contains the FLAG tag, we first assayed the Rad52 oligomerization status on anti-FLAG agarose beads. As shown in Figure 6A, the ratio of modified versus unmodified Rad52 protein does not change after anti-FLAG pull-down. This was further confirmed by size exclusion chromatography (Sephacryl S400, see ‘Materials and Methods’ section), which showed that SUMOylation has little or no effect on the oligomeric state of Rad52 (Figure 6B). In summary, the oligomeric state of Rad52 is not affected upon SUMOylation. Moreover, SUMO conjugation occurs to the same degree among the individual Rad52 oligomers (Figure 6A and B). However, we cannot exclude the possibility that SUMOylated and unmodified subunits re-distributed during the SUMOylation and pull-down assays. Electron microscopy of unmodified and SUMOylated Rad52 also revealed no difference in the oligomeric status of Rad52 (data not shown).
Figure 6.

Rad52 oligomerization and interaction with RPA and Rad51 are not affected by its SUMOylation. (A) SUMOylation of Rad52 does not influence its oligomerization status. Rad52 and Smt3-Rad52 (2.3 µM) in 40 µl of buffer S were incubated with 4 µl of anti-FLAG agarose in 10 µl of buffer T containing 200 mM KCl for 30 min at 4°C. The beads were washed and treated with 25 µl of SDS Laemmli buffer to elute bound proteins. The supernatant (S) that contained unbound Rad52 or Smt3-Rad52 protein, wash (W) and the SDS eluate (E) (10 µl each) were analysed on 12% gel SDS–PAGE followed by staining with Coomassie Blue. The arrows and symbol hash denote anti-FLAG IgG and higher order SUMOylated species, respectively. (B) Analysis of Rad52 oligomerization status by gel filtration. Purified Rad52 or Smt3-Rad52 proteins (9 µM) in 200 µl of buffer S were filtered through a sephacryl S400 column. The indicated fractions were run on 10% SDS–PAGE followed by staining with Coomassie blue. The elution positions of the size markers are indicated: TG, thyroglobulin (669 kDa) and CAT, catalase (223 kDa). (C) SUMOylation of Rad52 does not affect its interaction with Rad51. Purified Rad52(M + C) or Smt3-Rad52(M + C) (22 µM) were mixed with Affi-Rad51 beads (4,6 µM Rad51) in 25 µl of buffer K and incubated for 30 min at 4°C. The beads were washed and treated with 25 µl of SDS Laemmli buffer to elute bound proteins. The supernatant that contained unbound Rad52 or Smt3-Rad52 protein, wash, and the SDS eluate (5 µl each) were analysed by SDS–PAGE in 12% gel followed by western blotting using anti-Rad52 antibody. (D) SUMOylation of Rad52 does not affect its interaction with RPA. The interaction with RPA was tested using purified Rad52 (M) or Smt3-Rad52 (M) (230 pmol) and Affi-RPA (1 µM RPA) beads pre-incubated with ssDNA (1 µg of ФX174) for 10 min at 37°C. Both mixtures were combined in 45 µl of buffer T followed by an incubation for 30 min at 4°C and then analysed as in panel C.
Next, we checked the ability of SUMOylated Rad52 to interact with Rad51. For this purpose, we generated Affi-Rad51 beads to use as affinity matrix for testing interaction with unmodified and modified Rad52. We used a Rad52 fragment without the N-terminal oligomerization domain [Rad52 (M + C)] to minimize its effect on the role of SUMOylation on Rad51 interaction. The result from this experiment showed SUMOylated Rad52 is just as capable of Rad51 association as the unmodified protein (Figure 6C). To test the interaction with RPA, we generated Affi-RPA beads and performed a pull-down assay as described previously (5). Rad52 (M) fragment missing the N- and C-terminal DNA binding domains was used to monitor its ability to interact with DNA-bound RPA (5). Little or no change could be observed between Rad52 (M) and SUMOylated Rad52 (M) for RPA association (Figure 6D). Taken together, the above results allowed us to conclude that SUMOylation does not affect Rad52 self-association or its interaction with Rad51 and RPA.
