


Abstract
Selenium, an essential trace element, is incorporated into selenoproteins as selenocysteine (Sec), the 21st amino acid. In order to synthesize selenoproteins, a translational reprogramming event must occur since Sec is encoded by the UGA stop codon. In mammals, the recoding of UGA as Sec depends on the selenocysteine insertion sequence (SECIS) element, a stem-loop structure in the 3′ untranslated region of the transcript. The SECIS acts as a platform for RNA-binding proteins, which mediate or regulate the recoding mechanism. Using UV crosslinking, we identified a 110 kDa protein, which binds with high affinity to SECIS elements from a subset of selenoprotein mRNAs. The crosslinking activity was purified by RNA affinity chromatography and identified as nucleolin by mass spectrometry analysis. In vitro binding assays showed that purified nucleolin discriminates among SECIS elements in the absence of other factors. Based on siRNA experiments, nucleolin is required for the optimal expression of certain selenoproteins. There was a good correlation between the affinity of nucleolin for a SECIS and its effect on selenoprotein expression. As selenoprotein transcript levels and localization did not change in siRNA-treated cells, our results suggest that nucleolin selectively enhances the expression of a subset of selenoproteins at the translational level.
INTRODUCTION
Selenoproteins are a small but important subclass of proteins that contain the essential trace element selenium. The mammalian selenoproteins with known functions perform a variety of critical roles in anti-oxidant defense, thyroid hormone metabolism, male reproduction and development (1). Selenium is co-translationally incorporated into selenoproteins as selenocysteine (Sec), the 21st amino acid. The translation of selenoprotein mRNAs is complicated by the fact that Sec is encoded by the UGA codon, which typically signals the termination of protein synthesis. However, UGA will be recoded as Sec when the 3′ untranslated region (3′UTR) of the transcript contains a specific stem-loop structure called the Sec insertion sequence (SECIS) element (2). All eukaryotic SECIS elements form a similar structure composed of an apical loop and two stems separated by an internal loop. Although SECIS elements share little homology at the nucleotide level, they contain two highly conserved motifs, which are essential for recoding UGA as Sec. The SECIS core encompasses two sheared tandem G•A base pairs (3,4). The other essential sequence is the AAPurine (AAR) motif, which is found either in the apical loop (Type 1 SECIS) or adenosine bulge (Type 2 SECIS) (5,6). Sec incorporation also requires a novel Sec-charged tRNA, which has a UCA anticodon (7). The synthesis and utilization of the Sec-tRNASec requires several proteins, including a specialized elongation factor, EFsec, which is dedicated to Sec incorporation (8,9). Our group purified and cloned two other trans-acting factors in this pathway: SECIS binding protein 2 (SBP2) and ribosomal protein L30 (10,11). In vitro studies support the hypothesis that SBP2 and L30 compete for binding to the SECIS core and that the two proteins act sequentially during UGA recoding (10). However, there is debate in the field regarding the specific functions of SBP2 and L30, as well as the exact sequence of events that occur during Sec incorporation (12,13).
Although dramatic progress has been made in elucidating the mechanism of Sec incorporation, less is known about how this pathway is regulated. Selenoprotein synthesis is regulated by dietary selenium in vivo, but the effects vary between tissues and between individual selenoproteins. When selenium is limiting, the element is preferentially retained and utilized by the brain and endocrine organs at the expense of other tissues (14). There is also a hierarchy of expression of individual selenoproteins during selenium deficiency. For example, the synthesis of phospholipid hydroperoxide glutathione peroxidase (PHGPx or GPx4) is maintained, whereas the expression of glutathione peroxidase 1 (GPx1) is rapidly lost when selenium becomes limiting (15–17). Given that SBP2 and L30 have both been shown to be limiting factors for Sec incorporation, there may even be a hierarchy of selenoprotein expression under selenium-adequate conditions. Based on gene targeting studies in mice, certain selenoproteins, including PHGPx, are required for normal development and health (18,19), whereas others, such as GPx1, appear to have redundant or secondary functions (20,21). Thus, the limiting UGA recoding machinery may be preferentially recruited to a subset of transcripts, which encode selenoproteins that perform critical functions.
To the best of our knowledge, the 25 mammalian selenoproteins all depend on the same set of trans-acting factors for their synthesis. What is not well understood is how the cell prioritizes the utilization of these factors. SBP2 may dictate the expression pattern of the selenoproteome since it is an essential factor for Sec incorporation and binds various SECIS elements with different affinities in vitro and in vivo (22,23). However, the complex hierarchy of selenoprotein expression is likely to be maintained by the interplay between multiple trans-acting factors that bind to cis-acting sequences in the SECIS element and prioritize the utilization of selenium in a tissue-specific and selenoprotein-dependent manner. One such protein that links selenium status with differential selenoprotein expression is eukaryotic initiation factor 4a isoform 3 (eIF4a3). We recently showed that eIF4a3 is a selenium-regulated SECIS-binding protein that interacts selectively with a subset of selenoprotein mRNAs, including GPx1 (24). In vitro and cell culture studies support a model in which eIF4a3 translationally represses the expression of GPx1 by preventing the binding of SBP2 to the SECIS, thus inhibiting Sec incorporation (24).
In this study, we show that nucleolin selectively binds to SECIS elements from a subset of selenoprotein mRNAs, including PHGPx but not GPx1. Our studies support a model in which nucleolin acts as a positive regulator of selenoprotein mRNA translation.
MATERIALS AND METHODS
Cloning and mutagenesis
Information on the size and sequences of the SECIS elements and irrelevant RNAs is provided in Supplementary Table S1. The rat PHGPx and GPx1 3′UTRs are previously described (25). The rat Sel15 (BC060547) 3′UTR (1019–1217 nt) was cloned into the EcoRI–HindIII sites of pGEM3zf′(+) plasmid. The 3′UTRs of mouse GPx2 (BC039658), SelO (AK005048), SelR (AF195142), SelW (BC052719) and the long TR1 (AB027565) were prepared as PCR products containing a T7 promoter and were generous gifts of Heiko Mix and Vadim Gladyshev (University of Nebraska). The short TR1 SECIS of 207 nt was prepared as a PCR product containing a T7 promoter. The SBP2 constructs that encompass regions 1 and 2 correspond to 394–535 and 1–100 nt of the SBP2 3′ UTR and are described in ref. (26). The hypoxia stability region (HSR) from the human VEGF-A 3′ UTR (27) was a generous gift from Peng Yao and Paul Fox. The PHGPx SECIS basal stem deletion mutants are described in ref. (28). Additional mutations in the PHGPx SECIS were made using the primers listed in Supplementary Table S2 and the QuikChange Site-Directed Mutagenesis Kit (Stratagene). All mutations were confirmed by DNA sequencing.
In vitro transcription
32P-labeled wild-type and mutant PHGPx SECIS probes were prepared from linearized templates with T7 RNA polymerase using 10 mM GTP, 10 mM ATP, 10 mM CTP, 0.05 mM UTP and 25 µCi 32P-labeled UTP for 3 h at 37°C. Cold competitor RNAs were synthesized from linearized plasmid or PCR-amplified DNAs using T7 RNA polymerase (Ribomax T7; Promega). After incubation, all transcription reactions were treated with DNase I for 20 min and then phenol–chloroform-extracted. The aqueous phase was passed through a Micro Bio-Spin P30 column according to manufacturer’s instructions (Bio-Rad).
Preparation of extracts
McArdle 7777 cells were grown in DMEM/F12 media containing 10% fetal bovine serum (FBS). Nuclear and cytosolic extracts were isolated as previously described (26). Protein concentrations were determined using the Bio-Rad protein assay using IgG as a standard.
UV crosslinking assays
UV crosslinking assays were performed in crosslink buffer (10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) pH 7.9, 100 mM KCl, 12.5% glycerol, 0.75 mM MgCl2, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 0.25 mM dithiothreitol (DTT)], with 27 ng yeast tRNA (Invitrogen) and 12 U RNase inhibitor (Roche). Reactions contained 20 fmol 32P-labeled probe (22 000 cpm/fmol) and cell extracts or purified nucleolin (Vaxron) as indicated in the figure legends. Each reaction was treated with UV irradiation (Bio-Rad GS Genelinker) for 10 min at 254 nm in a 96-well tissue culture plate (Corning). Samples were digested using RNase A (1 mg/ml) for 1 h at 37°C, analyzed by 10% (w/v) SDS–PAGE and detected by autoradiography. Quantitation was performed using a Storm 800 Imager and ImageQuant software (Molecular Dynamics). For competition experiments, proteins were pre-incubated with various concentrations of unlabeled SECIS RNAs as indicated for 10 min at 37°C prior to the addition of the 32P-labeled PHGPx probe.
