

Abstract
Ethanol preconditioning (EtOH-PC) reduces postischemic neuronal injury in response to cerebral ischemia/reperfusion (I/R). We examined the mechanism underlying this protective effect by determining 1) whether it was associated with a decrease in I/R-induced leukocyte-endothelial adhesive interactions in postcapillary venules, and 2) whether the protective effects were mediated by activation of large conductance, calcium-activated potassium (BKCa) channels. Mice were administered ethanol by gavage or treated with the BKCa channel opener, NS1619, 24 hrs prior to I/R with or without prior treatment with the BKCa channel blocker, paxilline. Both common carotid arteries were occluded for 20 min followed by 2 and 3 hrs of reperfusion, and rolling (LR) and adherent (LA) leukocytes were quantified in pial venules using intravital microscopy. The extent of delayed neuronal death (DND), apoptosis and glial activation in hippocampus were assessed 4 days after I/R. Compared with sham, I/R elicited increases in LR and LA in pial venules and DND and apoptosis as well as glial activation in the hippocampus. These effects were attenuated by EtOH-PC or antecedent NS1619 administration, and this protection was reversed by prior treatment with paxilline. Our results support a role for BKCa channel activation in the neuroprotective effects of EtOH-PC in cerebral I/R.
Keywords: Large conductance, Ca2+-activated K+ channels, Ethanol preconditioning (EtOH-PC), cerebral ischemia/reperfusion (I/R), leukocyte rolling and adhesion, neuroinflammation, delayed neuronal death (DND), apoptosis
INTRODUCTION
Epidemiologic studies have demonstrated that consumption of moderate amounts of red wine is associated with significant reductions in cardiovascular and cerebrovascular morbidity and mortality [1–4], reducing both the incidence and severity of myocardial infarction and stroke [5, 6]. Nonethanolic components of wine such as resveratrol, a polyphenolic nutrient from the grape skin, have been shown to be cardioprotective as well as neuroprotective in myocardial and cerebral ischemia/reperfusion (I/R) models by our laboratory and others [7–13]. Other reports indicate that alcohol alone limits postischemic injury and inflammation [14–16].
Ethanol preconditioning (EtOH-PC) refers to a phenomenon whereby tissues are protected from the deleterious effects of prolonged I/R by antecedent ingestion of the alcohol at low to moderate levels [16, 17]. Doses of ethanol required for this effect produce a transient increase (i.e. 30–60 min) in plasma concentrations of ethanol to approximately 10 mM, levels similar to those measured in humans consuming 1–2 alcoholic beverages [16]. The beneficial actions of EtOH-PC become apparent within 2 hours of ingestion and remain effective for 2 hours before disappearing. However, a delayed or late phase of protection re-emerges 12–24 hours after consumption of moderate levels of ethanol [16]. Because preconditioning becomes apparent only after plasma levels of ethanol have returned to baseline, it has been postulated that ethanol consumption triggers a downstream signaling cascade which induces development of a protected phenotype that limits the detrimental effects of a subsequent I/R insult. Although we have recently demonstrated that ethanol ingestion 24 hours prior to induction of cerebral ischemia/reperfusion (I/R) reduces postischemic neuronal injury [18], the underlying mechanism for the protective effect of EtOH-PC is not clear.
Large conductance, Ca2+-activated K+ channels (BKCa) are localized to cell membranes throughout the body, including the central nervous system as well as mitochondrial membrane in cardiac myocytes [19, 20]. These channels are activated by increase in cytosolic calcium largely in response to calcium influx via voltage-gated Ca2+ channels by cell membrane depolarization. BKCa channel opening allows cytosolic K+ efflux, which promotes cell membrane repolarization. This, in turn, reduces Ca2+ entry by closing voltage-dependent Ca2+ channels [21]. The first study to show neuroprotection with BKCa channel opener was done by Busija’s group [22]. Increases in BKCa channel activity cause the depression of neuronal excitability with potential beneficial effects on cell survival [19]. Indeed, it is being increasingly recognized that activation of BKCa channels is cytoprotective. For example, agonists that promote opening of BKCa channels reduce infarct size after I/R in heart and brain [23, 24], and inhibit ROS production of isolated rat brain mitochondria [25], while administration of BKCa channel inhibitors increases pyramidal neuronal cell death in hippocampal slices subjected to oxygen/glucose deprivation, an in vitro model of ischemia [26]. Several other studies have demonstrated that BKCa channel inhibitors prevent the infarct-sparing effects induced by short bouts of ischemia (ischemic preconditioning) or pharmacological agents such as silfenadil and TNFα [27–31].
Evidence obtained in our previous studies indicates that EtOH-PC prevents postischemic leukocyte-endothelial cell adhesive interactions and protects against delayed neuronal death (DND)-induced by I/R [16, 18]. Since neutrophil infiltration plays an important role in the genesis of cerebral I/R injury [32–35] and BKCa channel activation has been implicated as an initiator for ischemic [23, 28] and some forms of pharmacological preconditioning [21, 23, 24, 31, 35, 36], we hypothesized that BKCa channel activation may play an important role in EtOH-PC, reducing I/R-induced leukocyte-endothelial adhesive interactions in pial postcapillary venules and delayed neuronal death in hippocampus. Our approach to this question was to use a mouse model of cerebral I/R to determine 1) whether selective pharmacologic activation of BKCa channels would reproduce the effects of EtOH-PC, and 2) whether treatment with BKCa channel inhibitors at the time of ethanol ingestion would abrogate the protective effects of EtOH-PC to attenuate cerebral microvascular dysfunction, neuroinflammation, and DND induced by cerebral I/R.
MATERIALS AND METHODS
Animal and groups
Wild-type (WT) male C57BL/6 mice (6–7 weeks) were obtained from Jackson Laboratories (Bar Harbor, ME). All mice were given free access to water and lab chow and maintained at 22 ± 2°C with a constant humidity under a 12:12 hrs light:dark cycle. They were used at 8–10 weeks. The experimental procedures described were performed according to the criteria outlined in the National Institutes of Health guidelines and were approved by the University of Missouri Institutional Animal Care and Use Committee.
Experimental protocols
The mice were randomly divided into the following 8 treatment groups and all drug dosages were selected from previous reports in the literature:
Sham
These mice were surgically prepared (see below) in an identical manner as I/R mice, i.e. both common carotid arteries (CCA) exposed, installation of cranial window, however CCA were not occluded.
Ischemia/reperfusion (I/R)
After exposure of CCA, both arteries were occluded for 20 min, using aneurysm clips. Perfusion was re-established by removing the clips.
Ethanol preconditioning followed by I/R (EtOH-PC+I/R)
Ethanol was administered by gavage, 24 hrs prior to surgery for production of I/R.
NS1619 preconditioning followed by I/R (NS-PC+I/R)
To determine whether preconditioning with BKCa channel opener would mimic the effects of EtOH-PC, another group of mice were treated with the BKCa channel opener [37], NS1619 (NS, 0.1mg/kg, i.p.) 24 hrs prior to I/R.
Ethanol and NS-1619 preconditioning followed by I/R (EtOH-PC+NS-PC+I/R)
To determine whether there is an synergized action of enhanced BKCa channel activation under ethanol and NS1619 preconditioning paradigm, NS1619 i.p injection as well as EtOH-PC were administrated at 24 hrs prior to I/R.
Pharmacological intervention with a BKCa channel inhibitor: To determine whether BKCa channels are involved in the putative neuroprotective effects of EtOH-PC and NS-PC, additional groups of mice were treated BKCa channel inhibitor, paxilline (PX, 2.5mg/kg, i.p.) [21, 38], as described below.
Paxilline pretreatment in EtOH-PC (PX+EtOH-PC+I/R)
Paxilline was administered by i.p. injection, 10 min prior to administration of ethanol, and 24 hrs prior to I/R.
