

Abstract
Oxidative damage is implicated in many neurological disorders including ischemic cerebral white matter injury. Oligodendrocyte precursors (preOLs) are intrinsically highly susceptible to various forms of oxidative stress. Here we report the identification of RIP1 kinase as a signaling molecule that mediates arachidonic acid- and glu-tathione depletion-induced oxidative death of preOLs. Blockade of RIP1 kinase activity with the specific allosteric inhibitor, necrostatin-1, rescued preOLs from arachidonic acid, cystine deprivation, and buthionine sulfoximine, but not hydrogen peroxide, induced necrosis. Arachidonic acid triggered robust production of reactive oxygen species (ROS) and sustained activation of the JNK pathway in preOLs, whereas inhibition of JNK significantly prevented cell death. Treatment of cells with necrostatin-1 efficiently abolished arachidonic acid-induced ROS production and JNK activation, indicating that RIP1 kinase activation is an upstream event. This study provides the first evidence that RIP1 kinase may play an active role in arachidonic acid- and glutathione depletion-mediated oxidative damage and suggests the therapeutic potential of necrostatin-1 in protecting undifferentiated OLs against oxidative injury.
Keywords: Arachidonic acid, oligodendrocyte precursors, programmed cell death, oxidative stress, RIP1, periventricu-lar leukomalacia, necroptosis, necrosis
Introduction
Oxidative stress-induced cell damage is implicated in many neurological disorders including stroke, spinal cord injury, and periventricular leukomalacia (PVL), the major underlying pathology for cerebral palsy and cognitive impairment associated with prematurity [1, 2]. Hy-poxia/ischemia and maternal/fetal infection are the two primary etiologies for PVL. Premyelinat-ing oligodendrocytes (preOLs) as well as axons [3] are damaged in PVL [4], resulting in subsequent myelination abnormalities and neurological deficits in long-term survivors. The exquisite susceptibility of preOLs to free radical-mediated oxidative damage in comparison to mature differentiated OLs appears to account for the vulnerability of developing cerebral white matter to hypoxic/ischemic insults [5-9]. Reactive oxygen/ nitrogen species, oxidants and arachidonate metabolites are all elevated in the cerebral spinal fluid of PVL patients [10] and in PVL white matter lesions [11]. Blockade of oxidative stress with N-acetylcysteine, an antioxidant and precursor for glutathione (GSH) synthesis, ameliorates preOL damage and hypomyelination in an inflammation animal model of PVL [12]. These results suggest that oxidative injury to preOLs plays an important role in the pathogenesis of PVL [13].
Increased production of free arachidonic acid occurs during brain ischemia and inflammation as a result of activation of phospholipases [14]. Arachidonic acid is a substrate for cycloxy-genases and lipoxygenases (LOX). Metabolism of arachidonic acid through these two enzyme systems is a potential source for free radicals and peroxides and thus oxidative damage. Altered arachidonic acid metabolism has been implicated in cerebral ischemic injury, neurode-generative diseases and psychiatric disorders [15]. Arachidonate metabolites and lipid peroxi-dation products such as 4-hydroxynonenal and isoprostanes are in fact found in premyelinating OLs in PVL lesions [10, 11]. In culture, when cells such as immature neurons and preOLs are exposed to free arachidonic acid or subjected to various GSH depletion paradigms, they undergo oxidative cell death in a 12-LOX dependent manner [6, 16-20]. Consistently, inhibition of 12/15-LOX has been shown to ameliorate hy-poxic-ischemic injury in animal models of stroke [21].
We previously demonstrated that arachidonic acid-induced death of preOLs is dependent on 12-LOX activation and reactive oxygen species (ROS) production and is independent of cas-pase activation [22]. Based on morphological characteristics of dying preOLs, such as cell swelling and nuclear condensation, we asked here whether arachidonic acid triggers programmed necrosis (also named necroptosis). Necroptosis is a unique type of cell death that, unlike classic necrosis, is controllable and involves specific signaling events. A small-molecule inhibitor of death receptor-induced necroptosis, necrostatin-1 (Nec-1), was identified a few years ago and has been shown to be specific for programmed necrotic cell death, but not apoptotic cell death [23]. Nec-1 specifically targets the serine/threonine kinase RIP1 (receptor-interacting protein 1) and inhibits its kinase activity [24]. The RIP kinase family members are essential sensors for cellular stress. RIP1 has emerged as a bifunctional molecule, being either a pro-survival scaffold protein or a kinase that promotes programmed necrosis [25]. The kinase activity of RIP1 and its interaction with RIP3 have been shown to be responsible for TNFα, Fas and TRAIL-induced necrosis [24, 26-29]. In this study, we report that programmed necrosis contributes to arachidonic acid-induced oxidative cell death in primary preOL cultures. Our finding of a role for RIP1 kinase in mediating arachidonic acid toxicity suggests a receptor-independent pathway for RIP1 kinase activation.