Rad52 SUMOylation regulates recombination in vivo
To address the effects of Rad52 SUMOylation in vivo, we examined the SUMO-deficient mutants (rad52-K43,44R, -K253R or -K43,44,253R) using several recombination assays. In agreement with previous work (15), we found that these mutations did not dramatically affect the rate of intra- or inter-chromosomal recombination during mitosis (Tables 1 and 2), but caused a shift from single-stranded annealing to gene conversion events (Table 1). In addition, these mutations resulted in slight hyper-recombination phenotype in rDNA recombination [Table 3, (16)].
Table 1.
Effect of rad52 SUMOylation-defective mutants on mitotic heteroallelic and direct-repeat recombination
| Genotype | Heteroallelic recombination rate (×10−8)a | Fold change relative to wild-typeb | Direct-repeat recombination (×10−6)a | Fold change relative to wild-type | P-value (direct-repeat)c | Fraction Ura+ GC | P-value (Ura+)d |
|---|---|---|---|---|---|---|---|
| RAD52 | 219 ± 29 | 1 | 51 ± 8 | 1 | n.a. | 0.33 | n.a. |
| rad52Δ | 0.7 ± 0.4 | 0.005 | 2.3 ± 0.5 | 0.03 | <0.001 | 0.0004 | <0.001 |
| K43,44R | 253 ± 48 | 1.2 | 107 ± 15 | 2.1 | 0.06 | 0.45 | <0.001 |
| K253R | 179 ± 30 | 0.8 | 70 ± 11 | 1.4 | 0.13 | 0.37 | 0.075 |
| K43,44,253R | 216 ± 36 | 1.0 | 74 ± 11 | 1.4 | 0.16 | 0.40 | <0.001 |
aRecombination rate is presented as events per cell per generation as described previously (37). For each strain, 15–19 trials were performed. Strains ML412 (wild-type), ML414 (rad52Δ), ML466 (rad52-K43,44R), ML467 (rad52-K253R), ML480 (rad52-K43,44,253R).
bThere is no significant difference in mitotic heteroallelic recombination between wild-type and the SUMOylation defective rad52 mutants.
cP-value for t-test applied to the direct-repeat recombination rate relative to wild-type.
dP-value for Pearson’s χ2 test relative to wild-type.
Schematic of the assays for spontaneous heteroallelic (i) and direct-repeat (ii) recombination between leu2-ΔEcoRI and leu2-ΔBstEII heteroalleles.
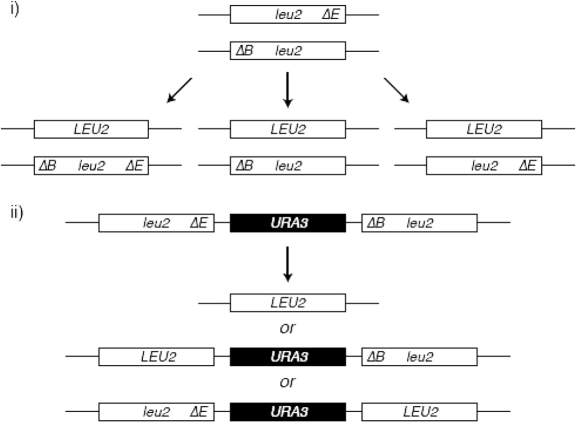
Table 2.
Effect of rad52-K43,44,253R on recombination rates of the inverted-repeat substratea
| Genotype | Rates (10−8)a | P-value (χ2)b | Fold change relative to wild-type |
|---|---|---|---|
| WT | 28 432 ± 4982 | n.a. | 1.00 |
| rad52Δ | 5 ± 1.4 | n.d | 0.0002 |
| rad51Δ | 5631 ± 1358 | n.a. | 0.20 |
| rad51Δ, rad52Δ | 0 | 0.002 | <0.00004 |
| rad51Δ, rad52K43,44,253R | 4986 ± 1617 | 0.31 | 0.18 |
aRates are per cell per generation as described previously (38). Results presented are a mean of 3–5 independent isolates for each genotype, out of a single representative experiments.
bThe χ2-test was applied to compare the recombination rates with that of rad51Δ strain.