Paxilline pretreatment in NS-PC (PX+NS-PC+I/R)
Paxilline was administered 10 min prior to NS1619 preconditioning, and 24 hrs prior to I/R.
Paxilline alone followed by I/R (PX+I/R)
Paxilline was i.p. administrated 24 hrs prior to I/R.
These treatment groups and experimental protocols are described in schematic form in Figure 1. Animals in Sham, I/R-alone, EtOH-PC+I/R and NS-PC+I/R groups received i.p. injections of saline (the vehicle for paxilline) at the same volume and time as the interventional groups.
Figure 1.

Diagram of the experimental protocols assigned to each group. The numbers at top in minutes refer to the timeline of the protocol on day 1, 24 hrs before ischemia/reperfusion (I/R), day 2, the day of I/R, and day 4, the day of brain perfusion and fixation. Black triangles indicate administration of saline or drugs. White triangles indicate gavages of water or EtOH. Solid black bars indicate the 20-min period of ischemia during which both CCA had no blood flow. Short hatched bars indicate digital video recording (10 min) and long hatched bars indicate perfusion transcardially. Two sets of experimental groups, each encompassing all treatments were used: one group was used for intravital microscopic studies of leukocyte rolling and adhesion in pial postcappilary venules at both 2 and 3 hrs of reperfusion. The other group was used for histological examination of hippocampal injury at 4 days of reperfusion. See text for further details and definition of groups.
Induction of ethanol preconditioning
EtOH-PC was induced by administering ethanol as a single intragastric bolus, 24 hrs before I/R. Using our dosing regimen, plasma ethanol increased to a peak value of ~45 mg/dl 30 min after gavage (equivalent to levels measured in humans consuming 1–2 alcoholic beverages) and returned to control levels within 60 min of alcohol administration [16]. The volume of 95% ethanol to be instilled (in μl) was calculated as follows: [body weight (g) × 0.6] + 0.3. This volume of ethanol was mixed in 0.3 ml of sterile distilled water just before bolus administration to the animals by gavage. Animals in the sham control (no I/R) and I/R alone (no EtOH-PC) groups received sterile distilled water without ethanol by gavage.
Induction of global forebrain ischemia
The C57BL/6 strain is highly susceptible to cerebral ischemia following bilateral common carotid occlusion [39]. At 24 hrs after gavage, transient global cerebral ischemia was induced by occlusion of both common carotid arteries (CCA) for 20 min followed by reperfusion [39]. For the morphological studies, animals were anesthetized with a mixture of 70% nitrous oxide, 30% oxygen and 2.5% isoflurane during preparation for surgical operation, after which isoflurane was reduced to 1% during surgery and ischemic insult. Although volatile anesthetics have been reported to induce preconditioning, any putative preconditioning effects would be similar in all groups. It is important to note that significant hippocampal injury occurred in our model, even in animals anesthetized as described above. Body temperature was kept at 37 °C by a warming pad. After exposure of both CCA, the arteries were clamped with aneurysm clips for 20 min and reperfusion was effected by removing the aneurysm clips. Sham-operated animals underwent similar procedures without occlusion of the CCA. For the intravital microcirculation studies, animals were anesthetized initially with a mixture of ketamine (150 mg/kg body wt i.p.) and xylazine (7.5 mg/kg body wt ip), then they were equipped with an infusion catheter (PE-10) installed in their right external jugular vein. Prior to making the cranial window, they were injected through their jugular cannula with Carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) to fluorescently label circulating leukocytes.
Monitoring of regional cerebral blood flow by laser Doppler flowmetry
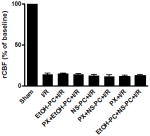
The presence of communicating arteries in mice was determined by monitoring the decrease in regional cerebral blood flow (rCBF) before and after clamping the bilateral CCA using a laser Doppler blood flow monitor (MBF3D, Moor Instruments, Devon, UK). Animals showing a decrease in rCBF of less than 80% of the initial value were excluded from subsequent analyses [12]. There is a significant decrease in rCBF after ischemia at less than 20% of the initial value of pre-ischemia (p<0.001). However, there was no significant differences among the ischemia/reperfusion with or without intervention (p>0.05) (See Table 1).
Table 1.
Forebrain rCBF by laser Doppler measurements immediately after global cerebral ischemia in mice (% value of pre-ischemia). There is a significant decrease in rCBF after ischemia at less than 20% of the initial value of pre-ischemia (p<0.01). However, there was no significant differences among the ischemia/reperfusion with or without intervention (p>0.05). (Data are expressed as mean ± S.E.M and n = 10).
| Sham | I/R | EtOH-PC+I/R | PX+EtOH-PC+I/R | NS-PC+I/R | PX+NS-PC+I/R | PX+I/R | EtOH-PC+NS- PC+I/R |
|---|---|---|---|---|---|---|---|
| 100±0.00 | 13.90±1.81 | 14.80±1.01 | 13.80±1.44 | 12.50±1.58 | 11.70±2.02 | 11.40±1.75 | 12.70±1.58 |
 | |||||||
Cranial Window
The head of each animal was fixed in a plastic frame in the sphinx position. The left parietal bone was exposed by a midline skin incision, followed by a craniotomy (diameter: 2.5 mm), 1 mm posterior to the bregma and 4 mm lateral to the midline, using a drill. Artificial cerebrospinal fluid (ACSF composition in mM: NaCl 128, KCl 3.0, CaCl2 1.3, MgCl2 1.0, Na2HPO4 21.0, NaH2PO4 1.3) was used to suffuse the tissue exposed by the craniotomies. A 12-mm glass coverslip was placed over the exposed brain tissue, and the artificial CSF was superfused beneath this viewing window [32, 34].
Intravital Fluorescence Microscopy and Video Analysis
The numbers of rolling (LR) and adherent (LA) leukocytes in pial postcapillary venules were quantified using intravital microscopy. Postcapillary venules (30 to 40 μm diameter) in the cerebral microcirculation were identified through the cranial window through a 20× objective lens, using a Nikon-E600FN upright microscope. Intravital fluorescence images of the vessel with CFDA-SE-labeled leukocytes (excitation wavelength, 420–490 nm; emission wavelength, 520 nm) were detected with a charge-coupled device (CCD) camera (XC-77; Hamamatsu Photonics), a CCD camera control unit (C2400; Hamamatsu Photonics) and an intensifier head (M4314; Hamamatsu Photonics) attached to the camera. Microfluorographs were projected on a television monitor (PVM-1953MD; Sony) and recorded on digital video using a digital video recorder (DMR-E50; Panasonic) for offline quantification of measured variables during playback of the recorded image. A video time-date generator (WJ810; Panasonic) displayed the stopwatch function on the monitor.
The interactions between leukocytes and vessel walls were recorded in 5 randomly selected venular segments which were 30 to 40 μm in diameter and at least 100 μm in length. Rolling and adherent leukocytes were classified according to the durationof their immobility on the venular wall, as follows [34]: (1) rolling cells were adherent for >2 and <30 seconds, and (2) adherent cells were stationary for >30 seconds, and they were expressed as the number of cells per mm2 of venular surface, calculated from diameter and length, with the assumption of cylindrical vessel shape.
Preparation of brain samples
Four days after ischemia, separate groups of mice, treated as described above for the intravital microscopy studies, were anesthetized with isoflurane, then transcardially perfused with 4% paraformaldehyde in 0.05 mol/L PBS. Brains were removed and post-fixed in the same fixative for 3 days. Brain tissues were embedded in paraffin and coronal sections (6 μm thicknesses) were cut from the dorsal hippocampal region.