Materials and Methods
Materials
Hank's Balanced Salt Solution (HBSS) and Earle's Balanced Salt Solution (EBSS) were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum and Dulbecco's Modified Eagle Medium (DMEM) were from Hyclone (Logan, UT). Human platelet-derived growth factor (PDGF) and basic fibroblast growth factor (b-FGF) were from PeproTech (Rocky Hill, NJ). 2,3,5-trimethyl-6-(12 -hydroxy-5,10-dodecadiynyl)-1,4-benzoquinone (AA861) was purchased from BioMol Research Laboratory (Plymouth, PA). SP600125 was from EMD Chemicals (Gibbstown, NJ). JNK and pJNK antibodies were obtained from Cell Signaling Technology (Danvers, MA). RIP1 and RIP3 antibodies were from R&D Systems (Minneapolis, MN) and IMGENEX (San Diego, CA), respectively. Unless specified otherwise, all other reagents were from Sigma (St. Louis, MO).
Primary cell cultures
Primary preOL cultures were prepared from the forebrains of 1 to 2-d-old Sprague-Dawley rat pups using a differential detachment method as described previously [30-32]. Briefly, forebrains free of meninges were digested with HBSS containing 0.01% trypsin and 10 μg/ml DNase, and triturated with DMEM containing 20% heat-inactivated fetal bovine serum and 1% penicillin -streptomycin. Dissociated cells were plated onto poly-d-lysine coated 75cm2 flasks and fed every other day for 7-10 days. Following 1 h preshake to remove microglia, the flasks were shaken overnight at 200 rpm to separate preOLs from the astrocyte layer. The cell suspension was then plated onto uncoated petri dishes for 1 h to further remove residual contaminating microglia/ astrocytes. Resulting preOLs were plated into poly-L-ornithine-coated culture plates, and maintained in a serum-free basal defined medium (BDM, 0.1% bovine serum albumin, 50 μg/ml human apo-transferrin, 50 μg/ml insulin, 30 nM sodium selenite, 10 nM D-biotin and 10 nM hydrocorti-sone in DMEM) supplemented with PDGF 10 ng/ ml and bFGF 10 ng/ml for 5-9 days at 37°C in a humid atmosphere of 5% CO2 and 95% air. Culture medium was half changed every other day, and cells were used between DIV7-9. The cultures contained primarily OL precursors (O4+, O1-) and are referred to as preOLs. Contamination by astrocytes and microglia was normally less than 2%–4%.
Cell treatments
To induce oxidative stress, unless specified otherwise, freshly prepared arachidonic acid (stock concentration 100 mM) in anhydrous dimethyl sulfox-ide (DMSO) was added to cells in their growth medium at concentrations specified in the figure legends. Nec-1 and AA861 were also made in anhydrous DMSO as a 1000* stock solution and added at the same time as arachidonic acid. The final concentration of DMSO in the culture medium was < 0.1%, and had no effect on cell viability, proliferation, or morphology. DMSO was used as vehicle controls in all experiments. For GSH depletion experiments, preOLs were treated with either buthionine sulfoximine (BSO) or subjected to cystine deprivation as detailed earlier [33]. Briefly, cells were washed two times with EBSS containing 1 mg/ml BSA and then exposed to growth medium containing no cystine. Omission of cystine, the precursor for GSH synthesis, in the culture medium resulted in gradual depletion of intra-cellular GSH and thus, oxidative cell death [5, 34, 35].
Cell viability assay
Cell viability was determined 20-24 h after treatment using Alamar Blue (Southern Biotechnology, Birmingham, AL), a tetrazolium dye that is reduced by living cells to a colored product as previously described [33]. This assay is similar in principle to the 3-(4,5-dimethyldiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) cell viability assay and has been previously validated as an accurate measure of preOL survival in our culture system [36]. All results of cell death assays were also confirmed by visual inspection of cells under a phase contrast light microscope. Survival assays were performed in triplicate and expressed as mean ± SD. In some cases, cell toxicity was also assessed by lactate dehydrogenase (LDH) release assay (Roche Diagnostics, Indianapolis, IN). Briefly, culture media were collected at the end of experiments and analyzed for LDH activity according to the manufacturer's protocol.
Intracellular free radical accumulation measurement and imaging
Intracellular free radical generation was evaluated with dichlorohydrofluorescin diacetate (DCFH-DA) and dihydrorhodamine 123 (Rho123; Invitrogen, Carlsbad, CA) as previously described [22, 33]. DCFH-DA and Rho123 were prepared as 100 mM and 10 mM stocks, respectively, in DMSO and stored in the dark at -20°C. After cells were treated with arachidonic acid for 5-18 h, DCFH-DA (100 μM) or dihydrorhodamine (10 μM) was added directly to the cells and incubated for 20 min at 37°C. Extracellular DCFH-DA or dihydrorhodamine was then removed by washing the cells twice with EBSS. The fluorescence of the cells loaded with DCFH-DA was measured with a fluorescence plate reader (FluoStar Optima, BMG Labtech) using excitation wavelength of 485 nm and emission wavelength of 530 nm. For live fluorescence imaging of oxidized Rho123, cells were immediately visualized using a fluorescence microscope (Olympus IX71) equipped with an Olympus DP70 digital camera. For image acquisition, the microscope settings, such as brightness, contrast, and exposure time, were held constant to compare the relative intensity of oxidized Rho123 across all treatment conditions.