Protein purification and peptide sequencing
Nuclear extracts from McArdle 7777 cells were precipitated sequentially with 0–20, 20–40 and 40–60% ammonium sulfate (AMS). As the bulk of the 110 kDa crosslinking activity was detected in the 40–60% AMS fraction, this fraction was further purified by RNA affinity chromatography. The rat PHGPx SECIS RNA was coupled to cyanogen bromide-activated Sepharose 4B as described (29). The 40–60% AMS fraction was adsorbed to the column in binding buffer, and the column was extensively washed. Bound proteins were step-eluted with binding buffer containing 0.5 M NaCl (29). The eluted proteins were concentrated, separated by SDS–PAGE, and visualized by Coomassie Blue staining. The bands were excised from the gel and submitted for peptide sequence analysis using a Finnegan LCQ ion trap mass spectrometer. The protein was reduced, alkylated and digested with trypsin. The resulting peptides were analyzed by capillary column LC-tandem mass spectrometry. The CID spectra were used to search against the non-redundant database using a mammalian taxonomy filter.
Immunodepletion studies
MagnaBind Protein A magnetic beads (Pierce) were washed three times with NET2 buffer [10 mM tris(hydroxymethyl)aminomethane (TRIS), 150 mM sodium chloride (NaCl), 0.05% (v/v) Nonidet-P40] and incubated with two different affinity-purified anti-nucleolin antibodies (Sigma [N2662] and Novus Biologicals [NB100-2239]) or purified mouse IgG in 0.1 M sodium phosphate buffer, pH 8.1 for 1 h at room temperature on a rotator. The samples were then placed on a magnetic stand to capture the bead-antibody complexes, and the beads were washed once with crosslink buffer. Nuclear extract from McArdle 7777 cells (30 µg in 25 µl total volume of crosslink buffer) was added to the beads and incubated at room temperature for 1 h on a rotator. After incubation, samples were placed on a magnetic stand to capture the complexes. The supernatants were removed and analyzed by UV crosslinking.
RNA electrophoretic mobility shift assays
The 32P-labeled PHGPx SECIS RNA (10 fmol) was heated at 95°C and slow cooled to room temperature. The RNA was then incubated with increasing amounts of nucleolin protein purified from HeLa cells (Vaxron), in 20 µl crosslink buffer. Incubations were done at 37°C for 30 min and the resulting RNA–protein complexes were resolved on 4% non-denaturing polyacrylamide gels. The gels were dried, and the signals were quantified using a Storm 800 Imager and ImageQuant software (Molecular Dynamics). Competition experiments with cold RNAs were performed as described above for the UV crosslinking assays.
Co-immunoprecipitation
Nuclear extracts from McArdle 7777 cells were incubated at 4 mg/ml in crosslink buffer containing 0.2 mM EDTA and 0.5 mM DTT, with the two affinity-purified anti-nucleolin antibodies described above or purified mouse IgG for 1 h at 4°C. The immune complexes were removed by incubation with recombinant Protein A-Sepharose (Zymed). The resin was washed five times with NET2 buffer, and RNA was extracted directly from the beads in 300 µl Trizol according to manufacturer’s protocol (Invitrogen). RNA was resuspended in 10 µl RNase-free water for subsequent cDNA synthesis.
Quantitative real-time PCR
Equal volumes of the co-immunoprecipitated RNAs were used as templates for cDNA synthesis. All cDNAs were synthesized using the TaqMan Reverse Transcription Reagents kit, based on the manufacturer’s instructions and suggested thermal cycling profile (Applied Biosystems). Quantitative real-time PCR (qRT-PCR) was performed using SYBR Green PCR Master Mix and the ABI PRISM 7000 Sequence Detection System (SDS; Applied Biosystems), under conditions recommended in the instructions. Primers used for qRT-PCR are listed in Supplementary Table S2. The results were analyzed with ABI PRISM SDS software from Applied Biosystems, using the IgG sample as a control for relative selenoprotein mRNA expression in the immunoprecipitates. Relative gene expression was calculated using the 2−ΔΔCT method (30). For siRNA experiments, 2 µg of RNA from the harvested cells was used as a template for cDNA synthesis using the same method described above. qRT-PCR and gene expression analysis were performed as described above and the results were normalized to GAPDH mRNA levels. For analyzing the subcellular distribution of specific transcripts, qRT-PCR was performed using RNA extracted from nuclear extracts, which were prepared as described above.
siRNA experiments
Four synthetic ON-TARGETplus siRNA duplexes against rat nucleolin were purchased from Dharmacon (Catalog #J-087768-05, J-087768-06, J-087768-07 and J-087768-08). Accell non-targeting siRNA #1 (Dharmacon) was used as a negative control, which we term NT siRNA. McArdle 7777 cells were seeded at a density of 3 × 105 in 6-well cell culture plates and grown in DMEM/F12 media supplemented with 10% FBS for 24 h. At 40% confluency, cells were transfected with 67 nM siRNAs using Dharmafect 4 reagent, according to the manufacturer’s instructions (Dharmacon). After 72–96 h, cells were harvested for RNA using Trizol according to manufacturer’s instructions (Invitrogen), or for total protein, as follows. Cells were washed in 1 × Dulbecco’s phosphate-buffered saline (D-PBS) and collected in a microcentrifuge tube. After centrifugation, the cell pellets were resuspended in two packed cell volumes of lysis buffer (0.3 M HEPES pH 7.4, 1.4 M KCl, 30 mM MgCl2, 0.05% NP-40) and incubated on ice for 30 min. The samples were then centrifuged at 14 000 r.p.m. for 20 min at 4°C. The supernatants were analyzed by western blotting for nucleolin expression and mRNA levels from the RNA extracts were analyzed using qRT-PCR. Maximal knockdown of nucleolin was achieved after 96 h using siRNAs J-087768-07 and J-087768-08, which will be referred to as siRNAs 7 and 8, respectively. These siRNAs and conditions were subsequently used to analyze the effects on selenoprotein expression.
Western blotting
Proteins were electrophoresed on SDS–PAGE gels and transferred to ImmunoBlot polyvinylidene fluoride (PVDF) membrane (BioRad). The primary antibodies used were α-NCL rabbit polyclonal antibody (Sigma, N2662), α-PHGPx rabbit polyclonal antibody, α-GPx1 rabbit polyclonal antibody (Abcam, ab22604), α-GAPDH (6C5) mouse monoclonal antibody (Abcam, ab8245), α-TXNRD1 (TR1) rabbit polyclonal antibody (ProteinTech Group, 11117-1-AP), α-SEP15 mouse polyclonal antibody (Abnova, H00009403-A01), α-histone H1 mouse monoclonal antibody (Santa Cruz, SC-56694) and β-tubulin isotype I and II antibody (Sigma, T8535). The secondary antibodies used were either α-rabbit-HRP or α-mouse-HRP (Jackson Immunochemicals). Proteins were detected using Immobilon Chemiluminescent HRP detection substrate (Millipore), and exposure to BioMax MR film (Kodak). Analysis was performed using ImageQuant RT ECL (GE Healthcare).
RESULTS
Identification of a 110 kDa SECIS-binding protein
In order to identify SECIS-binding proteins, UV crosslinking experiments were carried out using nuclear and cytosolic extracts from McArdle 7777 cells, a rat hepatoma cell line that expresses PHGPx, GPx1 and several other selenoproteins. The extracts were analyzed by western blotting using antibodies to histone H1 or β-tubulin as nuclear and cytosolic markers, respectively, to monitor the purity of the fractions. As shown in Figure 1A, the nuclear and cytosolic extracts were relatively free of contamination from the other subcellular compartment. The extracts (0–20 µg) were incubated with the 32P-labeled wild-type PHGPx SECIS, which is a Type 2 element. Proteins were crosslinked to the RNA by exposure to UV radiation. Following degradation of the unbound RNA by RNase A, the samples were separated by SDS–PAGE and analyzed by autoradiography (Figure 1B). A prominent band of 110 kDa, which was detected in both nuclear and cytosolic extracts, crosslinked to the PHGPx SECIS in a dose-dependent manner. To assess specificity, crosslinking experiments were also performed with an irrelevant RNA corresponding to region 1 of the SBP2 3′UTR (26). This RNA is predicted to form a stable stem-loop structure, with two stems, an internal loop and an apical loop, similar to a SECIS element. In contrast to the results obtained with the PHGPx SECIS, we did not detect a strong signal corresponding to the 110 kDa protein in nuclear or cytoplasmic extracts when the crosslinking experiments were performed with the 32P-labeled region 1 probe. The nuclear and cytosolic extracts both contained a protein of ∼50 kDa, which crosslinked to the PHGPx SECIS (Figure 1B) and the SBP2 region 1 RNA (Figure 1C). The identity of the 50 kDa protein (indicated by the asterisk in Figure 1) is not known but it may represent a non-specific RNA-binding protein.
Figure 1.