Schematic of the assay for spontaneous inverted-repeat recombination between ade2-5′Δ and ade2-ΔNdeI heteroalleles.
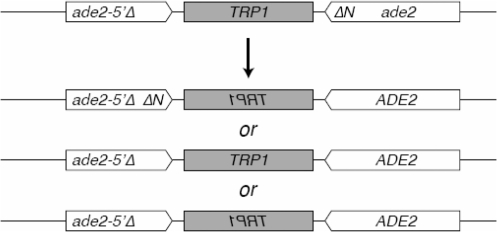
Table 3.
Effect of rad52 SUMOylation-defective mutants on rDNA recombination
| Genotype |
ADE2 lossa |
Fold change relative to wild-type | ||
|---|---|---|---|---|
| Rate of loss (×10−3) | P-valueb | Half-sectored/ total | ||
| RAD52 | 2.89 | n.a. | 112/38 700 | 1 |
| rad52Δ | 0.85 | <0.001 | 17/20 029 | 0.29 |
| K43,44R | 3.71 | 0.095 | 79/21 279 | 1.28 |
| K253R | 3.95 | 0.036 | 84/21 271 | 1.36 |
| K43,44,253R | 9.51 | <0.001 | 380/39 939 | 3.29 |
arDNA recombination in the first generation after plating was assayed by counting half-sectored colonies as described previously (24). Recombination was assayed in strains RMY180-5A (wild-type), NEB136-2B (rad52Δ), NEB187-10A (rad52-K43,44R), NEB168-11B (rad52-K253R), NEB63-2B (rad52-K43,44,253R).
bFisher’s exact test was applied to compare the recombination rate with that of the wild-type strain.
Schematic of the assay for spontaneous rDNA recombination resulting in ADE2 marker loss.
We also expanded the previous studies and examined the effect of rad52 SUMOylation mutants in additional mitotic and meiotic recombination assays. We found that rad52-K43,44,253R exhibited 60% of a wild-type level of BIR (Table 4), suggesting that Rad52 SUMOylation contributes positively to this type of recombination. In meiosis, although all three rad52 SUMO mutants exhibited wild-type levels of recombination at the leu2 locus (Table 5), rad52-K43,44,253R, but not rad52-K43,44R and rad52-K253R cells exhibited increased spore viability. This effect is not due to an alteration of the ability of these mutant proteins to bind ssDNA or dsDNA or to anneal complementary DNA, because Rad52-K43,44,253R, Rad52-K43,44R and Rad52-K253R proteins exhibit wild-type levels of activities in these reactions in vitro (Supplementary Figure S1). Thus, the effect on spore viability could be due to the fact that SUMOylation of Rad52 affects other meiotic events. Taken together, these results suggest that Rad52 SUMOylation facilitates certain recombination sub-pathways and disfavours the others and may contribute to meiotic processes that determine spore viability.
Table 4.
Effect of rad52-K43,44,253R mutation on the frequency of break-induced replicationa
| Genotype | Frequency of stable Ura+ (×10−2) with CFV/D8B-tg | P-value (χ2)b | Fold change relative to wild-type |
|---|---|---|---|
| RAD52 | 3.94 ± 0.75 | n.a. | 1 |
| rad52Δ | <0.0002 | n.d. | <0.0039 |
| K43,44,253R | 2.39 ± 0.44 | 0.0044 | 0.60 |
aThe frequency of break-induced replication is the number of stable Ura+ transformants per microgram with cut DNA divided by the number of transformants per microgram with uncut DNA transformed, as described previously (25). For each genotype, five to seven trials were performed.
bThe χ2-test was applied to compare the recombination rate with that of the wild-type strain.