Reverse transcription-PCR
PreOL RNA was extracted with Trizol according to the manufacturer's instructions (Sigma-Aldrich, St. Louis, MO). Samples were reverse transcribed to cDNA using a reverse transcription system kit (Promega, Madison, WI), and random primers in the presence or absence of reverse transcriptase for 10 min at 25°C, followed by 15 min at 45°C, 5 min at 95°C, and 5 min at 4°C. PCR was then performed to test for the presence of RIP1 usingspecific primers. The primers used in the study were RIP1, forward-GCACCAGCTGTCAGGGCCAG, reverse-GCCCAGCTTTCGGGCACAGT; and β-actin, forward- AGACTTCGAGCAGGAGATGG, reverse-CCATCATGAAGTGTGACGTTG. Briefly, after an initial denature step at 95°C for 10 min, 100 ng of cDNA per reaction was subjected to 30 cycles of PCR (95°C 15 sec, 56°C 15 sec, 72°C 30 sec) followed by a final elongation step at 72°C for 5 min. Products were electrophoresed on 2% agarose gels and visualized under UV light using a Bio-Rad Chemidoc XRS gel documentation system.
Western blotting analysis
Cell lysates were subjected to SDS-polyacrylamide gel electrophoresis followed by electrotransferring of separated proteins to PVDF membrane. Nonspecific binding was blocked with TBS-T (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1%Tween-20) containing 5% non-fat milk for 1 h at room temperature. Anti RIP1 (1: 1000 dilution, mouse IgG), RIP3 (1:1000 dilution, Rabbit IgG), JNK (1:1000 dilution), Phos-pho-JNK (1:1000 dilution), and β-actin (1:10,000 dilution) antibodies were diluted in TBS-T containing 2% non-fat milk and incubated overnight with the membrane at 4°C. After washing 3-5 times with TBS-T, the membrane was incubated with horseradish peroxidase conjugated secondary antibody (1:1000) for 1 h with agitation. Following 4-5 washes with TBS-T, protein bands were visualized by chemilumi-nescence using the SuperSignal detection kit (Thermo Scientific, Rockford, IL).
Statistical analysis
All cell culture treatments were performed in triplicate. GraphPad Prism software was used for data analyses. One-way ANOVA followed by Bonferroni's post-hoc multiple comparison test was used to determine statistical significance. Comparison between two experimental groups was based on two-tailed ttest. P<0.05 was considered statistically significant. Results are presented as mean ± SD unless otherwise indicated.
Results
RIP1 kinase inhibitor Necrostatin-1 prevents arachidonic acid-induced cell death
As we showed previously, challenging preOLs with free arachidonic acid triggers an oxidative cell death pathway that is dependent on activation of 12-LOX and production of ROS [6, 22]. We confirmed here that arachidonic acid-induced cytotoxicity was markedly prevented by antioxidant butylated hydroxyanisole (BHA) and the 12-LOX specific inhibitor AA861 (Figure 1A). Blockade of caspase activation with the pan caspase inhibitor z-VAD-fmk had no protective effect, indicating a non-apoptotic cell death pathway [22]. Morphologically, arachidonic acid-treated preOLs underwent cell swelling and nuclear condensation prior to cell lysis and death (data not shown). Since specific cell signaling events occur in arachidonic acid toxicity, we reasoned that a programmed necrotic cell death pathway may account for this toxicity and thus tested if blockade of programmed necrosis prevents arachidonic acid-induced cell death.
Figure 1.

Blockade of RIP1 kinase activity with necrostatin-1 abrogates arachidonic acid-induced death of preOLs and ROS production. (A, B). RIP1 kinase inhibitor Nec-1 prevents arachidonic acid (AA)-induced cell death. PreOLs were treated with AA (100μM), Nec-1 (20μM), antioxidant BHA (75μM), or 12-LOX inhibitor AA861 (10μM) as indicated for 20-24 h and cell viability was determined. Data represent mean ± SEM of 3-5 independent experiments. (C, D). AA-induced ROS production is significantly inhibited by Nec-1. ROS production was quantified with DCFH-DA (C) after overnight treatment with AA (100μM) in the presence or absence of Nec-1 (20μM) or visualized with dihy-drorhodamine 123 (Rho123) (D). Data are a representative of 2-4 independent experiments. Scale bars, 20μm. *, p<0.01; **, p<0.001; ns, not significant.
Nec-1 is a highly specific small molecule that allosterically targets RIP1 kinase [24] and has been shown to be a powerful tool to characterize the role of programmed necrosis in vitro and in vivo [23, 26-29]. When preOLs were treated with arachidonic acid in the presence of Nec-1, cell death was completely prevented (Figure 1A, B). Moreover, arachidonic acid-induced production of ROS was also significantly abolished by Nec-1 (Figure 1C, D). As we demonstrated before [22], 12-LOX inhibitor AA861 completely abrogated ROS production. These results indicate that arachidonic acid triggers a necrotic cell death pathway that requires RIP1 kinase activity.