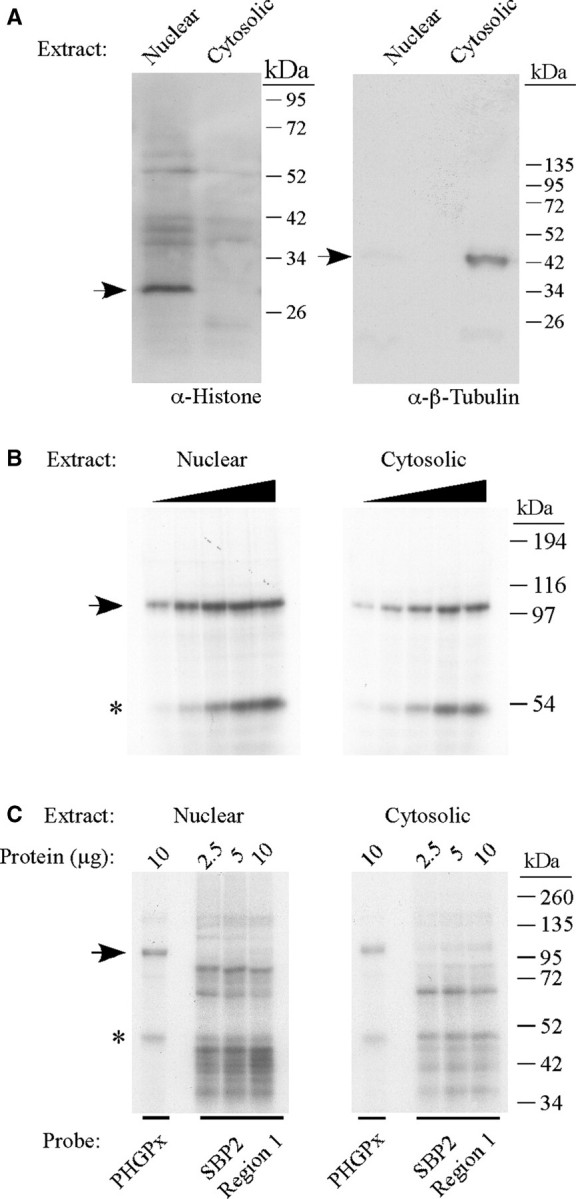
Identification of a 110 kDa SECIS-binding protein. (A) Nuclear and cytosolic extracts (30 µg protein) were analyzed by SDS–PAGE and western blotting using antibodies against histone H1 (nuclear marker) and β-tubulin (cytosolic marker) as indicated. The arrows indicate the positions of histone (left panel) and β-tubulin (right panel). (B) The 32P-labeled wild-type PHGPx SECIS probe was incubated with increasing amounts (2.5–20 µg) of nuclear or cytosolic extracts from McArdle 7777 cells in a UV crosslinking assay. The samples were analyzed by SDS–PAGE and autoradiography. The 110 kDa protein is indicated by the arrow. The band of ∼50 kDa, indicated by the asterisk, may represent a non-specific RNA-binding protein. (C) UV crosslinking experiments were performed with 2.5, 5 or 10 µg of nuclear or cytosolic extracts as described in (A) except that the 32P-labeled probe was either the PHGPx SECIS or an irrelevant RNA (region 1 from the SBP2 3′ UTR) that does not contain a SECIS but is predicted to form a stem-loop structure.
The 110 kDa protein requires the upper part of the basal stem for binding to the SECIS
As the specific activity of the 110 kDa crosslinking activity was higher in the nuclear fraction compared to the cytosol (Figure 1B), subsequent experiments were performed using the nuclear extract. To identify nucleotide sequences that are required for binding of the 110 kDa protein, we performed UV crosslinking/competition assays in which increasing amounts of wild-type or mutant PHGPx SECIS RNAs were added to the reaction. We first tested the importance of the AAR motif, which is essential for Sec incorporation but has no known function, by generating a loopless RNA that lacks the AAR motif and apical loop (Figure 2A). As shown in Figure 2B, the wild-type SECIS of 200 nt and the loopless RNA were both able to compete for crosslinking of the 110 kDa protein to the 32P-labeled PHGPx SECIS probe. We then analyzed mutant RNAs that ranged in size from 155 to 81 nt due to sequential deletion of the basal stem (Figure 2A). The 155, 129 and 102 nt RNAs were all effective competitors (Figure 2C). In contrast, the 81 nt SECIS did not compete at a 250- or 500-fold molar excess. Thus, binding of the 110 kDa protein to the PHGPx SECIS requires the upper part of the basal stem but not the AAR motif or apical loop.
Figure 2.
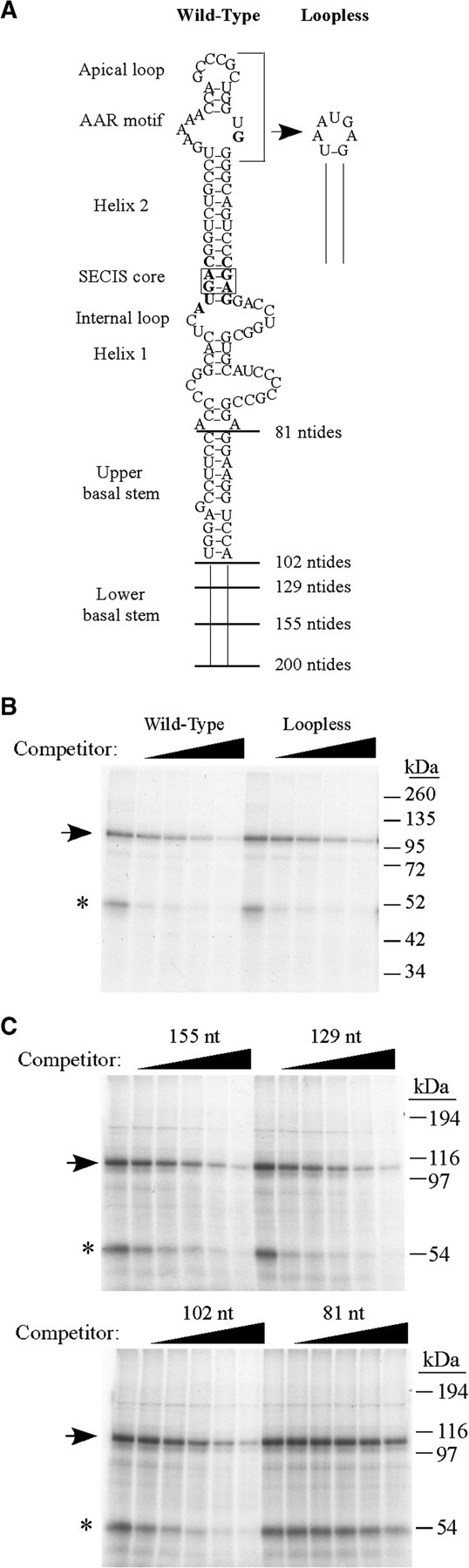
Analysis of mutant PHGPx SECIS RNAs to define the binding site requirements for the 110 kDa protein. (A) Schematic of the PHGPx wild-type SECIS (200 nt), the loopless mutant RNA, and the basal stem deletion mutants of 155, 129, 102 or 81 nt. (B) Nuclear extract from McArdle 7777 cells (2 µg) was preincubated with buffer or increasing amounts (0–200-fold molar excess over the probe) of unlabeled wild-type or loopless PHGPx SECIS RNAs for 10 min prior to the addition of the 32P-labeled PHGPx SECIS probe. The 110 kDa crosslinked product is indicated by the arrow and the asterisk indicates the 50 kDa protein of unknown identity. (C) Competition experiments were performed as in (B) using 5.5 µg nuclear extract and increasing amounts (0–500-fold molar excess) of cold basal stem deletion mutant RNAs of 155, 129, 102 and 81 nt, as indicated.
The 110 kDa protein distinguishes between SECIS elements
The 110 kDa protein may represent a general SECIS-binding protein or it may selectively bind to a subset of selenoprotein mRNAs. In order to distinguish between these two possibilities, mammalian SECIS elements were analyzed for their ability to compete for crosslinking of the 110 kDa protein. Figure 3A shows a typical experiment in which crosslinking of the 110 kDa protein to the 32P-labeled wild-type PHGPx SECIS is nearly eliminated in the presence of the cold PHGPx SECIS but not the GPx1 SECIS competitor RNA. A range of cold competitor concentrations was tested to calculate an IC50 as a measure of relative binding affinity of the 110 kDa protein. This value represents the fold molar excess of cold competitor over the labeled RNA that causes a 50% reduction in crosslinking signal. As shown in Figure 3A, the IC50 of the PHGPx SECIS was ∼ 24-fold lower than the IC50 of the GPx1 SECIS. Thus the 110 kDa protein binds to the PHGPx SECIS with higher affinity than to the GPx1 SECIS. We then analyzed an additional 10 mammalian SECIS elements as competitors in UV crosslinking assays. Figure 3B shows that there is a spectrum of IC50 values, which ranged from 7- to 470-fold molar excess over the labeled RNA. Similar results were obtained when UV crosslinking experiments were performed using nuclear extracts from HeLa cells (data not shown). We wondered whether there was a correlation between protein binding and the type of SECIS structure. The 110 kDa protein binds with high affinity to a subset of Type 2 SECIS elements, including TR1, PHGPx, and SelW (Figure 3B). However, this structure alone is not required for binding as evidenced by mouse SelR and SelN, which are Type 1 elements that contain the AAR motif in the apical loop, and by SelO, which has a non-canonical CCC motif in place of AAR. The type 2 structure is also not sufficient since the 110 kDa protein binds with low affinity to Sel15 and SelT, which are Type 2 elements (Figure 3B).