Schematic of the assay for BIR resulting in a stable Ura+ recombinant strain with the CFV/D8B-tg fragment.
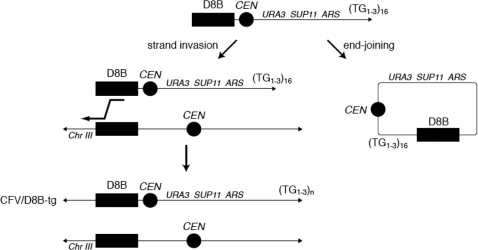
Table 5.
Effect of rad52 SUMOylation-defective mutants on meiotic recombination
| Genotype | Meiotic recombination frequency (×10−4)a | Fold change relative to wild-type | Sporulation efficiencyb, % | Germination efficiencyc, % | Relative viabilityd |
|---|---|---|---|---|---|
| RAD52 | 118 ± 31 | 1 | 41 | 96 | 1 |
| rad52Δ | 0.06 ± 0.06 | 0.001 | 1 | n.a. | 0.10 |
| K43,44R | 144 ± 38 | 1.2 | 58 | 69 | 0.95 |
| K253R | 77 ± 24 | 0.7 | 55 | 88 | 0.84 |
| K43,44,253R | 109 ± 27 | 0.9 | 53 | 96 | 2.39 |
aRecombination frequency is presented as events per colony forming unit. For each strain, three trials were performed. Strains ML412 (wild type), ML414 (rad52Δ), ML466 (rad52-K43,44R), ML467 (rad52-K253R), ML480 (rad52-K43,44,253R) were sporulated on solid medium at 30°C essentially as described previously (39). [See schematic in Table 1(i)].
bSporulation efficiency was determined as the fraction of three to four spore tetrads out of all cells after 3 days on solid SPO medium at 30°C.
cGermination efficiency was determined by tetrad dissection of four-spore tetrads after 3 days on solid SPO medium at 30°C.
dViability is the number of colony forming units per milliliter per OD600 after sporulated on solid medium at 30°C.
Finally, we also tested the in vivo localization of a Rad52 SUMO-deficient mutant by cytological analysis. The fusion of wild-type as well as the Rad52-K43,44,253R proteins to YFP revealed that the mutant protein forms foci similarly to wild-type, but the duration of the foci is significantly shorter (P = 0.01, t-test) (Figure 7A and B). The average intensity of the YFP signal for wild-type and the SUMO-deficient mutant foci is similar, suggesting that the difference in duration of foci is not due to a difference in the amount of proteins (data not shown).
Figure 7.

Rad52-K43,44,253R foci exhibits shorter duration than wild-type. Time-lapse of spontaneous wild-type Rad52 and mutant Rad52-K43,44,253R foci. Strains W5094-1C (RAD52-YFP) and ML228 (rad52-K43,44,253R-YFP) were examined by fluorescence microscopy at 5 min intervals as described in the ‘Materials and Methods’ section. (A) Representative examples of time-lapse image sequences. Arrowheads mark Rad52 foci. (B) Quantitative analysis of time-lapse analysis. The number of cell cycles analysed is n = 173 for wild-type Rad52 and n=252 for the Rad52-K43,44,253R mutant protein. The class of cells for which no foci were observed amounted to 62 and 63% for RAD52 and rad52-K43,44,253R, respectively (not shown in the graph).
DISCUSSION
HR is essential for genome maintenance and must be tightly controlled to avoid of loss of heterozygosity, chromosome translocations and gene deletions (1,3). Recent studies have documented several examples of HR proteins and associated factors being post-translationally modified by the SUMO protein. In particular, defects in SUMOylation of Rad52, Rad59 and RPA result in genomic context-dependent changes in HR and chromosomal rearrangements (31). Also, during S-phase, Srs2 (a helicase that blocks recombinational repair by disrupting Rad51 nucleoprotein filaments) is recruited to replication forks via interaction with SUMOylated PCNA (proliferating cell nuclear antigen), thereby preventing assembly of Rad51 nucleoprotein filaments on ssDNA associated with the replication forks (32).