Nec-1 protects against glutathione depletion induced oxidative damage
Since 12-LOX plays an essential role in arachidonic acid-induced preOL cell death, we next asked whether Nec-1 is also protective in other oxidative cell death paradigms where 12-LOX activation is shown to be necessary. GSH is a major antioxidant in cells and depletion of GSH results in oxidative stress. Experimentally, GSH can be depleted by either omitting cystine from culture medium, thereby depleting intracellular cysteine, the precursor for GSH synthesis, or with BSO, an inhibitor of γ-glutamylcysteine syn-thetase. In both cases, 12-LOX has been shown to be involved in ROS production and cell death [6, 16-20]. We found that Nec-1 was indeed able to rescue preOLs from both cystine deprivation and BSO-induced cell death (Figure 2).
Figure 2.
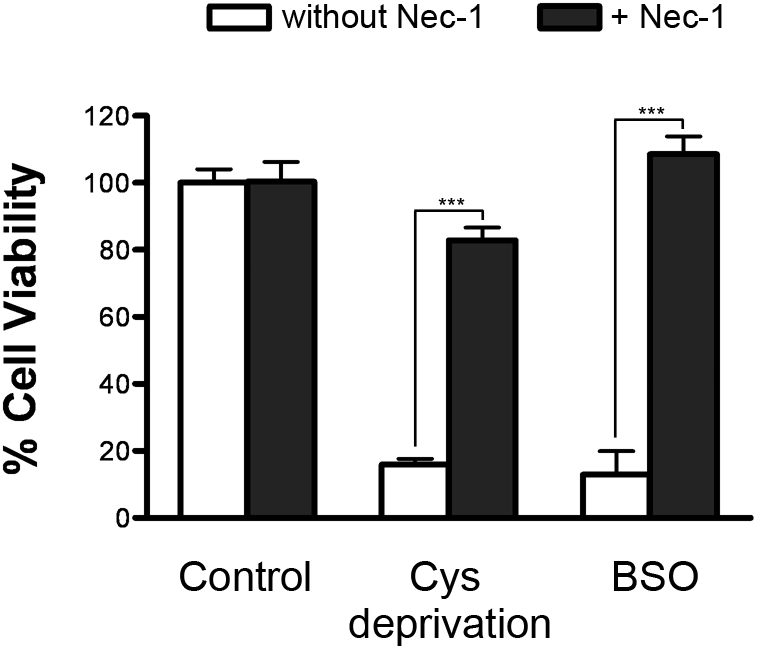
Nec-1 blocks oxidative cell death induced by glutathione-depleting agents. PreOLs were subjected to cystine deprivation or treated with BSO (5mM) in the presence and absence of Nec-1 (20 μM) for 24 h and cell viability was analyzed. BSO inhibits γ-glutamylcysteine synthetase and results in intracellular GSH depletion and oxidative cell death. Co-treatment with Nec-1 markedly blocked cystine deprivation and BSO-induced cell death. Results are shown as mean ± SD of a representative of 3 independent experiments. ***, p<0.001.
RIP1 and RIP3 are expressed in preOLs and their expression levels are not affected by arachidonic acid treatment
Given the profound protective effect of Nec-1 against arachidonic acid- and GSH depletion-mediated cell death, we examined whether RIP1, which is the target of Nec-1, is in fact expressed in preOLs and whether its level is affected by arachidonic acid treatment in culture. PreOLs express abundant RIP1 transcript and protein as determine by RT-PCR and Western blotting analysis (Figure 3). Treatment of cells with arachidonic acid and Nec-1 did not affect the protein level of RIP1 or RIP3 (Figure 3B). Since caspases are known to cleave RIP1, thereby ensuring apoptotic cell death, this result agrees with our previous observation that caspases are not involved in arachidonic acid-induced cell death [22]. Furthermore, this result reinforces the concept that it is RIP1 kinase activity that mediates arachidonic acid-induced programmed necrosis.
Figure 3.

RIP1 and RIP3 are expressed in preOLs and their expression level is not affected by arachidonic acid treatment. (A). RT-PCR analysis of RIP1 transcript in cultured preOLs. RT, reverse transcrip-tase. (B). Western blotting analyses of RIP1/3 protein levels. PreOLs were treated with AA (100μM) in the presence and absence of Nec-1 (20μM) for 1 or 5 h. AA treatment did not change RIP1/3 expression. Nec-1, which inhibits RIP1 kinase activity and prevents AA toxicity, also had no effect on RIP1/3 expression.