Figure 3.

The 110 kDa protein distinguishes among SECIS elements. (A) The inset shows a representative UV crosslinking competition experiment in which nuclear extracts and the 32P-labeled wild-type PHGPx SECIS were incubated in the presence of increasing amounts of cold PHGPx or GPx1 SECIS RNAs. The arrow indicates the position of the 110 kDa protein. The graph shows the quantitation of competition experiments. The IC50 values were calculated as described in ‘Materials and Methods' section, and the results represent the data from three or more experiments (B) UV crosslinking competition experiments were performed as described in the legend to Figure 2B. Crosslinking of the 110 kDa protein to the 32P-labeled PHGPx SECIS was competed using increasing amounts of unlabeled SECIS RNAs from 12 mammalian selenoproteins. The IC50 values were calculated as described in ‘Materials and Methods' section. The data for each SECIS element represent the mean from three or more experiments ± SD.
Purification and identification of the 110 kDa protein as nucleolin
For purification of the 110 kDa protein, nuclear extracts from McArdle 7777 cells were precipitated with 0–20, 20–40 and 40–60% AMS. The bulk of the crosslinking activity was retained in the 40–60% fraction. This fraction was then purified by RNA affinity chromatography using the PHGPx SECIS as a ligand, as described in ‘Materials and Methods' section. Bound proteins were step-eluted with 0.5 M NaCl. The eluate was concentrated for separation by SDS–PAGE and visualization by Coomassie Blue staining. The eluted fraction contained several proteins, including a 110 kDa protein and a more prominent band of 98 kDa (indicated by the arrows in Figure 4A). The bands were excised from the gel and submitted for peptide sequence analysis by mass spectrometry. Both bands were identified as nucleolin, with six peptides covering 14% of the sequence for the 110 kDa protein and 29 peptides covering 38% of the sequence for the 98 kDa protein. The 98 kDa protein presumably represents a degradation product that was generated during purification.
Figure 4.

Purification and identification of the crosslinking activity. (A) The fraction eluted from the PHGPx RNA affinity column was analyzed by SDS–PAGE and Coomassie Blue staining. The 110 and 98 kDa proteins indicated by the arrows were submitted for peptide sequence analysis by mass spectrometry and both bands were identified as nucleolin. (B) Nuclear and cytosolic extracts (30 µg) were analyzed by western blotting using an anti-nucleolin antibody. The arrow indicates the position of nucleolin. (C) Nuclear extracts from McArdle 7777 cells (30 µg) were immunodepleted with purified IgG or affinity purified anti-nucleolin antibodies (α-NCL-1, from Sigma; or α-NCL-2, from Novus Biologicals). Aliquots of starting material (Start) and the depleted extracts were analyzed by UV crosslinking with the 32P-labeled wild-type PHGPx SECIS probe as described in the legend to Figure 1. The 110 kDa crosslinked product is indicated by the arrow. The asterisk indicates an unidentified protein of ∼50 kDa. (D) The 32P-labeled wild-type PHGPx SECIS (top) or loopless RNA (bottom) probes were incubated in the presence of varying amounts of nucleolin purified from HeLa cells as indicated. The samples were analyzed by UV crosslinking. The arrow indicates the position of the crosslinked product. (E) REMSA assay using the 32P-labeled wild-type PHGPx SECIS probe, which was incubated with increasing amounts of nucleolin purified from HeLa cells, as indicated. The reactions were analyzed by non-denaturing polyacrylamide gel electrophoresis and autoradiography. The nucleolin:SECIS complexes (NCL:SECIS) are indicated by a bracket. The asterisk may represent a slowly migrating conformation of probe.
Nucleolin is a highly conserved protein that contains multiple domains: an N-terminal region rich in acidic amino acids; a central domain with four non-identical RNA recognition motifs (RRM) that function in RNA-binding; and a C-terminal region with multiple RGG repeats that mediates protein–protein interactions (31). Based on the cDNA sequence, nucleolin is predicted to be 77 kDa, but the protein migrates anomalously at 110 kDa on SDS–PAGE due to the highly acidic N-terminus. Western blot analysis using an anti-nucleolin antibody detected the protein in nuclear and cytosolic extracts from McArdle 7777 cells (Figure 4B). We also performed immunodepletion experiments to confirm that the 110 kDa band detected by crosslinking is nucleolin. Nuclear extracts from McArdle 7777 cells were incubated with two different affinity-purified anti-nucleolin antibodies or purified IgG. The immune complexes were removed by binding to protein A magnetic beads and the unbound fractions were assayed by UV crosslinking using the 32P-labeled PHGPx SECIS probe. As shown in Figure 4C, the 110 kDa crosslinking band was substantially depleted by the anti-nucleolin antibodies but not by the IgG control.
In order to determine whether nucleolin binds directly to the SECIS or whether this activity depends on other cellular factors, we carried out UV crosslinking experiments using nucleolin purified from HeLa cells. The purified protein crosslinked to the 32P-labeled PHGPx SECIS and to the loopless RNA in a dose-dependent manner, with a signal detected using as little as 3 ng of protein (Figure 4D). We also analyzed nucleolin–SECIS interactions using RNA electrophoretic mobility shift assay (REMSA). The 32P-labeled PHGPx SECIS was incubated with purified nucleolin and the samples were analyzed by electrophoresis on native gels. As shown in Figure 4E, the probe shifted upwards in a dose-dependent manner in the presence of nucleolin. The nucleolin–SECIS complex appeared as a discrete band within a smear, presumably due to the dissociation of the complex during electrophoresis. Approximately 50% of the probe was shifted by 40–50 ng of protein. This corresponds to an apparent KD of ∼30 nM, which is similar to the affinity of nucleolin for other known targets.
Purified nucleolin shows selective SECIS-binding activity
We next assessed the specificity of nucleolin binding by performing competition experiments, which were analyzed by REMSA. As shown in Figure 5, the purified nucleolin exhibits sequence-specificity similar to the crosslinking activity detected in crude extracts. Binding of nucleolin was effectively competed by the wild-type PHGPx SECIS but not by the 81 nt basal stem deletion mutant, even at a 200-fold molar excess (Figure 5A). We also tested three non-SECIS RNAs that are predicted to form stem-loop structures, including the HSR of the VEGF-A 3′ UTR (27), region 1 from the SBP2 3′ UTR which was used in Figure 1C, and an RNA corresponding to region 2 from the SBP2 3′ UTR. A 20-fold molar excess of the cold PHGPx SECIS RNA reduced the REMSA signal by 70% whereas the three irrelevant RNAs were not effective competitors at the same concentration (Figure 5B). In terms of selectivity, our crosslinking experiments with crude extracts suggested that nucleolin bound with the highest affinity to the TR1 SECIS. However, the RNA used for this experiment was ∼1550 nt long, compared to an average size of 250 nt for the other SECIS elements that were tested (Supplementary Table S1). Therefore we generated a short TR1 SECIS RNA of 207 nt. As shown in Figure 5C, both the long and short TR1 SECIS RNAs were very effective competitors, reducing the signal by 75–80% at 2.5-fold molar excess. Thus RNA length alone is not sufficient to explain high affinity binding.
Figure 5.

Purified nucleolin shows selective SECIS-binding activity. (A) REMSA competition experiments were performed using nucleolin purified from HeLa cells (25 ng), the 32P-labeled PHGPx SECIS probe, and the wild-type PHGPx SECIS or the 81 nt basal stem deletion mutant RNAs as described in the legend to Figure 2B. Cold RNAs were added at 0, 25, 50, 100 or 200-fold molar excess over the probe. The brackets indicate the position of the nucleolin:SECIS complex (NCL:SECIS) and the arrow indicates the free probe. (B) REMSA competition experiments were performed as described in (A) except the competitor RNAs included the wild-type PHGPx SECIS and three irrelevant RNAs [HSR from the VEGF-A 3′ UTR (27), region 1 from the SBP2 3′ UTR which was tested in Figure 1C, and an RNA corresponding to region 2 of the SBP2 3′ UTR (see ‘Materials and Methods' section and Supplementary Table S1 for details)]. The RNAs were added at 0, 5, 10 and 20-fold molar excess over the probe. (C) REMSA competition experiments were performed using cold long (1550 nt) and short (207 nt) TR1 SECIS RNAs, which were added at 0, 2.5, 5, 10 and 20-fold molar excess. (D) UV crosslinking competition experiments in which purified nucleolin (20 ng) was incubated with 32P-labeled wild-type PHGPx SECIS probe. Various amounts of unlabeled SECIS RNAs (PHGPx, SelR, Sel15 or GPx1) were added to the binding reaction in molar excess over the labeled probe as indicated. The arrow indicates the position of the crosslinked product. No other bands were detectable on the autoradiogram.