Assessment of neuronal damage and apoptosis
Cresyl violet and Fluoro-Jade B histochemical staining were used to evaluate ischemic neuronal damage and neurodegeneration, respectively. Fluoro-Jade B is a novel polyanionic fluorescein derivitave which sensitively and specifically binds to degenerating neurons (both apoptotic and necrotic). Its high affinity to degenerating neurons with green fluorescence has made it a definitive marker of neurodegeneration. Fluoro-Jade B staining to examine ongoing damage was performed according to the protocol described by Schmued et al [40].
Neuronal apoptotic death staining was performed using a terminal deoxynucleotide transferase dUTP Nick End Labeling (TUNEL) assay, using an in situ cell death detection kit (Phoenix Flow Systems, San Diego, CA) according to the manufacturer’s instructions. The deparaffinized and hydrated sections were covered with proteinase K (1:100 diluted in 10 mM Tris pH 8) and incubated for 20 min at room temperature, then washed with PBS, and incubated for 5 min with 3% H2O2 diluted in methanol. The slides were washed again with PBS and incubated with reaction buffer for 20 min at room temperature. After blotting the reaction buffer, the sections were incubated for 1.5 hours at 37°C with DNA Labeling Solution, then washed with PBS and incubated for 10 min with blocking buffer. The latter was blotted and the sections were incubated with antibody solution in the dark for 1.5 hours at room temperature, then washed with PBS and covered with blocking buffer for 10 min. After blotting the buffer, the sections were incubated at room temperature for 30 min with conjugate solution, then washed with PBS and incubated for 15 min with a DAB (3, 3′-Diaminobenzidine) solution, washed again with water and counterstained with methyl green for 3 min, then dipped in ethanol, blotted and mounted in Permount.
Assessment of neuroinflammatory activation
GFAP (Glial fibrillary acidic protein) and isolectin-B4 immunohistochemistry staining were used to demonstrate activation of astrocytes and microglial cells. Brain sections were stained with GFAP for astrocytes and isolectin-B4 for microglial cells. Rabbit anti-human GFAP (1:100 dilutions, Sigma, St. Louis, MO) antibody was used as primary antibody and microglial cells were identified using HRP (Horseradish peroxidase)-labeled isolectin B4 (20 μg/ml, Sigma, St. Louis, MO) as described previously [41] with modifications [12].
Quantitative assessment of hippocampal injury/inflammation
Neuronal damage, degeneration, apoptosis, and glial activation were quantified by counting the number of live neurons and fluorescent or immune-positive cells of Fluoro-Jade B, TUNEL, GFAP, Isolectin-B4 and in the middle of a defined CA1 area (0.17 μm × 0.54 μm) in both left and right hippocampus per high magnification field (magnification 400x) using the Mata Imaging Serials (Version 6.1, Molecular Devices Corporation, Downingtown, PA). The average values of live neurons and above positive staining cells were obtained from left and right hippocampus of each animal.
Statistical analysis
Data (mean ± S.E.M.) were analyzed by one-way ANOVA followed by the Newman-Keuls post-hoc test. A p-value of less than 0.05 was considered statistically significant.
RESULTS
BKCa channel inhibitor treatment attenuates the ability of EtOH-PC and NS-PC to decrease cerebral I/R-induced leukocyte-endothelial adhesive interactions
Twenty min of bilateral CCA occlusion followed by 2 and 3 hrs of reperfusion resulted in significant (p<0.001) increases in LR and LA in pial venules (Fig. 2). In contrast to the increased postischemic adhesive events in venules, no LR or LA were observed in arterioles at any time during these experiments. Preconditioning with ethanol 24 hrs before I/R significantly decreased (p<0.001) the postischemic increases in LR and LA, effects that were attenuated (p<0.001 and p<0.01 for LR and LA, respectively) by coincident treatment with paxilline (PX), a BKCa channel inhibitor (Fig. 2). Preconditioning with the BKCa channel opener, NS1619, was as effective as ethanol in reducing I/R-induced LR and LA (all p<0.001). The anti-adhesive effects of NS-PC were also attenuated (p<0.001 and p<0.01 for LR and LA, respectively) by PX (Fig. 2). Preconditioning with both EtOH plus NS1619 did not showed any synergistic effect to further reduce I/R-induced LR and LA compared with EtOH-PC and NS1619 alone (Fig. 2) (all p>0.05). Taken together, these data support the hypothesis that BKCa channel activation may play a critical role in initiating the development of an anti-inflammatory phenotype after EtOH-PC.
Figure 2.

Role of BKCa channels in EtOH-PC 24 hrs prior to ischemia/reperfusion (I/R) on postischemic leukocyte rolling (upper panel) and adhesion (lower panel) determined after 2 and 3 hrs of reperfusion in wild-type C57BL/6J mice. BKCa channel opener, NS1619 was administrated i. p. 24 hours prior to I/R. BKCa channel inhibitor, paxilline was administered 10 minutes prior to EtOH-PC and NS1619 administration. Solid and open bars represent data obtained at 2 and 3 hrs of reperfusion, respectively. Values are mean ± S.E.M. for 6 mice per group. * denotes values statistically different from Sham (P<0.05). # denotes values statistically different from I/R (P<0.05). a denotes values statistically different from EtOH PC+I/R and b denotes values statistically different from NS-PC+I/R(P<0.05).
Paxilline reverses EtOH-PC and NS-PC-elicited decreases in cerebral I/R-induced neuronal injury and inflammation
Cresyl violet staining revealed healthy neurons in the hippocampal CA1 area in the sham control group (Fig. 3A) and largely dead neurons in I/R group (Fig. 3B). Ethanol or NS1619 administered 24 hrs before CCA occlusion both significantly (p< 0.001) decreased DND (based on neuron count) in the hippocampal CA1 area as compared with I/R group (without EtOH-PC and NS-PC (Fig. 3C, 3E vs. 3B). Similar to the findings with leukocyte-endothelial rolling and adhesion, paxilline administration 10 min prior to treatment with either ethanol or NS1619 significantly (p<0.05 and p<0.01) attenuated the neuroprotective effects of EtOH-PC and NS-PC in I/R (Fig. 3D vs. 3C, 3F vs. 3E and Fig. 4A).
Figure 3.

Representative micrographs depicting I/R-induced DND (Cresyl violet) and neuronal degeneration (Fluoro-Jade B) and apoptosis (TUNEL) in the hippocampal CA1 subfield of mice subjected to sham operation (A, I and Q), 20 min occlusion of the CCA followed by 4 days reperfusion (I/R) (B, J and R), EtOH-PC 24 hrs prior to I/R (EtOH-PC+I/R) (C, K and S), NS1619 administration 24 hrs prior to I/R (NS-PC+I/R)(E, M and U), paxilline treatment prior to ethanol or NS1619 administration, 24 hrs prior to I/R (PX+EtOH-PC+I/R; PX+NS-PC+I/R) (D, L, T and F, N, V), paxilline alone 24 hrs prior to I/R (PX+I/R) ( G, O, W), and both EtOH-PC and NS-PC 24 hrs prior to I/R(EtOH-PC+NS-PC+I/R) (H, P, X). (Magnification, 200x).
Figure 4.

Quantification of viable neurons (Cresyl violet, Panel A), neuronal degeneration (Fluoro-Jade B, Panel B) and apoptosis (TUNEL, Panel C) in the hippocampal CA1 region of mice subjected to sham control, I/R alone (I/R), ethanol preconditioning and NS1619 administration 24 hrs prior to I/R (EtOH-PC+I/R and NS-PC+I/R), and paxilline treatment coincident with ethanol and NS1619 administration 24 hrs prior to I/R (PX+EtOH-PC+I/R and PX+NS-PC+I/R), as well as paxilline alone 24 hrs prior to I/R (PX+I/R) and both EtOH-PC and NS-PC 24 hrs prior to I/R(EtOH-PC+NS-PC+I/R). Values are means ± S.E.M. for 10 mice per group. *, #, a and b = values statistically different from corresponding values in sham, I/R, and EtOH-PC+I/R groups, respectively, at p < 0.05.