JNK pathway is activated by arachidonic acid in preOLs and is inhibited by Nec-1
Persistent activation of Jun N-terminal kinase (JNK), a family of stress-activated protein kinases, has been implicated in programmed necrosis in at least some cell types [25]. The JNK family comprises three members, JNK1, JNK2 and JNK3. The first two are ubiquitously distributed, whereas JNK3 is confined to the central nervous system and cardiac myocytes. To determine whether arachidonic acid-triggered oxidative damage to preOLs is mediated through the JNK pathway, we used a potent and specific JNK kinase inhibitor, SP600125 [37]. As demonstrated in Figure 4A and B, blockade of JNK activity with SP600125 significantly eliminated arachidonic acid-induced cell death. It should be noted that at the concentration of 7.5uM, SP600125 was itself mildly toxic to preOLs, suggesting that physiological activation of JNK may be required for maintaining preOLs in the mitotic state [38, 39]. Nevertheless, blocking JNK activity increased cell viability to the same level as cells treated with SP600125 alone. Western blotting analyses confirmed activation of JNK upon arachidonic acid treatment (Figure 4C, D). Furthermore, co-treatment with Nec-1 significantly prevented arachidonic acid-induced phosphoryla-tion of JNK. Interestingly, whereas SP600125 prevented arachidonic acid-induced cell death, it did not block ROS production as determined by the DCF measurement, suggesting that JNK activation is downstream of ROS production. Taken together, our results suggest that arachidonic acid-induced activation of RIP1 kinase functions upstream of ROS production and JNK activation.
Figure 4.

The JNK pathway is involved in arachidonic acid-triggered oxidative death and is blocked by Nec-1. (A). JNK inhibitor SP600125 significantly prevented AA-induced cell death. PreOLs were treated with AA (100μM), Nec-1 (20μM) and SP600125 (7.5μM) as indicated. Cell viability was analyzed 20-24h later. *, p<0.05; ***, p<0.001. Results represent 3 independent experiments. (B). Representative phase contrast images of cells treated as in (A). (C). Nec-1 inhibits AA-induced activation of JNK. PreOLs were treated with vehicle, AA (100μM) or AA + Nec-1 (20μM) for 6 h and then subjected to western blotting analyses. AA treatment caused JNK activation, which was inhibited by Nec-1. (D). Quantification of pJNK levels of preOLs treated as indicated. Results are mean from two independent experiments. (E). JNK inhibitor SP600125 attenuates AA-induced cell death but not AA-induced ROS production as determined by DCF measurement. ***, p<0.001 when compared with the control; ns, not significant. Data are a representative of 2-3 independent experiments.
Necrostatin-1 does not prevent hydrogen peroxide-induced oxidative damage
Based on our above findings on an essential role for RIP1 in oxidative damage induced by arachidonic acid and GSH depletion, we questioned whether this RIP1's involvement can be generalized to other oxidative stress conditions. Hydrogen peroxide (H2O2) is a major oxidant naturally produced in cells and is decomposed catalytically by catalase and glutathione peroxi-dase [40]. H2O2 causes oxidative damage in many cell types and has been widely used to study oxidative stress. As expected and consistent with our previous data [40], preOLs treated with increasing concentrations of H2O2showed dose-dependent loss of cell viability (Figure 5). However, blockade of RIP1 kinase with Nec-1 or 12-LOX with AA861 did not offer any significant protection against H2O2 toxicity. This is in stark contrast to their protective effect against GSH depletion-mediated oxidative death, further demonstrating that specific signaling pathways are involved in various forms of oxidative damage.
Figure 5.

Nec-1 does not prevent hydrogen peroxide-induced oxidative cell death. Hydrogen peroxide causes oxidative cell death of preOLs in a dose-dependent manner. Co-treatment with Nec-1 (20μM) or AA861 (10μM) did not prevent cell death. PreOLs were treated with increasing concentrations of H2O2 in the presence or absence of Nec-1 and AA861 for 24h. Cell toxicity was evaluated by LDH release assay (A), and cell viability was assessed by Alamar-Blue analyses (B). Data are representative of three independent experiments.
Discussion
OL precursors are highly sensitive to oxidative stress-induced damage, in part due to a weaker antioxidant defense system as compared to mature OLs [41]. This developmental stage specific vulnerability of preOLs to oxidative stress and to AMPA/kainate excitotoxicity predisposes them to various types of injury that lead to mye-lination abnormalities [5-9, 41]. In this study, we investigated key signaling events involved in arachidonic acid and GSH depletion, we questioned whether this RIP1's involvement can be generalized to other oxidative stress conditions. Hydrogen peroxide (H2O2) is a major oxidant naturally produced in cells and is decomposed catalytically by catalase and glutathione peroxi-dase [40]. H2O2 causes oxidative damage in many cell types and has been widely used to study oxidative stress. As expected and consistent with our previous data [40], preOLs treated with increasing concentrations of H2O2showed dose-dependent loss of cell viability (Figure 5). However, blockade of RIP1 kinase with Nec-1 or 12-LOX with AA861 did not offer any significant protection against H2O2 toxicity. This is in stark contrast to their protective effect against GSH depletion-mediated oxidative death, further demonstrating that specific signaling pathways are involved in various forms of oxidative damage.
Discussion
OL precursors are highly sensitive to oxidative stress-induced damage, in part due to a weaker antioxidant defense system as compared to mature OLs [41]. This developmental stage specific vulnerability of preOLs to oxidative stress and to AMPA/kainate excitotoxicity predisposes them to various types of injury that lead to mye-lination abnormalities [5-9, 41]. In this study, we investigated key signaling events involved in arachidonic acid-induced oxidative death of preOLs in culture. We demonstrate that both arachidonic acid and GSH depletion trigger a programmed necrotic death pathway that is regulated by the kinase activity of RIP1. Specific inhibition of RIP1 kinase activity with Nec-1 markedly prevents cell death via blocking ROS production and JNK activation.