We also analyzed the selectivity of nucleolin binding using UV crosslinking/competition assays since the signal is easier to quantify with this method. As shown in Figure 5D, a 10-fold molar excess of the PHGPx SECIS reduced the signal by ∼40% whereas the crosslinking signal was decreased by ∼60% with a 5-fold molar excess of the SelR SECIS. In contrast, a 40-fold molar excess of Sel15 SECIS RNA or 160-fold molar excess of GPx1 SECIS RNA (Figure 5D) was required to compete away ∼50% of the labeled PHGPx SECIS probe (Figure 5D). Thus, nucleolin can discriminate between SECIS elements in the absence of SBP2, L30 or other cellular proteins.
Co-immunoprecipitation of nucleolin–RNA complexes from cells
To establish in vivo relevance, we wanted to determine whether nucleolin is associated with selenoprotein mRNAs in cells. This was of particular interest as nucleolin showed selective SECIS-binding activity in our in vitro assays, but a recent study reported that all selenoprotein mRNAs were co-immunoprecipitated with similar efficiency by an anti-nucleolin antibody compared to the IgG control (22). However, it is important to note that the latter study examined only selenoprotein mRNAs. We performed co-immunoprecipitation experiments using extracts from McArdle 7777 cells and the two anti-nucleolin antibodies that were effective in the immunodepletion experiments described above or purified IgG as the negative control. RNAs were isolated from the immune complexes and used as templates in cDNA reactions, which were analyzed by qRT-PCR using gene-specific primers. The mean crossing points were calculated for PCR products derived from three independent immunoprecipitations (two performed with one anti-nucleolin antibody and one performed with the other). The results were expressed as the change in mean crossing points for the anti-nucleolin samples over the IgG controls (Figure 6). We found that the anti-nucleolin antibodies immunoprecipitated the PHGPx, TR1, Sel15 and GPx1 mRNAs, with mean crossing point changes that ranged from 8.6 to 9.8 compared to the IgG control. These results are similar to those reported by Squires et al. (22). However, β-actin and GAPDH were also preferentially immunoprecipitated to a similar extent by the anti-nucleolin antibodies, with mean crossing point changes of 9.6 and 8.5, respectively, over the IgG control (Figure 6). These results suggest that the co-immunoprecipitation experiments presented here and in ref. (22) may not be detecting true in vivo interactions.
Figure 6.

Co-immunoprecipitation of selenoprotein mRNAs with nucleolin:RNA complexes from cells. Nucleolin:RNA complexes were immunoprecipitated from McArdle 7777 cell nuclear extracts using two different anti-nucleolin antibodies or a purified IgG control as described in ‘Materials and Methods' section. Total RNA was isolated from the immune complexes, and analyzed by qRT-PCR analysis using gene-specific primers. The results are expressed as change in crossing points (mean ± SD) for PCR products derived from the anti-nucleolin samples compared to the IgG controls. The data represent the combined results from three independent immunoprecipitation experiments using the two different anti-nucleolin antibodies.
siRNA knockdown of nucleolin selectively inhibits PHGPx and TR1 expression
In order to elucidate the role of nucleolin in selenoprotein synthesis, we used siRNAs to knockdown nucleolin expression in McArdle 7777 cells. The conditions for maximal knockdown were optimized as described in ‘Materials and Methods' section. Cells were transfected for 96 h with two different siRNAs against nucleolin (siRNAs 7 and 8) or a non-targeting (NT) siRNA as a negative control. The cell lysates from three independent transfections were analyzed by western blotting. As shown in Figure 7A and B, each of the nucleolin siRNAs reduced nucleolin expression by ∼6-fold, whereas the NT control siRNA had no effect. There was a 2.3- and 2.7-fold reduction in PHGPx protein levels in cells treated with siRNAs 7 and 8, respectively, which was not observed in the NT siRNA-treated cells (Figure 7C and D). We also observed a modest but statistically significant decrease in TR1 protein levels in the nucleolin siRNA-treated cells, with siRNAs 7 and 8 causing a 1.2- and 1.4-fold reduction, respectively (Figure 7C and D). The decrease that we observed for TR1 is likely to be an underestimate as this selenoprotein contains the Sec residue near the C-terminus. Thus the full-length TR1 and the truncated protein that lacks Sec cannot be distinguished by SDS–PAGE. The effect of the nucleolin siRNAs on PHGPx and TR1 expression was specific, as the knockdown of nucleolin did not alter the levels of GPx1 and Sel15, which are encoded by mRNAs containing SECIS elements that are bound by nucleolin with low affinity, or of GAPDH, which is not a selenoprotein (Figure 7C and D). Thus, there is a good correlation between the affinity of nucleolin for a SECIS element and its ability to enhance selenoprotein expression.
Figure 7.

Selective inhibition of PHGPx and TR1 expression due to siRNA knockdown of nucleolin. (A) McArdle 7777 cells were treated with siRNAs directed against nucleolin (siRNA 7 and 8) or a NT siRNA for 96 h as described in 'Materials and Methods' section. Cell lysates were analyzed by western blotting using the indicated antibodies. (B) Quantitation of the western blot data from three independent experiments. The results were normalized to GAPDH and are represented as mean ± SEM. Asterisks indicate significant change in protein expression for the nucleolin siRNA treated cells relative to the cells treated with the NT siRNA (single asterisk indicates P < 0.001). (C) As described in (A), cell lysates were analyzed by western blotting using the indicated antibodies. The PHGPx and GPx1 antibodies also recognize two non-specific bands of 34 and 52 kDa, which are not shown in this figure but did not change with siRNA treatment. The bands shown in the figure correspond to the specific bands for PHGPx and GPx1 proteins at 20 and 26 kDa, respectively. (D) Quantitation and statistics were performed as described in (B) (single asterisk indicates P < 0.001, double asterisk indicates P < 0.05). (E) Effect of nucleolin manipulation on PHGPx and TR1 mRNA levels. Total RNA was isolated from McArdle 7777 cells treated with nucleolin siRNAs 7 and 8 or the NT control for 96 h. The levels of β-actin, PHGPx and TR1 mRNAs were quantified by qRT-PCR. The results were normalized to GAPDH and expressed as fold-change. The data represent the mean from at least three independent analysis ± SEM. (F) Transcript levels in nuclear extracts were quantified by qRT-PCR as described in (E).
In terms of mechanism, nucleolin has been shown to stabilize a number of cellular mRNAs by binding to the 3′UTR of the transcript (32–35). Therefore, we analyzed transcript levels in the siRNA-treated cells by qRT-PCR. The results were normalized to GAPDH. As shown in Figure 7E, the knockdown of nucleolin did not reduce PHGPx, TR1 or β-actin mRNA levels. Thus, the decrease in PHGPx and TR1 protein that was observed in the nucleolin siRNA-treated cells was not due to a reduction in transcript abundance. We also tested the possibility that nucleolin may be involved in mRNA export by performing qRT-PCR analysis of RNA isolated from nuclear extracts of siRNA-treated cells. As shown in Figure 7F, siRNA knockdown of nucleolin did not alter the nuclear levels of the transcripts encoding PHGPx, TR1 and β-actin. Thus, nucleolin does not appear to stimulate selenoprotein expression by facilitating nuclear export of the transcript. Taken together, these results suggest that nucleolin may selectively enhance the expression of PHGPx and TR1 at the translational level.
DISCUSSION
The co-translational incorporation of Sec into selenoproteins is a complex process that requires both cis-acting sequences in the transcript and multiple trans-acting factors. As the UGA recoding machinery is limiting in cells, it is likely that selenoprotein synthesis needs to be tightly regulated in vivo in order to ensure the proper expression of the selenoproteome. The SECIS element plays an essential role in this mechanism by acting as a platform for RNA-binding proteins. Although mammalian SECIS elements all contain a stem-loop, they vary in their structure and composition, which raises the possibility that each SECIS may recruit a different spectrum of proteins. In the current study, we present several lines of evidence that strongly support the hypothesis that nucleolin selectively binds with high affinity to SECIS elements from PHGPx and other selenoprotein mRNAs to enhance their translation in cells.