Fluoro-Jade B staining was used as a marker of neurodegeneration. In the sham-operated group, very few degenerated neurons were observed in the hippocampal CA1 region (Fig. 3I). Positive staining of degenerated neurons significantly (p<0.001) increased 4 days after I/R (Fig. 3J vs. 3I). Preconditioning with EtOH or NS1619 24 hrs before I/R significantly (p<0.001) reduced the staining of degenerated neurons as compared with those in non-preconditioned, postischemic brain (Fig. 3K, 3M vs. 3J). Again, paxilline significantly reversed (p<0.05 and p<0.01 for EtOH-PC and NS-PC, respectively) the protective effects of EtOH-PC and NS-PC (Fig. 3L vs. 3K, 3N vs. 3M and Fig. 4B).
TUNEL staining was used to determine apoptotic neuronal death. In the sham-operated group, very few TUNEL-positive cells were observed in the hippocampal CA1 region (Fig. 3Q). TUNEL-positive cells increased significantly (p<0.001) 4 days after I/R (Fig. 3R). Preconditioning with EtOH and NS1619 24 hrs prior to I/R significantly (p<0.01 and p<0.001, respectively) reduced the number of TUNEL-positive cells as compared to those in non-preconditioned, postischemic brain (Fig. 3S, 3U vs. 3R). This protective effect was significantly attenuated by prior administration of paxilline just prior to EtOH-PC or NS-PC (all p< 0.05) (Fig. 3T vs. 3S, 3V vs. 3U and Fig. 4C).
Neuroinflammatory mechanisms play a critical role in the brain damage produced by cerebral ischemia, an effect found to be dependent upon recruitment of glial cells (astrocyte and microglia) [42, 43]. Immunohistochemical staining with the glial cell-specific stain, GFAP, showed astrocytes with small cell bodies and fine cytoplasmic processes distributed around the hippocampal CA1 area. Few GFAP-positive astrocytes were found in the sham control group (Fig. 5A). However, animals subjected to I/R showed a large increase in reactive astrocytes, which were found in multiple hippocampal layers (Fig. 5B). Counting of GFAP-positive astrocytes within a fixed area in the hippocampus showed that I/R produced a significant (p<0.001) increase in such cells (Fig. 5B vs. 5A and Fig. 6A). Preconditioning with either EtOH or NS1619 resulted in significantly fewer (all p < 0.001) reactive astrocytes as compared with the I/R group (Fig. 5C, 5E vs. 5B). Paxilline significantly attenuated the effects of EtOH-PC and NS-PC (p<0.05 and p<0.01, respectively) to reduce postischemic GFAP-positive cells (Fig. 5D vs. 5C, 5F vs. 5E and Fig. 6A).
Figure 5.

Representative micrographs depicting I/R-induced activation of astrocytes (GFAP) and microglial cells (Isolectin-B4) in the hippocampal CA1 subfield of mice subjected to sham (A, I), I/R (B, J), ethanol and NS 1619 administration 24 hrs prior to I/R (EtOH-PC+I/R and NS-PC+I/R) (C, K and E, M), paxilline treatment coincident with ethanol and NS1619 administration, 24 hrs prior to I/R (PX+EtOH-PC+I/R and PX+NS-PC+I/R) (D, L and F, N), paxilline alone 24 hrs prior to I/R (PX+I/R) ( G, O), and both EtOH-PC and NS-PC 24 hrs prior to I/R(EtOH-PC+NS-PC+I/R) (H, P). (Magnification, 200x).
Figure 6.

Quantification of astrocytic (GFAP, Panel A) and microglial activation (Isolectin-B4, Panel B) in the hippocampal CA1 region of mice subjected to sham control, I/R alone (I/R), ethanol and NS1619 administration 24 hrs prior to I/R (EtOH-PC+I/R and NS-PC+I/R), and paxilline treatment coincident with ethanol and NS1619 administration 24 hrs prior to I/R (PX+EtOH-PC+I/R and PX+NS-PC+I/R), as well as paxilline alone 24 hrs prior to I/R (PX+I/R) and both EtOH-PC and NS-PC 24 hrs prior to I/R(EtOH-PC+NS-PC+I/R). Values are means ± S.E.M. for 10 mice per group. *, #, a and b = values statistically different from corresponding values in sham, I/R, and EtOH-PC+I/R groups, respectively, at p < 0.05.
When brain sections were stained with isolectin B4 to identify microglial cells, very few of these cells were observed in the sham control group (Fig. 5I). However, I/R resulted in a substantial increase (p<0.001) in microglial cells in the CA1 area where pyramidal neuron death was apparent (Fig. 5J vs. 5I). EtOH-PC and NS-PC 24 hrs before I/R resulted in a significant (p<0.001) reduction in isolectin B4-positive cells as compared to that noted after I/R (Fig. 5K, 5M vs. 5J). The decreases in the number of microglial cells in EtOH and NS1619 preconditioned mice were significantly (p< 0.05 and p<0.01) attenuated by coincident treatment with paxilline (Fig. 5L vs. 5K, 5N vs. 5M and Fig. 6B).
As noted for the data depicted in Figure 2, there is no additive effect induced by coincident treatment with both EtOH and NS-1619 with regard to protection against postischemic neuronal damage and inflammation (Fig. 3–6).
DISCUSSION
The resistance of the brain and other tissues to ischemic injury can be transiently augmented by prior exposure to non-injurious preconditioning stimuli, most of which trigger two distinct time frames of increased tolerance to the subsequent ischemia [16, 44]. The early or acute phase of protection (early EtOH-PC) is apparent 1–2 hrs after ethanol exposure, disappears 2 or 3 hrs later, and induces the development of a less powerful protected phenotype than the late or delayed phase of EtOH-PC. The latter phase re-emerges 24 hrs after the preconditioning stimulus [16]. Since available evidence indicates that the delayed phase of preconditioning is characterized by a more profound protection than the acute phase [45, 46], we chose to focus our present work on this late phase.
Our results clearly demonstrate that moderate ethanol ingestion 24 hrs prior to cerebral I/R exerts preconditioning-like protective effects on the brain as assessed by 1) indices of neuronal degeneration, inflammation, and damage, and 2) cerebral microvascular leukocyte-endothelial adhesive interactions. Preconditioning with the BKCa channel opener, NS1619, 24 hrs before I/R, mimicked these protective actions of EtOH-PC. The beneficial effects of EtOH-PC and NS-PC to reduce neuronal degeneration, inflammation, and damage were attenuated by prior treatment with paxilline, a BKCa channel inhibitor. These latter observations suggest that the neuronal and neurovascular protective effects of EtOH-PC may be initiated, in part, by BKCa channel activation.