RIP1 belongs to the RIP family of serine/ threonine kinases that are involved in innate and adaptive immunity. RIP1 contains an N-terminal kinase domain, an intermediate domain, a RIP homotypic motif that enables its interaction with RIP3, and a C-terminal death domain (DD), through which it interacts with death receptor TNFR1 and also with DD-containing adaptor proteins such as TRADD and FADD [25]. Recent studies identified RIP1 and RIP3 as key regulators of programmed necrosis induced by TNF, Fas, Toll-like receptor 3 (TLR3) and 4. RIP1 has now emerged as a tightly regulated signaling molecule with bifurcated functions, the kinase-independent proinflammatory function and the kinase-dependent pro-necrosis function, under the control of caspases and ubiquitination. The identification of Nec-1 as a small-molecule that allosterically inhibits the kinase activity of RIP1 without affecting RI P1-mediated activation of NFkB allows one to dissect the role of RIP1 in necrotic cell death [23, 24]. It should be noted that Nec-1 is not an anti-oxidant [23, 42]. Our finding that arachidonic acid-induced oxidative death of preOLs is prevented by Nec-1 reveals that programmed necrosis may be a common pathway for certain oxidative cell death and suggests that receptor-independent activation of RIP1 kinase may occur under GSH depletion paradigms. At present, it is unclear how arachidonic acid or GSH depletion activates RIP1 kinase. The possibility that arachidonic acid induces RIP1 kinase activation indirectly through TNF production is highly unlikely in our experimental conditions since direct addition of TNF to preOLs fails to cause significant loss of preOL viability [43]. Furthermore, Nec-1 does not prevent lipopolysaccha-ride-induced, TNF-dependent killing of preOLs in mixed glial cultures (Li, unpublished data). Thus, a receptor-independent RIP1 activation seems operative in arachidonic acid-challenged or GSH -depleted preOLs. Since RIP1 is regulated by ubiquitination, and deubiquitination enables its dissociation from the TNF receptor complex and subsequent interaction with RIP3 to form necro-some [25], one potential mechanism is that the redox status of preOLs regulates RIP1 kinase activation indirectly through affecting the ubiquitination pathway. The ubiquitination machinery has in fact been previously shown to be under redox control [44]. Intracellular GSH depletion dose-dependently reduces the levels of endogenous ubiquitinated protein conjugates. A20, a RIP1 deubiquitination enzyme, is upregu-lated by oxidative stress and promotes necrotic cell death [45]. Recently, in a genome-wide siRNA screen for regulators of programmed necrosis induced by the caspase inhibitor zVAD, Hitomi et al [46] identified the involvement of the GSH metabolic pathway in RIP1 kinase-dependent necrosis. Reducing glutathione per-oxidase and glutathione S-transferases via siRNA, and presumably the subsequent increase of free GSH level, protected L929 cells against zVAD-induced necrosis [46]. This observation is consistent with our finding that arachidonic acid and GSH depletion trigger a receptor-independent, RIP1 kinase-dependent necrosis, and is in line with our speculation that RIP1 kinase activation may be under redox control. A similar protective effect of Nec-1 has also been found in glutamate-induced GSH depletion and oxidative death of HT-22 cells [42]. Moreover, nitric oxide-induced endothelial cell death has recently been shown to be mediated by RIP 1/3 and blocked by over-expression of mitochondrial superoxide dismutase [47]. Interestingly, nitric oxide toxicity has been linked to GSH depletion and 12-LOX activation in primary midbrain cultures [19]. Apparently, further work is needed to unravel the mechanism of receptor-independent activation of RIP1 kinase and its yet-to-be identified downstream substrate.
Our results revealed a causal role for RIP1 kinase in arachidonic acid-, cystine deprivation-, and BSO-induced oxidative damage to preOLs. In all three cell death paradigms, 12-LOX activation is essential. Blocking either 12-LOX or RIP1 kinase confers significant protection. For oxidative cell death that is not dependent on 12-LOX, such as that induced by H2O2, Nec-1 is ineffective. Furthermore, our preliminary data suggest that Nec-1 has no effect on kainate-triggered excitotoxicity to preOLs, which is 12-LOX independent (Li unpublished data). The functional relationship between 12-LOX and RIP1 kinase activation is currently unknown and requires further investigation. Our identification of an essential role for RIP1 kinase in arachidonic acid-induced oxidative damage to primary preOLs has important implications for conditions like hypoxic-ischemic injury where increased arachidonic acid and oxidative stress are key pathogenic events. Necro-statins have been shown to reduce tissue damage in transient ischemic brain injury in mice [23], traumatic brain injury in mice [48], myo-cardial ischemia-reperfusion injury [49], and retinal ischemia/reperfusion injury [50]. It is of note that blockade of 12/15-LOX or 12/15-LOX deficiency also protects mice against transient focal ischemia in a mouse model of stroke [21]. Given our current findings and the known intrinsic vulnerability of preOLs to oxidative stress, blocking RIP1 kinase activity with necrostatins may offer beneficial effects in white matter injury. Therefore, it would be interesting to test whether RIP1-dependent programmed necrosis pathway contributes to hypoxic/ischemic white matter injury and other pathological conditions such as multiple sclerosis and spinal cord injury.