Nucleolin is an abundant multi-functional protein found in a variety of subcellular locations (31,36), As the major protein in the nucleolus, nucleolin plays a critical role in ribosome biogenesis by interacting with both pre-ribosomal RNA and ribosomal proteins. The protein is also found in the nucleus where it promotes the transcription of rRNA genes and modulates chromatin structure. Although nucleolin shuttles between the nucleus and cytoplasm, the protein is not found on mature ribosomes. Instead cytoplasmic nucleolin has been shown to regulate the stability or translation of several cellular and viral mRNAs (32–35,37,38). Nucleolin is also found on the cell surface where it binds a number of ligands (31,36). In light of the newly discovered functions of nucleolin outside of the nucleolus, it is reasonable to consider that this protein may be involved in the regulation of selenoprotein synthesis. In fact, nucleolin was previously identified as a potential SECIS-binding protein based on ligand screening of a bacterial expression library with a radiolabeled GPx1 SECIS element (39). However, the affinity of nucleolin for other SECIS elements and the relevance of nucleolin to selenoprotein synthesis were not investigated in this earlier study.
Our in vitro binding assays showed that nucleolin binds with high affinity to SECIS elements from a subset of selenoprotein mRNAs. Interestingly co-immunoprecipitation studies perform by Squires et al. (22) and our group (Figure 6) failed to detect selective nucleolin–SECIS interactions in cells. The most likely explanation for the discrepancy between the in vitro binding and cell culture results is that UV crosslinking experiments are performed with shorter RNAs that encompass the SECIS element, not the entire transcript. In contrast, the co-immunoprecipitation experiments can detect interactions between nucleolin and full-length endogenous mRNAs, which may contain many high or low affinity binding sites. Nucleolin may have associated with cellular mRNAs after the cells were lysed or during the immunoprecipitation step. Indeed a non-specific association between nucleolin and GAPDH mRNA in co-immunoprecipitation experiments has been previously reported (37). There is also evidence in other systems that post-lysis reassortment of some RNA–protein interactions can occur (40).
The fact that nucleolin exhibits selective binding activity in vitro raises the question as to how it distinguishes among SECIS elements. As nucleolin contains four non-identical RRM domains, it is not surprising that the protein interacts with a variety of different targets. The RRM is a well-characterized RNA-binding domain of 80–90 amino acids that is found in many proteins involved in RNA processing or metabolism (41). For proteins that contain multiple RRMs, each RRM may bind a different sequence, or a subset of the RRM domains may be required for sequence-specific binding (42). In the case of nucleolin, the protein binds to two mutually exclusive binding sites in pre-rRNAs: the nucleolin recognition element (NRE) and the evolutionarily conserved motif (ECM). The NRE is a short single-stranded sequence (U/GCCCGA/G) found within the loop of a stem-loop structure in pre-rRNAs (43,44). The nucleolin–NRE interaction is mediated by RRMs 1 and 2 (45). In contrast, binding of nucleolin to the ECM, an 11-nt motif (GATCGATGTGG), requires all four RRMs (46). In addition to the NRE and ECM, nucleolin interacts with a variety of distinct sequences in the 5′ or 3′UTRs of cellular transcripts, including AU-rich elements in the Bcl-2 and Bcl-XL mRNAs (47). The binding sites for nucleolin in the 3′UTR of the amyloid precursor protein mRNA and the 5′UTR of the interleukin-2 mRNA have not been finely mapped but they both contain the sequence CUCUCUUUA (35). A similar CU-rich region is present in the 3′UTR of another nucleolin target, the CD154 mRNA (34). In contrast, the determinant for nucleolin binding to the 3′UTR of the β-globin mRNA is a G-rich sequence in a stem structure opposite a CU-rich region (32). In addition to the known binding sites for full-length nucleolin, a 64 kDa form of the protein, presumably generated by autocatalytic cleavage, binds to the sequences UAAAUC or AAAUCU in the 5′UTR of the collagen prolyl 4-hydroxylase-α(I) mRNA (48). Based on nucleotide sequence analysis, we have not yet been able to identify a motif that is common to SECIS elements that are bound by nucleolin with high affinity. Our results suggest that binding of nucleolin to the PHGPx SECIS requires the upper part of the basal stem (Figure 2C and 5A) but not the AAR motif or apical loop (Figure 2B and 4D). Whether nucleolin binds directly to the upper part of the basal stem and whether this interaction depends on the nucleotide sequence and/or secondary structure of the SECIS is currently under investigation.
Our siRNA experiments found that the knockdown of nucleolin resulted in a decrease in PHGPx and TR1 protein levels but had no effect on Sel15 and GPx1 expression. Importantly, the levels and subcellular localization of the transcripts did not change in the siRNA-treated cells. These results suggest that nucleolin acts as a positive regulator for the translation of a subset of selenoprotein mRNAs, including PHGPx and TR1. Do the transcripts that are bound by nucleolin with high affinity share a common biological function or biosynthetic pathway? We have not detected any correlation between binding of nucleolin and the following parameters: known enzymatic activity of the selenoprotein; tissue-specific expression pattern; dependence on the Um34 isoform of the Sec-tRNASec for synthesis (49); or presence of the second stop codon redefinition element in the coding region (50). However, we did note that nucleolin binds with higher affinity to SECIS elements from selenoproteins that exhibit a severe phenotype when deleted in mice and/or rank high in the hierarchy of expression during selenium deficiency. Nucleolin binds the PHGPx and TR1 SECIS elements with high affinity, and targeted disruption of either of these genes in mice is embryonic lethal in mice (18,19). Furthermore, these two selenoproteins, as well as SelW, rank high in the hierarchy of selenoprotein expression (51,52). Although the SelN gene has not been disrupted in mice, naturally occurring mutations in the human gene lead to a group of early-onset neuromuscular disorders referred to as SEPN1-related myopathy (53). In contrast, nucleolin binds the GPx1 SECIS element with low affinity, and the GPx1 knockout mouse model expresses a normal phenotype (20,21). In addition, GPx1 ranks low in the hierarchy of selenoprotein expression (51). Notably, nucleolin binds the Dio2, GPx2 and Dio1 SECIS elements with moderate affinity. The expression of these selenoproteins is preserved when selenium is limiting (54,55), but the knockout mice have either no phenotype or a modest phenotype, which suggests that these selenoproteins perform important but non-essential functions (56–58). The exception to this trend is SelR (also known as MsrB1), whose SECIS is bound by nucleolin with very high affinity. Although the SelR knockout mice exhibit signs of oxidative stress in the liver and kidney, they have no obvious phenotype (59). Knockout models for SelO, SelT and Sel15 have not been reported and it is not known where these selenoproteins rank in the hierarchy.
An emerging theme in the field is that the regulation of selenoprotein expression is much more complex than previously anticipated. The affinity of SBP2 for different SECIS elements varies both in vitro (23) and in vivo (22). These differences in the relative binding affinity of SBP2 determine the expression pattern of the selenoproteome (23). In addition to the components of the basal recoding machinery, other regulatory factors modulate the expression of individual selenoproteins. We recently reported that eIF4a3 binds to the GPx1 and SelR SECIS elements and represses their translation by preventing the binding of SBP2 when selenium is limiting (24). This effect is specific, as eIF4a3 did not bind to the PHGPx and TR1 SECIS elements. In contrast, nucleolin appears to be a selective positive regulator of selenoprotein expression, binding to the PHGPx and TR1 SECIS elements, but not to the GPx1 SECIS. Intriguingly, SelR appears to be an exception as both nucleolin (this study) and eIF4a3 (24) bind to this SECIS element with high affinity. As the two proteins have opposing effects on Sec incorporation, these results suggest that the expression of SelR may be tightly regulated at the translational level in different cell types or in response to environmental cues. There is a rapid increase in eIF4A3 protein when selenium becomes limiting and we proposed that this up-regulation serves to link selenium status with the hierarchy of selenoprotein expression (24). Interestingly, selenium supplementation has no effect on nucleolin activity and protein levels in McArdle 7777 cells. Our results suggest that nucleolin is required for optimal expression of PHGPx and TR1 but it is tempting to speculate that nucleolin may play an additional regulatory role since its RNA-binding activity and subcellular localization respond to a variety of cell stressors (37,47,60,61).
Given the variety of activities that nucleolin can carry out, there are multiple mechanisms by which this protein could modulate selenoprotein mRNA translation. There is precedent for nucleolin acting as a substrate remodeling factor. Nucleolin has a strand annealing activity (62), which could be involved in refolding the SECIS structure into a more favorable substrate for protein–SECIS interactions. Nucleolin also participates in protein–protein interactions, often through the RGG domains located in its C-terminus (63). Thus, the SECIS-bound nucleolin could directly recruit factors involved in Sec incorporation. Interestingly, a recent study identified nucleolin as a cell surface receptor for the bacterial pathogen Francisella tularensis (64). The ligand for nucleolin was bacterial elongation factor Tu, which was expressed on the surface of the bacteria. The finding that nucleolin can interact with an elongation factor raises the possibility that binding of nucleolin to the SECIS may facilitate the recruitment of the EFsec-Sec-tRNASec complex. Finally, nucleolin may not affect Sec incorporation directly but instead may modulate another step in protein synthesis. It has been proposed that nucleolin enhances the translational efficiency of the matrix-metalloproteinase-9 mRNA by binding to the 3′UTR and recruiting the translationally inactive mRNA to polysomes (38). Given that the 5′- and 3′-ends of a transcript interact during translation, there may be a SECIS-dependent initiation event that is mediated or regulated by nucleolin. The investigation of the mechanism of action of nucleolin is an important direction for future research.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (grant numbers RO1 DK078591 to D.M.D. and F32 DK083154 to M.E.B.); American Heart Association (Postdoctoral Fellowship to L.M.M.). Funding for open access charge: National Institutes of Health (grant numbers RO1 DK078591).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Mike Kinter and Dr Belinda Willard from the Lerner Research Institute Proteomics Laboratory for the mass spectrometry analysis; Dr Heiko Mix, Dr Vadim Gladyshev, Dr Peng Yao and Dr Paul Fox for providing PCR-amplified templates for in vitro transcription; and Dr Jodi Bubenik, Dr Tracey Ferrara and Dr Sanghamitra Bhattacharyya for reading the manuscript.