Delayed neuronal death in the hippocampus following global ischemia/reperfusion has been attributed largely to apoptosis [47–49]. Our findings were consistent with this conclusion, since most of the CA1 pyramidal neurons after I/R demonstrated TUNEL-positive staining. These changes were inhibited in a paxilline-reversible manner by EtOH-PC and NS1619 administration, indicating that opening of BKCa channels may mediate the anti-apototic effects of EtOH-PC. The precise mechanism whereby BKCa channel opening may protect against delayed neuronal death is not clear. However, it has been shown that excitatory amino acid release, neuronal calcium overload and reactive oxygen species (ROS) production play a major role in delayed neuronal death following cerebral I/R [47, 50, 51]. BKCa channels are expressed in neurons throughout the vertebrate brain [52–54] and a coupling of BKCa channels with NMDA receptors has been demonstrated in neurons [55]. Most recently, it is reported that BKCa channel opening inhibits ROS production of isolated rat brain mitochondria and enhances neuronal survival [25]. The opening of neuronal BKCa channels after EtOH-PC may hyperpolarize the neurons and reduce NMDA receptors mediated Ca 2+ overload and ROS production during I/R [19, 52], thus BKCa channels act as a brake on the brain to prevent neuronal overexcitation and excessive calcium influx and ROS generation during cerebral I/R. In addition, BKCa channels have been identified in the inner mitochondrial membrane and the mitochondrial permeability transition pore (PTP) in the inner mitochondrial membrane is considered to trigger the pathway of apoptosis [56]. It was reported that the ischemic preconditioning opened brain mitochondrial BKCa channels which could close brain mitochondrial PTP [56]. Therefore, it is possible that the opening of neuronal mitochondrial BKCa channels after EtOH-PC may keep mitochondrial PTP closed thus protecting against DND from apoptosis after cerebral I/R. While this is an attractive hypothesis, the existence of mitochondrial BKCa channels is controversial. Thus, it is clear that much additional work will be necessary to determine the role of modulation of neuronal cellular calcium levels and mitochondrial PTP by BKCa channel activation in the protection against I/R-induced DND by EtOH-PC.
In addition to neuronal BKCa channels, endothelial BKCa channels in the cerebral microvasculature may be another cellular and molecular target of EtOH-PC that could contribute to neuronal protection. Recent findings have implicated leukocytes in propagating tissue damage after I/R in stroke [35, 44]. Therefore, opening of microvascular endothelial BKCa channels by EtOH-PC, by limiting inflammatory responses in postcapillary venules, could play either a primary or contributory role in the protection against DND secondary to dysfunctional microcirculation after I/R.
Using intravital microscopy, we found that cerebral I/R induced large increases in leukocyte rolling and adhesion in pial venules compared with sham. The increased LR and LA were attenuated by EtOH-PC or antecedent NS1619 administration, and this protection was reversed by prior treatment with paxilline. Since it has been shown that BKCa channels are expressed in vascular endothelial cells, but not leukocytes [57–59], our results suggest that activation of endothelial BKCa channels may play an important role in mediating the protective effect of EtOH-PC in limiting cerebral I/R-induced leukocyte-endothelial adhesive interactions. However, the presence of BKCa channels in native endothelial cells has recently been challenged [60]. In this regard, it is important to note that astrocytes and neurons, together with endothelial cells, form a functional neurovascular unit that regulates barrier function and vasomotor activity in brain microvessels. Since astrocytes and microglia also express BKCa channels [56, 61], it is possible that they are a target for EtOH or NS1619.
Resident astrocytes and microglia, like neurons and vascular endothelial cells, also responded to EtOH-PC, and such responses were crucial to the foundation of the overall cerebroprotective phenotype. Moderate EtOH-PC interfered with various glial-mediated neurotoxic responses in the slices exposed to neurotoxic A-beta and HIV-1 protein gp120 [62–64]. BKCa channels have been identified in both astrocytes and microglial cells [56, 61]. Astrocytes are one of the components of neurovascular unit and they have been considered as an integral part of the cellular mechanisms that regulate cerebral microcirculation and cerebral blood flow [65]. Therefore, it is possible that the opening of glial BKCa channels after EtOH-PC regulates cerebral microvascular function to further protect against neuronal damage induced by I/R.
A general consequence of brain inflammation is activation and proliferation of astrocytes and microglia and reactive gliosis [66] and accumulation of activated microglia and reactive astrocytes is observed around degenerating neurons in various inflammatory or degenerative disorders in the central nervous system [67]. The production of inflammatory cytokines in activated glial cells contributed to neuronal damage in the progression of neurodegeneration [68, 69]. In our study we found that cerebral I/R produced extensive activation of astrocytes and microglial cells, abundant in the CA1 layer with prominent dying neurons; this was prevented by both EtOH-PC and NS1619, protective effects that were reversed by prior paxilline administration. It is thought that microglial cells are derived from the circulating monocytes through the disrupted blood brain barrier (BBB) in which the number of tight junctions has marked decreased [70]. Therefore, the observation that astrocyte and microglial activation occurs around degenerating neurons in CA1 after I/R is consonant with I/R-induced leukocyte-endothelial adhesive interactions in brain microcirculation and both support the anti-inflammatory property of EtOH-PC in ischemic brain.
Recently, it was reported that NS1619 could induce immediate and delayed preconditioning in primary rat neurons exposed to oxygen-glucose deprivation, hydrogen peroxide, or glutamate excitotoxicity, but the preconditioning protection was not blocked by BKCa channel inhibitor paxilline [71, 72]. This finding in cultured neurons differs from our present in vivo findings. In the central nervous system, BKCa channels are expressed, not only in neurons, but also in resident glial and vascular endothelial cells [56–59, 61]. Thus, the apparent discrepancy between our results and those of Gaspar, et al [71, 72] might be explained by BKCa channels in non-neuronal cells mediating the preconditioning effect. Other possible explanations could be differences due to injury model (I/R vs. simulated I/R), interventional environments (in vivo vs. cell culture), or species differences between mouse vs. rat. In addition, while it is clear that NS1619 activates BKCa channels, this compound also displays additional nonspecific effects at higher concentrations, such as inhibiting some Ca2+ currents and voltage-activated K+ and Na+ channels[37, 73]. However, the dose of NS1619 we used (0.1 mg/kg body weight) was lower than that reported to induce these off-target effects. Moreover, the effects of NS1619 to induce preconditioning were blocked by treatment with paxilline, suggesting that the effects of NS1619 are due to activation of BKCa channels in our model.
The molecular mechanisms underlying the effect of ethanol to activate BKCa channels are not clearly understood. However, discrete binding pockets for ethanol have recently been identified in potassium channels, which may be involved in channel activation [74]. Other studies have found evidence for a modulatory effect of ethanol on certain BKCa channel subunits [75–78]. Finally, it has been suggested that ethanol may modify ion channel activity indirectly via alterations in membrane fluidity [79, 80]. It is also unclear how BKCa channel activation 24 hrs prior to induction of I/R attenuates postischemic cerebral inflammation and injury. Rather than a direct effect, it seems likely that ethanol-induced BKCa channel activation stimulates as yet unidentified downstream effectors, which in turn, mediate the protective actions of EtOH-PC during I/R 24 hrs later. Although beyond the scope of the present study, likely candidates include iNOS, HO-1 and other downstream mediators implicated in other forms of preconditioning.
CONCLUSIONS
In summary, this study demonstrated that moderate ethanol consumption produces a protective phenotype in brain microvasculature and neurons against neurovascular inflammation and glial activation induced by cerebral I/R by a BKCa channel-dependent mechanism in either or both neurons and cerebral microvascular endothelial cells. Moderate consumption of ethanol may induce the development of a protected state wherein brain injury after stroke is minimized. Other pharmacological agents or physiologic stressors that activate BK channels may have therapeutic value for suppressing stroke-mediated damage in brain.
Acknowledgments
This work was supported by grant AA R01-014945 and HL-082186 from the National Institutes of Health. We thank Dr. D. Neil Granger and Ms. Shantel Vital for help in establishing intravital cerebral microcirculation.