Acknowledgments
This work was supported by NIH grants R01NS060017 (JL), NS38475 (PAR) and HD18655 (Children's Hospital), by the research grant RG3975 from the National Multiple Sclerosis Society (JL), and by the start-up funds from Texas A&M University. We thank Drs. Junying Yuan and Alexei Degterev for providing necro-statin-1 to test during an initial preliminary phase of this study.
Glossary
Abbreviations:
- LDH
lactate dehydrogenase
- MS
multiple sclerosis
- OLs
oligodendrocytes
- preOLs
oligodendrocyte precursors
- PVL
periventricular leukomalacia
- RIP1
receptor-interacting protein 1
- RIP3
receptor-interacting protein 3
References
- 1.Volpe JJ. Neurobiology of periventricular leuko-malacia in the premature infant. Pediatr Res. 2001;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Volpe JJ. Cerebral white matter injury of the premature infant-more common than you think. Pediatrics. 2003;112:176–180. doi: 10.1542/peds.112.1.176. [DOI] [PubMed] [Google Scholar]
- 3.Haynes RL, Billiards SS, Borenstein NS, Volpe JJ, Kinney HC. Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr Res. 2008;63:656–661. doi: 10.1203/PDR.0b013e31816c825c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, Volpe JJ, Kinney HC. Nitrosative and oxidative injury to premyeli-nating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62:441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- 5.Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, Volpe JJ, Rosenberg PA. 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur J Neurosci. 2004;20:2049–2058. doi: 10.1111/j.1460-9568.2004.03650.x. [DOI] [PubMed] [Google Scholar]
- 7.Gerstner B, DeSilva TM, Genz K, Armstrong A, Brehmer F, Neve RL, Felderhoff-Mueser U, Volpe JJ, Rosenberg PA. Hyperoxia causes maturation-dependent cell death in the developing white matter. J Neurosci. 2008;28:1236–1245. doi: 10.1523/JNEUROSCI.3213-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng W, Rosenberg PA, Volpe JJ, Jensen FE. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc Natl Acad Sci U S A. 2003;100:6801–6806. doi: 10.1073/pnas.1136624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inder T, Mocatta T, Darlow B, Spencer C, Volpe JJ, Winterbourn C. Elevated free radical products in the cerebrospinal fluid of VLBW infants with cerebral white matter injury. Pediatr Res. 2002;52:213–218. doi: 10.1203/00006450-200208000-00013. [DOI] [PubMed] [Google Scholar]
- 11.Back SA, Luo NL, Mallinson RA, O'Malley JP, Wallen LD, Frei B, Morrow JD, Petito CK, Roberts CT, Jr, Murdoch GH, Montine TJ. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann Neurol. 2005;58:108–120. doi: 10.1002/ana.20530. [DOI] [PubMed] [Google Scholar]
- 12.Paintlia MK, Paintlia AS, Barbosa E, Singh I, Singh AK. N-acetylcysteine prevents endotoxin-induced degeneration of oligodendrocyte progenitors and hypomyelination in developing rat brain. J Neurosci Res. 2004;78:347–361. doi: 10.1002/jnr.20261. [DOI] [PubMed] [Google Scholar]
- 13.Haynes RL, Baud O, Li J, Kinney HC, Volpe JJ, Folkerth DR. Oxidative and nitrative injury in periventricular leukomalacia: a review. Brain Pathol. 2005;15:225–233. doi: 10.1111/j.1750-3639.2005.tb00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katsuki H, Okuda S. Arachidonic acid as a neurotoxic and neurotrophic substance. Prog Neurobiol. 1995;46:607–636. doi: 10.1016/0301-0082(95)00016-o. [DOI] [PubMed] [Google Scholar]
- 15.Phillis JW, Horrocks LA, Farooqui AA. Cyclooxygenases, lipoxygenases, and epoxy-genases in CNS: Their role and involvement in neurological disorders. Brain Res Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glu-tathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 17.Kwon K, Jung Y, Lee S, Moon C, Baik E. Arachidonic acid induces neuronal death through lipoxygenase and cytochrome P450 rather than cyclooxygenase. J Neurosci Res. 2005;81:73–84. doi: 10.1002/jnr.20520. [DOI] [PubMed] [Google Scholar]
- 18.Kramer BC, Yabut JA, Cheong J, Jnobaptiste R, Robakis T, Olanow CW, Mytilineou C. Toxicity of glutathione depletion in mesencephalic cultures: a role for arachidonic acid and its lipoxygenase metabolites. Eur J Neurosci. 2004;19:280–286. doi: 10.1111/j.1460-9568.2004.03111.x. [DOI] [PubMed] [Google Scholar]