REFERENCES
- 1.Hatfield DL, Carlson BA, Xu XM, Mix H, Gladyshev VN. Selenocysteine incorporation machinery and the role of selenoproteins in development and health. Prog. Nucleic Acid Res. Mol. Biol. 2006;81:97–142. doi: 10.1016/S0079-6603(06)81003-2. [DOI] [PubMed] [Google Scholar]
- 2.Berry MJ, Banu L, Chen YY, Mandel SJ, Kieffer JD, Harney JW, Larsen PR. Recognition of UGA as a selenocysteine codon in type I deiodinase requires sequences in the 3' untranslated region. Nature. 1991;353:273–276. doi: 10.1038/353273a0. [DOI] [PubMed] [Google Scholar]
- 3.Walczak R, Carbon P, Krol A. An essential non-Watson-Crick base pair motif in 3'UTR to mediate selenoprotein translation. RNA. 1998;4:74–84. [PMC free article] [PubMed] [Google Scholar]
- 4.Walczak R, Westhof E, Carbon P, Krol A. A novel RNA structural motif in the selenocysteine insertion element of eukaryotic selenoprotein mRNAs. RNA. 1996;2:367–379. [PMC free article] [PubMed] [Google Scholar]
- 5.Grundner-Culemann E, Martin G.W., Harney JW, Berry MJ. Two distinct SECIS structures capable of directing selenocysteine incorporation in eukaryotes. RNA. 1999;5:625–635. doi: 10.1017/s1355838299981542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fagegaltier D, Lescure A, Walczak R, Carbon P, Krol A. Structural analysis of new local features in SECIS RNA hairpins. Nucleic Acids Res. 2000;28:2679–2689. doi: 10.1093/nar/28.14.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diamond A, Dudock B, Hatfield D. Structure and properties of a bovine liver UGA suppressor serine tRNA with a tryptophan anticodon. Cell. 1981;25:497–506. doi: 10.1016/0092-8674(81)90068-4. [DOI] [PubMed] [Google Scholar]
- 8.Tujebajeva RM, Copeland PR, Xu XM, Carlson BA, Harney JW, Driscoll DM, Hatfield DL, Berry MJ. Decoding apparatus for eukaryotic selenocysteine insertion. EMBO Rep. 2000;1:158–163. doi: 10.1093/embo-reports/kvd033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fagegaltier D, Hubert N, Yamada K, Mizutani T, Carbon P, Krol A. Characterization of mSelB, a novel mammalian elongation factor for selenoprotein translation. EMBO J. 2000;19:4796–4805. doi: 10.1093/emboj/19.17.4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chavatte L, Brown BA, Driscoll DM. Ribosomal protein L30 is a component of the UGA-selenocysteine recoding machinery in eukaryotes. Nat. Struct. Mol. Biol. 2005;12:408–416. doi: 10.1038/nsmb922. [DOI] [PubMed] [Google Scholar]
- 11.Copeland PR, Fletcher JE, Carlson BA, Hatfield DL, Driscoll DM. A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J. 2000;19:306–314. doi: 10.1093/emboj/19.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allmang C, Wurth L, Krol A. The selenium to selenoprotein pathway in eukaryotes: More molecular partners than anticipated. Biochim. Biophys. Acta. 2009;1790:1415–1423. doi: 10.1016/j.bbagen.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Donovan J, Caban K, Ranaweera R, Gonzalez-Flores JN, Copeland PR. A novel protein domain induces high affinity selenocysteine insertion sequence binding and elongation factor recruitment. J. Biol. Chem. 2008;283:35129–35139. doi: 10.1074/jbc.M806008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Behne D, Hilmert H, Scheid S, Gessner H, Elger W. Evidence for specific selenium target tissues and new biologically important selenoproteins. Biochim. Biophys. Acta. 1988;966:12–21. doi: 10.1016/0304-4165(88)90123-7. [DOI] [PubMed] [Google Scholar]
- 15.Weiss Sachdev S, Sunde RA. Selenium regulation of transcript abundance and translational efficiency of glutathione peroxidase-1 and -4 in rat liver. Biochem. J. 2001;357:851–858. doi: 10.1042/0264-6021:3570851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lei XG, Evenson JK, Thompson KM, Sunde RA. Glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase are differentially regulated in rats by dietary selenium. J. Nutr. 1995;125:1438–1446. doi: 10.1093/jn/125.6.1438. [DOI] [PubMed] [Google Scholar]
- 17.Bermano G, Nicol F, Dyer JA, Sunde RA, Beckett GJ, Arthur JR, Hesketh JE. Selenoprotein gene expression during selenium-repletion of selenium-deficient rats. Biol. Trace Elem. Res. 1996;51:211–223. doi: 10.1007/BF02784076. [DOI] [PubMed] [Google Scholar]
- 18.Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, Motta L, Richardson A, Prolla TA. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003;34:496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 19.Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M, Kitamoto T, Hayasaka M, Hanaoka K, Nakagawa Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 2003;305:278–286. doi: 10.1016/s0006-291x(03)00734-4. [DOI] [PubMed] [Google Scholar]
- 20.Ho YS, Magnenat JL, Bronson RT, Cao J, Gargano M, Sugawara M, Funk CD. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem. 1997;272:16644–16651. doi: 10.1074/jbc.272.26.16644. [DOI] [PubMed] [Google Scholar]
- 21.Cheng WH, Ho YS, Ross DA, Valentine BA, Combs GF, Lei XG. Cellular glutathione peroxidase knockout mice express normal levels of selenium-dependent plasma and phospholipid hydroperoxide glutathione peroxidases in various tissues. J. Nutr. 1997;127:1445–1450. doi: 10.1093/jn/127.8.1445. [DOI] [PubMed] [Google Scholar]
- 22.Squires JE, Stoytchev I, Forry EP, Berry MJ. SBP2 binding affinity is a major determinant in differential selenoprotein mRNA translation and sensitivity to nonsense-mediated decay. Mol. Cell Biol. 2007;27:7848–7855. doi: 10.1128/MCB.00793-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bubenik JL, Driscoll DM. Altered RNA binding activity underlies abnormal thyroid hormone metabolism linked to a mutation in selenocysteine insertion sequence-binding protein 2. J. Biol. Chem. 2007;282:34653–34662. doi: 10.1074/jbc.M707059200. [DOI] [PubMed] [Google Scholar]
- 24.Budiman ME, Bubenik JL, Miniard AC, Middleton LM, Gerber CA, Cash A, Driscoll DM. Eukaryotic initiation factor 4a3 is a selenium-regulated RNA-binding protein that selectively inhibits selenocysteine incorporation. Mol. Cell. 2009;35:479–489. doi: 10.1016/j.molcel.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lesoon A, Mehta A, Singh R, Chisolm GM, Driscoll DM. An RNA-binding protein recognizes a mammalian selenocysteine insertion sequence element required for cotranslational incorporation of selenocysteine. Mol. Cell Biol. 1997;17:1977–1985. doi: 10.1128/mcb.17.4.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bubenik JL, Ladd AN, Gerber CA, Budiman ME, Driscoll DM. Known turnover and translation regulatory RNA-binding proteins interact with the 3' UTR of SECIS-binding protein 2. RNA Biol. 2009;6:73–83. doi: 10.4161/rna.6.1.7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL. A stress-responsive RNA switch regulates VEGFA expression. Nature. 2008;457:915–919. doi: 10.1038/nature07598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Copeland PR, Driscoll DM. Purification, redox sensitivity, and RNA binding properties of SECIS-binding protein 2, a protein involved in selenoprotein biosynthesis. J. Biol. Chem. 1999;274:25447–25454. doi: 10.1074/jbc.274.36.25447. [DOI] [PubMed] [Google Scholar]
- 29.Gerber CA, Relich A, Driscoll DM. Isolation of an mRNA-binding protein involved in C-to-U editing. Methods Mol. Biol. 2004;265:239–249. doi: 10.1385/1-59259-775-0:239. [DOI] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Ginisty H, Sicard H, Roger B, Bouvet P. Structure and functions of nucleolin. J. Cell Sci. 1999;112(Pt 6):761–772. doi: 10.1242/jcs.112.6.761. [DOI] [PubMed] [Google Scholar]