ABBREVIATIONS
- BKCa, Large conductance
Ca2+-activated K+ channels
- EtOH-PC
Ethanol preconditioning
- I/R
ischemia/reperfusion
- CCA
common carotid arteries
- rCBF
regional cerebral blood flow
- CFDA-SE
Carboxyfluorescein diacetate succinimidyl ester
- LR
leukocyte rolling
- LA
leukocyte adhesion
- DND
delayed neuronal death
- GFAP
glial fibrillary acidic protein
- HRP
Horseradish peroxidase
- DAB
3,3′-Diaminobenzidine
- TUNEL
terminal dUTP nick-end labeling
- PX
paxilline
- NS
NS1619
- CSF
cerebrospinal fluid
- BBB
blood brain barrier
- EEA
excitatory amino acids
- ROS
reactive oxygen species
- PTP
permeability transition pore
References
- 1.Sun AY, Simonyi A, Sun GY. The “French Paradox” and beyond: neuroprotective effects of polyphenols. Free Radic Biol Med. 2002;32:314–8. doi: 10.1016/s0891-5849(01)00803-6. [DOI] [PubMed] [Google Scholar]
- 2.Truelsen T, Gronbaek M. Wine consumption and cerebrovascular disease mortality in Spain. Stroke. 1999;30:186–8. doi: 10.1161/01.str.30.1.186. [DOI] [PubMed] [Google Scholar]
- 3.Collins MA, Neafsey EJ, Mukamal KJ, Gray MO, Parks DA, Das DK, Korthuis RJ. Alcohol in moderation, cardioprotection, and neuroprotection: epidemiological considerations and mechanistic studies. Alcohol Clin Exp Res. 2009;33:206–19. doi: 10.1111/j.1530-0277.2008.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.German JB, Walzem RL. The health benefits of wine. Annu Rev Nutr. 2000;20:561–93. doi: 10.1146/annurev.nutr.20.1.561. [DOI] [PubMed] [Google Scholar]
- 5.Berger K, Ajani UA, Kase CS, Gaziano JM, Buring JE, Glynn RJ, Hennekens CH. Light-to-moderate alcohol consumption and risk of stroke among U.S. male physicians. N Engl J Med. 1999;341:1557–64. doi: 10.1056/NEJM199911183412101. [DOI] [PubMed] [Google Scholar]
- 6.Reynolds K, Lewis B, Nolen JD, Kinney GL, Sathya B, He J. Alcohol consumption and risk of stroke: a meta-analysis. Jama. 2003;289:579–88. doi: 10.1001/jama.289.5.579. [DOI] [PubMed] [Google Scholar]
- 7.Bradamante S, Barenghi L, Villa A. Cardiovascular protective effects of resveratrol. Cardiovasc Drug Rev. 2004;22:169–88. doi: 10.1111/j.1527-3466.2004.tb00139.x. [DOI] [PubMed] [Google Scholar]
- 8.Das DK, Sato M, Ray PS, Maulik G, Engelman RM, Bertelli AA, Bertelli A. Cardioprotection of red wine: role of polyphenolic antioxidants. Drugs Exp Clin Res. 1999;25:115–20. [PubMed] [Google Scholar]
- 9.Ray PS, Maulik G, Cordis GA, Bertelli AA, Bertelli A, Das DK. The red wine antioxidant resveratrol protects isolated rat hearts from ischemia reperfusion injury. Free Radic Biol Med. 1999;27:160–9. doi: 10.1016/s0891-5849(99)00063-5. [DOI] [PubMed] [Google Scholar]
- 10.Wang Q, Simonyi A, Li W, Sisk BA, Miller RL, Macdonald RS, Lubahn DE, Sun GY, Sun AY. Dietary grape supplement ameliorates cerebral ischemia-induced neuronal death in gerbils. Mol Nutr Food Res. 2005;49:443–51. doi: 10.1002/mnfr.200500019. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Sun AY, Simonyi A, Miller DK, Smith RE, Luchtefeld RG, Korthuis RJ, Sun GY. Oral administration of grape polyphenol extract ameliorates cerebral ischemia/reperfusion-induced neuronal damage and behavioral deficits in gerbils: comparison of pre-and post-ischemic administration. J Nutr Biochem. 2009;20:369–77. doi: 10.1016/j.jnutbio.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Q, Xu J, Rottinghaus GE, Simonyi A, Lubahn D, Sun GY, Sun AY. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002;958:439–47. doi: 10.1016/s0006-8993(02)03543-6. [DOI] [PubMed] [Google Scholar]
- 13.Shigematsu S, Ishida S, Hara M, Takahashi N, Yoshimatsu H, Sakata T, Korthuis RJ. Resveratrol, a red wine constituent polyphenol, prevents superoxide-dependent inflammatory responses induced by ischemia/reperfusion, platelet-activating factor, or oxidants. Free Radic Biol Med. 2003;34:810–7. doi: 10.1016/s0891-5849(02)01430-2. [DOI] [PubMed] [Google Scholar]
- 14.Miyamae M, Diamond I, Weiner MW, Camacho SA, Figueredo VM. Regular alcohol consumption mimics cardiac preconditioning by protecting against ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 1997;94:3235–9. doi: 10.1073/pnas.94.7.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guiraud A, de Lorgeril M, Boucher F, Berthonneche C, Rakotovao A, de Leiris J. Cardioprotective effect of chronic low dose ethanol drinking: insights into the concept of ethanol preconditioning. J Mol Cell Cardiol. 2004;36:561–6. doi: 10.1016/j.yjmcc.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi T, Dayton C, Shigematsu T, Carter P, Yoshikawa T, Gute DC, Korthuis RJ. Preconditioning with ethanol prevents postischemic leukocyte-endothelial cell adhesive interactions. Am J Physiol Heart Circ Physiol. 2002;283:H1019–30. doi: 10.1152/ajpheart.00173.2002. [DOI] [PubMed] [Google Scholar]
- 17.Korthuis RJ. Introduction to the special topics issue on alcohol and cardioprotection. Pathophysiology. 2004;10:81–2. doi: 10.1016/j.pathophys.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Wang Q, Sun AY, Simonyi A, Kalogeris TJ, Miller DK, Sun GY, Korthuis RJ. Ethanol preconditioning protects against ischemia/reperfusion-induced brain damage: role of NADPH oxidase-derived ROS. Free Radic Biol Med. 2007;43:1048–60. doi: 10.1016/j.freeradbiomed.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faber ES, Sah P. Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 2003;9:181–94. doi: 10.1177/1073858403009003011. [DOI] [PubMed] [Google Scholar]
- 20.Sato T, Saito T, Saegusa N, Nakaya H. Mitochondrial Ca2+-activated K+ channels in cardiac myocytes: a mechanism of the cardioprotective effect and modulation by protein kinase A. Circulation. 2005;111:198–203. doi: 10.1161/01.CIR.0000151099.15706.B1. [DOI] [PubMed] [Google Scholar]
- 21.Stowe DF, Aldakkak M, Camara AK, Riess ML, Heinen A, Varadarajan SG, Jiang MT. Cardiac mitochondrial preconditioning by Big Ca2+-sensitive K+ channel opening requires superoxide radical generation. Am J Physiol Heart Circ Physiol. 2006;290:H434–40. doi: 10.1152/ajpheart.00763.2005. [DOI] [PubMed] [Google Scholar]
- 22.Veltkamp R, Domoki F, Bari F, Busija DW. Potassium channel activators protect the N-methyl-D-aspartate-induced cerebral vascular dilation after combined hypoxia and ischemia in piglets. Stroke. 1998;29:837–42. doi: 10.1161/01.str.29.4.837. discussion 842–3. [DOI] [PubMed] [Google Scholar]
- 23.Gribkoff VK, Starrett JE, Jr, Dworetzky SI, Hewawasam P, Boissard CG, Cook DA, Frantz SW, Heman K, Hibbard JR, Huston K, Johnson G, Krishnan BS, Kinney GG, Lombardo LA, Meanwell NA, Molinoff PB, Myers RA, Moon SL, Ortiz A, Pajor L, Pieschl RL, Post-Munson DJ, Signor LJ, Srinivas N, Taber MT, Thalody G, Trojnacki JT, Wiener H, Yeleswaram K, Yeola SW. Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat Med. 2001;7:471–7. doi: 10.1038/86546. [DOI] [PubMed] [Google Scholar]