- 19.Canals S, Casarejos MJ, de Bernardo S, Rodriguez-Martin E, Mena MA. Nitric oxide triggers the toxicity due to glutathione depletion in midbrain cultures through 12-lipoxygenase. J Biol Chem. 2003;278:21542–21549. doi: 10.1074/jbc.M213174200. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E, Rosenberg PA. Peroxynitrite-induced neuronal apoptosis is mediated by intra-cellular zinc release and 12-lipoxygenase activation. J Neurosci. 2004;24:10616–10627. doi: 10.1523/JNEUROSCI.2469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Leyen K, Kim HY, Lee SR, Jin G, Arai K, Lo EH. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke. 2006;37:3014–3018. doi: 10.1161/01.STR.0000249004.25444.a5. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Wang H, Rosenberg PA. Vitamin K prevents oxidative cell death by inhibiting activation of 12-lipoxygenase in developing oligodendrocytes. J Neurosci Res. 2009;87:1997–2005. doi: 10.1002/jnr.22029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 24.Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3 doi: 10.1126/scisignal.3115re4. re4- [DOI] [PubMed] [Google Scholar]
- 26.Zhang D-W, Shao J, Lin J, Zhang N, Lu B-J, Lin S-C, Dong M-Q, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 27.Cho Y, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK-M. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 29.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 30.McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by induc-ible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci U S A. 2005;102:9936–9941. doi: 10.1073/pnas.0502552102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, Balasubramaniyan V, Peng J, Hurlock EC, Tallquist M, Li J, Lu QR. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat Protocols. 2007;2:1044–1051. doi: 10.1038/nprot.2007.149. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Lin JC, Wang H, Peterson JW, Furie BC, Furie B, Booth SL, Volpe JJ, Rosenberg PA. Novel role of vitamin K in preventing oxidative injury to developing oligodendrocytes and neurons. J Neurosci. 2003;23:5816–5826. doi: 10.1523/JNEUROSCI.23-13-05816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ. Cystine deprivation induces oligodendroglial death: rescue by free radical scavengers and by a diffusible glial factor. J Neurochem. 1996;67:566–573. doi: 10.1046/j.1471-4159.1996.67020566.x. [DOI] [PubMed] [Google Scholar]
- 36.Back SA, Khan R, Gan X, Rosenberg PA, Volpe JJ. A new Alamar Blue viability assay to rapidly quantify oligodendrocyte death. J Neurosci Methods. 1999;91:47–54. doi: 10.1016/s0165-0270(99)00062-x. [DOI] [PubMed] [Google Scholar]
- 37.Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hart IK, Richardson WD, Raff MC. PDGF increases the expression of Fos and Jun in newly formed oligodendrocytes that have become resistant to the mitogenic effect of PDGF. Glia. 1992;6:310–313. doi: 10.1002/glia.440060409. [DOI] [PubMed] [Google Scholar]
- 39.Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R, Jessen KR. c-Jun is a negative regulator of myelination. J Cell Biol. 2008;181:625–637. doi: 10.1083/jcb.200803013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baud O, Greene AE, Li J, Wang H, Volpe JJ, Rosenberg PA. Glutathione peroxidase-catalase cooperativity is required for resistance to hydrogen peroxide by mature rat oligodendrocytes. J Neurosci. 2004;24:1531–1540. doi: 10.1523/JNEUROSCI.3989-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baud O, Haynes RF, Wang H, Folkerth RD, Li J, Volpe JJ, Rosenberg PA. Developmental up-regulation of MnSOD in rat oligodendrocytes confers protection against oxidative injury. Eur J Neurosci. 2004;20:29–40. doi: 10.1111/j.0953-816X.2004.03451.x. [DOI] [PubMed] [Google Scholar]
- 42.Xu X, Chua CC, Kong J, Kostrzewa RM, Kumaraguru U, Hamdy RC, Chua BHL. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem. 2007;103:2004–2014. doi: 10.1111/j.1471-4159.2007.04884.x. [DOI] [PubMed] [Google Scholar]
- 43.Li J, Ramenaden ER, Peng J, Koito H, Volpe JJ, Rosenberg PA. Tumor necrosis factor {alpha} mediates lipopolysaccharide-induced microglial toxicity to developing oligodendrocytes when astrocytes are present. J Neurosci. 2008;28:5321–5330. doi: 10.1523/JNEUROSCI.3995-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Obin M, Shang F, Gong X, Handelman G, Blumberg J, Taylor A. Redox regulation of ubiquitin -conjugating enzymes: mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998;12:561–569. doi: 10.1096/fasebj.12.7.561. [DOI] [PubMed] [Google Scholar]
- 45.Storz P, Doppler H, Ferran C, Grey ST, Toker A. Functional dichotomy of A20 in apoptotic and necrotic cell death. Biochem J. 2005;387:47–55. doi: 10.1042/BJ20041443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis CW, Hawkins BJ, Ramasamy S, Irrinki KM, Cameron BA, Islam K, Daswani VP, Doonan PJ, Manevich Y, Madesh M. Nitration of the mito-chondrial complex I subunit NDUFB8 elicits RIP1 - and RIP3-mediated necrosis. Free Radic Biol Med. 2010;48:306–317. doi: 10.1016/j.freeradbiomed.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:1564–1573. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostata potentially novel cardioprotective agent? Cardio-vasc Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 50.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]