- 32.Jiang Y, Xu XS, Russell JE. A nucleolin-binding 3′ untranslated region element stabilizes beta-globin mRNA in vivo. Mol. Cell Biol. 2006;26:2419–2429. doi: 10.1128/MCB.26.6.2419-2429.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sengupta TK, Bandyopadhyay S, Fernandes DJ, Spicer EK. Identification of nucleolin as an AU-rich element binding protein involved in bcl-2 mRNA stabilization. J. Biol. Chem. 2004;279:10855–10863. doi: 10.1074/jbc.M309111200. [DOI] [PubMed] [Google Scholar]
- 34.Singh K, Laughlin J, Kosinski PA, Covey LR. Nucleolin is a second component of the CD154 mRNA stability complex that regulates mRNA turnover in activated T cells. J. Immunol. 2004;173:976–985. doi: 10.4049/jimmunol.173.2.976. [DOI] [PubMed] [Google Scholar]
- 35.Chen CY, Gherzi R, Andersen JS, Gaietta G, Jurchott K, Royer HD, Mann M, Karin M. Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation. Genes Dev. 2000;14:1236–1248. [PMC free article] [PubMed] [Google Scholar]
- 36.Srivastava M, Pollard HB. Molecular dissection of nucleolin's role in growth and cell proliferation: new insights. FASEB J. 1999;13:1911–1922. [PubMed] [Google Scholar]
- 37.Zhang Y, Bhatia D, Xia H, Castranova V, Shi X, Chen F. Nucleolin links to arsenic-induced stabilization of GADD45alpha mRNA. Nucleic Acids Res. 2006;34:485–495. doi: 10.1093/nar/gkj459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fahling M, Steege A, Perlewitz A, Nafz B, Mrowka R, Persson PB, Thiele BJ. Role of nucleolin in posttranscriptional control of MMP-9 expression. Biochim. Biophys. Acta. 2005;1731:32–40. doi: 10.1016/j.bbaexp.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 39.Wu R, Shen Q, Newburger PE. Recognition and binding of the human selenocysteine insertion sequence by nucleolin. J. Cell Biochem. 2000;77:507–516. doi: 10.1002/(sici)1097-4644(20000601)77:3<507::aid-jcb15>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 40.Mili S, Steitz JA. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004;10:1692–1694. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clery A, Blatter M, Allain FH. RNA recognition motifs: boring? Not quite. Curr. Opin. Struct. Biol. 2008;18:290–298. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 42.Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 43.Ghisolfi-Nieto L, Joseph G, Puvion-Dutilleul F, Amalric F, Bouvet P. Nucleolin is a sequence-specific RNA-binding protein: characterization of targets on pre-ribosomal RNA. J. Mol. Biol. 1996;260:34–53. doi: 10.1006/jmbi.1996.0380. [DOI] [PubMed] [Google Scholar]
- 44.Serin G, Joseph G, Faucher C, Ghisolfi L, Bouche G, Amalric F, Bouvet P. Localization of nucleolin binding sites on human and mouse pre-ribosomal RNA. Biochimie. 1996;78:530–538. doi: 10.1016/0300-9084(96)84759-6. [DOI] [PubMed] [Google Scholar]
- 45.Bouvet P, Jain C, Belasco JG, Amalric F, Erard M. RNA recognition by the joint action of two nucleolin RNA-binding domains: genetic analysis and structural modeling. EMBO J. 1997;16:5235–5246. doi: 10.1093/emboj/16.17.5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ginisty H, Amalric F, Bouvet P. Two different combinations of RNA-binding domains determine the RNA binding specificity of nucleolin. J. Biol. Chem. 2001;276:14338–14343. doi: 10.1074/jbc.M011120200. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Tsaprailis G, Bowden GT. Nucleolin stabilizes Bcl-X L messenger RNA in response to UVA irradiation. Cancer Res. 2008;68:1046–1054. doi: 10.1158/0008-5472.CAN-07-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fahling M, Mrowka R, Steege A, Nebrich G, Perlewitz A, Persson PB, Thiele BJ. Translational control of collagen prolyl 4-hydroxylase-alpha(I) gene expression under hypoxia. J. Biol. Chem. 2006;281:26089–26101. doi: 10.1074/jbc.M604939200. [DOI] [PubMed] [Google Scholar]
- 49.Carlson BA, Xu XM, Gladyshev VN, Hatfield DL. Selective rescue of selenoprotein expression in mice lacking a highly specialized methyl group in selenocysteine tRNA. J. Biol. Chem. 2005;280:5542–5548. doi: 10.1074/jbc.M411725200. [DOI] [PubMed] [Google Scholar]
- 50.Howard MT, Aggarwal G, Anderson CB, Khatri S, Flanigan KM, Atkins JF. Recoding elements located adjacent to a subset of eukaryal selenocysteine-specifying UGA codons. EMBO J. 2005;24:1596–1607. doi: 10.1038/sj.emboj.7600642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sunde RA. In: Selenium: Its Molecular Biology and Role in Human Health. Hatfield DL, editor. Norwell, MA: Kluwer Academic Publishers; 2001. pp. 81–98. [Google Scholar]
- 52.Jeong DW, Kim EH, Kim TS, Chung YW, Kim H, Kim IY. Different distributions of selenoprotein W and thioredoxin during postnatal brain development and embryogenesis. Mol. Cells. 2004;17:156–159. [PubMed] [Google Scholar]
- 53.Lescure A, Rederstorff M, Krol A, Guicheney P, Allamand V. Selenoprotein function and muscle disease. Biochim. Biophys. Acta. 2009;1790:1569–1574. doi: 10.1016/j.bbagen.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Kohrle J. Selenium and the control of thyroid hormone metabolism. Thyroid. 2005;15:841–853. doi: 10.1089/thy.2005.15.841. [DOI] [PubMed] [Google Scholar]
- 55.Wingler K, Bocher M, Flohe L, Kollmus H, Brigelius-Flohe R. mRNA stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. Eur. J. Biochem. 1999;259:149–157. doi: 10.1046/j.1432-1327.1999.00012.x. [DOI] [PubMed] [Google Scholar]
- 56.Schneider MJ, Fiering SN, Pallud SE, Parlow AF, St Germain DL, Galton VA. Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol. Endocrinol. 2001;15:2137–2148. doi: 10.1210/mend.15.12.0740. [DOI] [PubMed] [Google Scholar]
- 57.Schneider MJ, Fiering SN, Thai B, Wu SY, St Germain E, Parlow AF, St Germain DL, Galton VA. Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology. 2006;147:580–589. doi: 10.1210/en.2005-0739. [DOI] [PubMed] [Google Scholar]
- 58.Esworthy RS, Aranda R, Martin MG, Doroshow JH, Binder SW, Chu FF. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;281:G848–G855. doi: 10.1152/ajpgi.2001.281.3.G848. [DOI] [PubMed] [Google Scholar]
- 59.Fomenko DE, Novoselov SV, Natarajan SK, Lee BC, Koc A, Carlson BA, Lee TH, Kim HY, Hatfield DL, Gladyshev VN. MsrB1 (methionine-R-sulfoxide reductase 1) knock-out mice: roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J. Biol. Chem. 2009;284:5986–5993. doi: 10.1074/jbc.M805770200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang C, Maiguel DA, Carrier F. Identification of nucleolin and nucleophosmin as genotoxic stress-responsive RNA-binding proteins. Nucleic Acids Res. 2002;30:2251–2260. doi: 10.1093/nar/30.10.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daniely Y, Borowiec JA. Formation of a complex between nucleolin and replication protein A after cell stress prevents initiation of DNA replication. J. Cell Biol. 2000;149:799–810. doi: 10.1083/jcb.149.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanakahi LA, Bu Z, Maizels N. The C-terminal domain of nucleolin accelerates nucleic acid annealing. Biochemistry. 2000;39:15493–15499. doi: 10.1021/bi001683y. [DOI] [PubMed] [Google Scholar]
- 63.Bouvet P, Diaz JJ, Kindbeiter K, Madjar JJ, Amalric F. Nucleolin interacts with several ribosomal proteins through its RGG domain. J. Biol. Chem. 1998;273:19025–19029. doi: 10.1074/jbc.273.30.19025. [DOI] [PubMed] [Google Scholar]
- 64.Barel M, Hovanessian AG, Meibom K, Briand JP, Dupuis M, Charbit A. A novel receptor - ligand pathway for entry of Francisella tularensis in monocyte-like THP-1 cells: interaction between surface nucleolin and bacterial elongation factor Tu. BMC Microbiol. 2008;8:145. doi: 10.1186/1471-2180-8-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.