- 24.Xu W, Liu Y, Wang S, McDonald T, Van Eyk JE, Sidor A, O’Rourke B. Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science. 2002;298:1029–33. doi: 10.1126/science.1074360. [DOI] [PubMed] [Google Scholar]
- 25.Kulawiak B, Kudin AP, Szewczyk A, Kunz WS. BK channel openers inhibit ROS production of isolated rat brain mitochondria. Exp Neurol. 2008;212:543–7. doi: 10.1016/j.expneurol.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Runden-Pran E, Haug FM, Storm JF, Ottersen OP. BK channel activity determines the extent of cell degeneration after oxygen and glucose deprivation: a study in organotypical hippocampal slice cultures. Neuroscience. 2002;112:277–88. doi: 10.1016/s0306-4522(02)00092-1. [DOI] [PubMed] [Google Scholar]
- 27.Gao Q, Zhang SZ, Cao CM, Bruce IC, Xia Q. The mitochondrial permeability transition pore and the Ca2+-activated K+ channel contribute to the cardioprotection conferred by tumor necrosis factor-alpha. Cytokine. 2005;32:199–205. doi: 10.1016/j.cyto.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 28.Cao CM, Xia Q, Gao Q, Chen M, Wong TM. Calcium-activated potassium channel triggers cardioprotection of ischemic preconditioning. J Pharmacol Exp Ther. 2005;312:644–50. doi: 10.1124/jpet.104.074476. [DOI] [PubMed] [Google Scholar]
- 29.Shintani Y, Node K, Asanuma H, Sanada S, Takashima S, Asano Y, Liao Y, Fujita M, Hirata A, Shinozaki Y, Fukushima T, Nagamachi Y, Okuda H, Kim J, Tomoike H, Hori M, Kitakaze M. Opening of Ca2+-activated K+ channels is involved in ischemic preconditioning in canine hearts. J Mol Cell Cardiol. 2004;37:1213–8. doi: 10.1016/j.yjmcc.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Fisher PW, Xi L, Kukreja RC. Essential role of mitochondrial Ca2+-activated and ATP-sensitive K+ channels in sildenafil-induced late cardioprotection. J Mol Cell Cardiol. 2008;44:105–13. doi: 10.1016/j.yjmcc.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Yin C, Xi L, Kukreja RC. Opening of Ca2+-activated K+ channels triggers early and delayed preconditioning against I/R injury independent of NOS in mice. Am J Physiol Heart Circ Physiol. 2004;287:H2070–7. doi: 10.1152/ajpheart.00431.2004. [DOI] [PubMed] [Google Scholar]
- 32.Ishikawa M, Zhang JH, Nanda A, Granger DN. Inflammatory responses to ischemia and reperfusion in the cerebral microcirculation. Front Biosci. 2004;9:1339–47. doi: 10.2741/1330. [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa M, Cooper D, Arumugam TV, Zhang JH, Nanda A, Granger DN. Platelet-leukocyte-endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J Cereb Blood Flow Metab. 2004;24:907–15. doi: 10.1097/01.WCB.0000132690.96836.7F. [DOI] [PubMed] [Google Scholar]
- 34.Ishikawa M, Cooper D, Russell J, Salter JW, Zhang JH, Nanda A, Granger DN. Molecular determinants of the prothrombogenic and inflammatory phenotype assumed by the postischemic cerebral microcirculation. Stroke. 2003;34:1777–82. doi: 10.1161/01.STR.0000074921.17767.F2. [DOI] [PubMed] [Google Scholar]
- 35.Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–68. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida H, Kusama Y, Kodani E, Yasutake M, Takano H, Atarashi H, Kishida H, Takano T. Pharmacological preconditioning with bradykinin affords myocardial protection through NO-dependent mechanisms. Int Heart J. 2005;46:877–87. doi: 10.1536/ihj.46.877. [DOI] [PubMed] [Google Scholar]
- 37.Holland M, Langton PD, Standen NB, Boyle JP. Effects of the BKCa channel activator, NS1619, on rat cerebral artery smooth muscle. Br J Pharmacol. 1996;117:119–29. doi: 10.1111/j.1476-5381.1996.tb15163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanchez M, McManus OB. Paxilline inhibition of the alpha-subunit of the high-conductance calcium-activated potassium channel. Neuropharmacology. 1996;35:963–8. doi: 10.1016/0028-3908(96)00137-2. [DOI] [PubMed] [Google Scholar]
- 39.Yang G, Kitagawa K, Matsushita K, Mabuchi T, Yagita Y, Yanagihara T, Matsumoto M. C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res. 1997;752:209–18. doi: 10.1016/s0006-8993(96)01453-9. [DOI] [PubMed] [Google Scholar]
- 40.Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 41.Streit WJ. An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I-B4) J Histochem Cytochem. 1990;38:1683–6. doi: 10.1177/38.11.2212623. [DOI] [PubMed] [Google Scholar]
- 42.Becker K, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the alpha4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke. 2001;32:206–11. doi: 10.1161/01.str.32.1.206. [DOI] [PubMed] [Google Scholar]
- 43.Villa P, Bigini P, Mennini T, Agnello D, Laragione T, Cagnotto A, Viviani B, Marinovich M, Cerami A, Coleman TR, Brines M, Ghezzi P. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med. 2003;198:971–5. doi: 10.1084/jem.20021067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–48. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 45.Kamada K, Dayton CB, Yamaguchi T, Korthuis RJ. Antecedent ethanol ingestion prevents postischemic microvascular dysfunction. Pathophysiology. 2004;10:131–7. doi: 10.1016/j.pathophys.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 46.Nandagopal K, Dawson TM, Dawson VL. Critical role for nitric oxide signaling in cardiac and neuronal ischemic preconditioning and tolerance. J Pharmacol Exp Ther. 2001;297:474–8. [PubMed] [Google Scholar]
- 47.Kirino T. Delayed neuronal death. Neuropathology. 2000;20(Suppl):S95–7. doi: 10.1046/j.1440-1789.2000.00306.x. [DOI] [PubMed] [Google Scholar]
- 48.Hara A, Mori H, Niwa M. Novel apoptotic evidence for delayed neuronal death in the hippocampal CA1 pyramidal cells after transient ischemia. Stroke. 2000;31:236–8. doi: 10.1161/01.str.31.1.231-e. [DOI] [PubMed] [Google Scholar]
- 49.Wang Q, Sun AY, Simonyi A, Jensen MD, Shelat PB, Rottinghaus GE, MacDonald RS, Miller DK, Lubahn DE, Weisman GA, Sun GY. Neuroprotective mechanisms of curcumin against cerebral ischemia-induced neuronal apoptosis and behavioral deficits. J Neurosci Res. 2005;82:138–48. doi: 10.1002/jnr.20610. [DOI] [PubMed] [Google Scholar]
- 50.Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 51.Fujimura M, Tominaga T, Chan PH. Neuroprotective effect of an antioxidant in ischemic brain injury: involvement of neuronal apoptosis. Neurocrit Care. 2005;2:59–66. doi: 10.1385/NCC:2:1:059. [DOI] [PubMed] [Google Scholar]
- 52.Tseng-Crank J, Foster CD, Krause JD, Mertz R, Godinot N, DiChiara TJ, Reinhart PH. Cloning, expression, and distribution of functionally distinct Ca(2+)-activated K+ channel isoforms from human brain. Neuron. 1994;13:1315–30. doi: 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- 53.Sausbier U, Sausbier M, Sailer CA, Arntz C, Knaus HG, Neuhuber W, Ruth P. Ca2+-activated K+ channels of the BK-type in the mouse brain. Histochem Cell Biol. 2006;125:725–41. doi: 10.1007/s00418-005-0124-7. [DOI] [PubMed] [Google Scholar]