In this study we have strived to determine the effects of SUMOylation on the Rad52 protein. Using in vitro biochemical assays, we have found that Rad52 is SUMOylated in a manner that is stimulated by ssDNA and dependent on the carboxyl-terminal DNA binding domain. The stimulation is specific for ssDNA, as dsDNA has no effect on the level of SUMOylation. In addition, the CD analysis revealed that binding of ssDNA by Rad52 is accompanied by a conformational change. By replacing each of the SUMOylation-targeted lysines with arginine, we observed that increasing SUMOylation in the presence of ssDNA is directed to residue K253. In addition, we have found that RPA-bound, but not Rad51-bound, ssDNA promotes Rad52 SUMOylation, suggesting that Rad52 SUMOylation is favoured prior to the formation of Rad51 nucleoprotein filament during HR in cells. Significantly, while Rad52 SUMOylation has no effect on its oligomerization and interaction with RPA and Rad51, a small percentage (<10%) of SUMOylated Rad52 markedly decreases its affinity to ssDNA and dsDNA, and causes a reduction of its DNA annealing activity.
Collectively, our biochemical results suggest that upon binding to resected ssDNA tails coated with RPA, Rad52 undergoes conformational change that can promote its efficient SUMOylation. Because SUMOylation does not affect Rad52 interaction with RPA and Rad51 or its oligomerization, this modification does not appear to function by altering protein–protein interactions. Instead, our results suggest that SUMOylation attenuates Rad52 strand annealing activity and prompts its disassociation from DNA. This can provide a mechanism either for favouring appropriate pathways over others or for dynamic exchange of Rad52 on DNA. Consistently with previous reports (15,16), we find that SUMOylation of Rad52 in vivo suppresses rDNA recombination and favours single-strand annealing over gene conversion in direct repeat recombination at the LEU2 locus. In addition, we show that both the N-terminal (K43,44) and central (K253) SUMOylation sites contribute to these effects. Further, we show in this study that Rad52 SUMOylation reduces meiotic spore viability and appears to favour BIR events. While the mechanism for these effects remains to be determined, it may reflect a requirement of Rad52 dynamics in these pathways or a role of SUMO in facilitating events unique to these pathways.
Finally, it is noteworthy that the effects of SUMO on Rad52 activity are somewhat reminiscent of that on thymine-DNA glycosylase, SUMOylation of which induce its dissociation from the abasic site (33). However, different from thymine-DNA glycosylase, Rad52 functions in concert with several additional mediator and recombination proteins, such that its regulation is more complex and defects in any single form of regulation can be buffered by other mechanisms. Our in vivo results suggest that this is likely the case, as the lack of Rad52 SUMOylation does not dramatically affect several mitotic or meiotic recombination processes examined here. However, the observed alteration of recombination pathway usage as well as the changes of duration times of recombination foci in rad52 SUMOylation defective mutants indicate the modification of Rad52 can indeed influence recombination pathway choice or efficiency. An intriguing possibility is that SUMOylation, in conjunction with other Rad52 functions such as antagonizing Srs2 (34,35), helps to fine-tune the efficiency of recombinational repair. In addition, as other recombination proteins and Rad52-interacting factors are also subject to SUMOylation, the presence and modification of these factors may collectively provide a quality control mechanism to direct HR pathway choice depending on substrate types and the chromosomal environment (36). This work brings initial characterization of the role of SUMOylation during HR; additional studies will need to further our understanding of the underlying molecular mechanism.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Wellcome Trust International Senior Research Fellowship (WT076476); Czech Science Foundation (grants 301/09/317 and 203/09/H046); the Ministry of Education Youth and Sport of the Czech Republic [grants ME10048; MSM0021622413 and LC06030 (to L.K.), MSM0021622412 (to J.D.) and LC06010 (to R.Ch)]; the Danish Agency for Science, Technology and Innovation (to M.L.); the Villum Kann Rasmussen Foundation (to M.L.); the Lundbeck Foundation (to N.E.B.); RO1ES07061 (to P.S.); R01GM080670 grant (to X.Z.). Funding for open access charges: Wellcome Trust (WT047276).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank E. Johnson and B. Schulman for providing protein expression plasmids and all of the members of our laboratories for helpful discussions.
REFERENCES
- 1.Krogh BO, Symington LS. Recombination proteins in yeast. Ann. Rev. Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 2.Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 4.Sung P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem. 1997;272:28194–28197. doi: 10.1074/jbc.272.45.28194. [DOI] [PubMed] [Google Scholar]
- 5.Seong C, Sehorn MG, Plate I, Shi I, Song B, Chi P, Mortensen U, Sung P, Krejci L. Molecular anatomy of the recombination mediator function of Saccharomyces cerevisiae Rad52. J. Biol. Chem. 2008;283:12166–12174. doi: 10.1074/jbc.M800763200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sung P, Krejci L, Van Komen S, Sehorn MG. Rad51 recombinase and recombination mediators. J. Biol. Chem. 2003;278:42729–42732. doi: 10.1074/jbc.R300027200. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shinohara A, Shinohara M, Ohta T, Matsuda S, Ogawa T. Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells. 1998;3:145–156. doi: 10.1046/j.1365-2443.1998.00176.x. [DOI] [PubMed] [Google Scholar]
- 9.Mortensen UH, Bendixen C, Sunjevaric I, Rothstein R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl Acad. Sci. USA. 1996;93:10729–10734. doi: 10.1073/pnas.93.20.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 11.Milne GT, Weaver DT. Dominant negative alleles of RAD52 reveal a DNA repair/recombination complex including Rad51 and Rad52. Genes Dev. 1993;7:1755–1765. doi: 10.1101/gad.7.9.1755. [DOI] [PubMed] [Google Scholar]
- 12.Hays SL, Firmenich AA, Massey P, Banerjee R, Berg P. Studies of the interaction between Rad52 protein and the yeast single-stranded DNA binding protein RPA. Mol. Cell Biol. 1998;18:4400–4406. doi: 10.1128/mcb.18.7.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plate I, Hallwyl SC, Shi I, Krejci L, Muller C, Albertsen L, Sung P, Mortensen UH. Interaction with RPA is necessary for Rad52 repair center formation and for its mediator activity. J. Biol. Chem. 2008;283:29077–29085. doi: 10.1074/jbc.M804881200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krejci L, Song B, Bussen W, Rothstein R, Mortensen UH, Sung P. Interaction with Rad51 is indispensable for recombination mediator function of Rad52. J. Biol. Chem. 2002;277:40132–40141. doi: 10.1074/jbc.M206511200. [DOI] [PubMed] [Google Scholar]
- 15.Sacher M, Pfander B, Hoege C, Jentsch S. Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat. Cell Biol. 2006;8:1284–1290. doi: 10.1038/ncb1488. [DOI] [PubMed] [Google Scholar]
- 16.Torres-Rosell J, Sunjevaric I, De Piccoli G, Sacher M, Eckert-Boulet N, Reid R, Jentsch S, Rothstein R, Aragon L, Lisby M. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat. Cell Biol. 2007;9:923–931. doi: 10.1038/ncb1619. [DOI] [PubMed] [Google Scholar]
- 17.Sherman F, Fink GR, Hicks JB. Methods in Yeast Genetics. NY: Cold Spring Harbor; 1986. [Google Scholar]
- 18.Thomas BJ, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- 19.Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 20.Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265:1241–1243. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- 21.Petukhova G, Stratton S, Sung P. Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature. 1998;393:91–94. doi: 10.1038/30037. [DOI] [PubMed] [Google Scholar]
- 22.Smith J, Rothstein R. A mutation in the gene encoding the Saccharomyces cerevisiae single-stranded DNA-binding protein Rfa1 stimulates a RAD52-independent pathway for direct-repeat recombination. Mol. Cell Biol. 1995;15:1632–1641. doi: 10.1128/mcb.15.3.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lea DE, Coulson CA. The distribution in the numbers of mutants in bacterial populations. J. Genetics. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- 24.Merker RJ, Klein HL. hpr1Delta affects ribosomal DNA recombination and cell life span in Saccharomyces cerevisiae. Mol. Cell Biol. 2002;22:421–429. doi: 10.1128/MCB.22.2.421-429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis AP, Symington LS. RAD51-dependent break-induced replication in yeast. Mol. Cell Biol. 2004;24:2344–2351. doi: 10.1128/MCB.24.6.2344-2351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lisby M, Rothstein R, Mortensen UH. Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl Acad. Sci. USA. 2001;98:8276–8282. doi: 10.1073/pnas.121006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parsons CA, Baumann P, Van Dyck E, West SC. Precise binding of single-stranded DNA termini by human RAD52 protein. Embo J. 2000;19:4175–4181. doi: 10.1093/emboj/19.15.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson NP, Lindstrom J, Baase WA, von Hippel PH. Double-stranded DNA templates can induce alpha-helical conformation in peptides containing lysine and alanine: functional implications for leucine zipper and helix-loop-helix transcription factors. Proc. Natl Acad. Sci. USA. 1994;91:4840–4844. doi: 10.1073/pnas.91.11.4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shinohara A, Ogawa T. Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature. 1998;391:404–407. doi: 10.1038/34943. [DOI] [PubMed] [Google Scholar]
- 30.Sugiyama T, New JH, Kowalczykowski SC. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc. Natl Acad. Sci. USA. 1998;95:6049–6054. doi: 10.1073/pnas.95.11.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burgess RC, Rahman S, Lisby M, Rothstein R, Zhao X. The Slx5-Slx8 complex affects sumoylation of DNA repair proteins and negatively regulates recombination. Mol. Cell Biol. 2007;27:6153–6162. doi: 10.1128/MCB.00787-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, Ulrich HD. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell. 2005;19:123–133. doi: 10.1016/j.molcel.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 33.Steinacher R, Schar P. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr. Biol. 2005;15:616–623. doi: 10.1016/j.cub.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 34.Burgess RC, Lisby M, Altmannova V, Krejci L, Sung P, Rothstein R. Localization of recombination proteins and Srs2 reveals anti-recombinase function in vivo. J. Cell Biol. 2009;185:969–981. doi: 10.1083/jcb.200810055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seong C, Colavito S, Kwon Y, Sung P, Krejci L. Regulation of Rad51 recombinase presynaptic filament assembly via interactions with the Rad52 mediator and the Srs2 anti-recombinase. J. Biol. Chem. 2009;284:24363–24371. doi: 10.1074/jbc.M109.032953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marini V, Krejci L. Srs2: the “Odd-Job Man” in DNA repair. DNA repair. 2010;9:268–275. doi: 10.1016/j.dnarep.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mortensen UH, Erdeniz N, Feng Q, Rothstein R. A molecular genetic dissection of the evolutionarily conserved N terminus of yeast Rad52. Genetics. 2002;161:549–562. doi: 10.1093/genetics/161.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rattray AJ, Symington LS. Use of a chromosomal inverted repeat to demonstrate that the RAD51 and RAD52 genes of Saccharomyces cerevisiae have different roles in mitotic recombination. Genetics. 1994;138:587–595. doi: 10.1093/genetics/138.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lisby M, Rothstein R, Mortensen UH. Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl Acad. Sci. USA. 2001;98:8276–8282. doi: 10.1073/pnas.121006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}