- 54.Wanner SG, Koch RO, Koschak A, Trieb M, Garcia ML, Kaczorowski GJ, Knaus HG. High-conductance calcium-activated potassium channels in rat brain: pharmacology, distribution, and subunit composition. Biochemistry. 1999;38:5392–400. doi: 10.1021/bi983040c. [DOI] [PubMed] [Google Scholar]
- 55.Isaacson JS, Murphy GJ. Glutamate-mediated extrasynaptic inhibition: direct coupling of NMDA receptors to Ca(2+)-activated K+ channels. Neuron. 2001;31:1027–34. doi: 10.1016/s0896-6273(01)00428-7. [DOI] [PubMed] [Google Scholar]
- 56.Cheng Y, Gu XQ, Bednarczyk P, Wiedemann FR, Haddad GG, Siemen D. Hypoxia increases activity of the BK-channel in the inner mitochondrial membrane and reduces activity of the permeability transition pore. Cell Physiol Biochem. 2008;22:127–36. doi: 10.1159/000149790. [DOI] [PubMed] [Google Scholar]
- 57.Wang XL, Ye D, Peterson TE, Cao S, Shah VH, Katusic ZS, Sieck GC, Lee HC. Caveolae targeting and regulation of large conductance Ca(2+)-activated K+ channels in vascular endothelial cells. J Biol Chem. 2005;280:11656–64. doi: 10.1074/jbc.M410987200. [DOI] [PubMed] [Google Scholar]
- 58.Essin K, Salanova B, Kettritz R, Sausbier M, Luft FC, Kraus D, Bohn E, Autenrieth IB, Peschel A, Ruth P, Gollasch M. Large-conductance calcium-activated potassium channel activity is absent in human and mouse neutrophils and is not required for innate immunity. Am J Physiol Cell Physiol. 2007;293:C45–54. doi: 10.1152/ajpcell.00450.2006. [DOI] [PubMed] [Google Scholar]
- 59.Dong DL, Zhang Y, Lin DH, Chen J, Patschan S, Goligorsky MS, Nasjletti A, Yang BF, Wang WH. Carbon monoxide stimulates the Ca2(+)-activated big conductance k channels in cultured human endothelial cells. Hypertension. 2007;50:643–51. doi: 10.1161/HYPERTENSIONAHA.107.096057. [DOI] [PubMed] [Google Scholar]
- 60.Sandow SL, Grayson TH. Limits of isolation and culture: intact vascular endothelium and BKCa. Am J Physiol Heart Circ Physiol. 2009;297:H1–7. doi: 10.1152/ajpheart.00042.2009. [DOI] [PubMed] [Google Scholar]
- 61.Schilling T, Eder C. Ion channel expression in resting and activated microglia of hippocampal slices from juvenile mice. Brain Res. 2007;1186:21–8. doi: 10.1016/j.brainres.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 62.Belmadani A, Kumar S, Schipma M, Collins MA, Neafsey EJ. Inhibition of amyloid-beta-induced neurotoxicity and apoptosis by moderate ethanol preconditioning. Neuroreport. 2004;15:2093–6. doi: 10.1097/00001756-200409150-00019. [DOI] [PubMed] [Google Scholar]
- 63.Belmadani A, Neafsey EJ, Collins MA. Human immunodeficiency virus type 1 gp120 and ethanol coexposure in rat organotypic brain slice cultures: Curtailment of gp120-induced neurotoxicity and neurotoxic mediators by moderate but not high ethanol concentrations. J Neurovirol. 2003;9:45–54. doi: 10.1080/13550280390173409. [DOI] [PubMed] [Google Scholar]
- 64.Belmadani A, Zou JY, Schipma MJ, Neafsey EJ, Collins MA. Ethanol pre-exposure suppresses HIV-1 glycoprotein 120-induced neuronal degeneration by abrogating endogenous glutamate/Ca2+-mediated neurotoxicity. Neuroscience. 2001;104:769–81. doi: 10.1016/s0306-4522(01)00139-7. [DOI] [PubMed] [Google Scholar]
- 65.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–76. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 66.Nedergaard M, Dirnagl U. Role of glial cells in cerebral ischemia. Glia. 2005;50:281–6. doi: 10.1002/glia.20205. [DOI] [PubMed] [Google Scholar]
- 67.Suzumura A, Takeuchi H, Zhang G, Kuno R, Mizuno T. Roles of glia-derived cytokines on neuronal degeneration and regeneration. Ann N Y Acad Sci. 2006;1088:219–29. doi: 10.1196/annals.1366.012. [DOI] [PubMed] [Google Scholar]
- 68.Ravizza T, Rizzi M, Perego C, Richichi C, Veliskova J, Moshe SL, De Simoni MG, Vezzani A. Inflammatory response and glia activation in developing rat hippocampus after status epilepticus. Epilepsia. 2005;46(Suppl 5):113–7. doi: 10.1111/j.1528-1167.2005.01006.x. [DOI] [PubMed] [Google Scholar]
- 69.Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26:349–54. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 70.Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75:388–97. doi: 10.1189/jlb.0303114. [DOI] [PubMed] [Google Scholar]
- 71.Gaspar T, Domoki F, Lenti L, Katakam PV, Snipes JA, Bari F, Busija DW. Immediate neuronal preconditioning by NS1619. Brain Res. 2009 doi: 10.1016/j.brainres.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gaspar T, Katakam P, Snipes JA, Kis B, Domoki F, Bari F, Busija DW. Delayed neuronal preconditioning by NS1619 is independent of calcium activated potassium channels. J Neurochem. 2008;105:1115–28. doi: 10.1111/j.1471-4159.2007.05210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Olesen SP, Munch E, Moldt P, Drejer J. Selective activation of Ca(2+)-dependent K+ channels by novel benzimidazolone. Eur J Pharmacol. 1994;251:53–9. doi: 10.1016/0014-2999(94)90442-1. [DOI] [PubMed] [Google Scholar]
- 74.Aryal P, Dvir H, Choe S, Slesinger PA. A discrete alcohol pocket involved in GIRK channel activation. Nat Neurosci. 2009 doi: 10.1038/nn.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brodie MS, Scholz A, Weiger TM, Dopico AM. Ethanol interactions with calcium-dependent potassium channels. Alcohol Clin Exp Res. 2007;31:1625–32. doi: 10.1111/j.1530-0277.2007.00469.x. [DOI] [PubMed] [Google Scholar]
- 76.Feinberg-Zadek PL, Martin G, Treistman SN. BK channel subunit composition modulates molecular tolerance to ethanol. Alcohol Clin Exp Res. 2008;32:1207–16. doi: 10.1111/j.1530-0277.2008.00704.x. [DOI] [PubMed] [Google Scholar]
- 77.Wynne PM, Puig SI, Martin GE, Treistman SN. Compartmentalized beta subunit distribution determines characteristics and ethanol sensitivity of somatic, dendritic, and terminal large-conductance calcium-activated potassium channels in the rat central nervous system. J Pharmacol Exp Ther. 2009;329:978–86. doi: 10.1124/jpet.108.146175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martin GE, Hendrickson LM, Penta KL, Friesen RM, Pietrzykowski AZ, Tapper AR, Treistman SN. Identification of a BK channel auxiliary protein controlling molecular and behavioral tolerance to alcohol. Proc Natl Acad Sci U S A. 2008;105:17543–8. doi: 10.1073/pnas.0801068105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Crowley JJ, Treistman SN, Dopico AM. Cholesterol antagonizes ethanol potentiation of human brain BKCa channels reconstituted into phospholipid bilayers. Mol Pharmacol. 2003;64:365–72. doi: 10.1124/mol.64.2.365. [DOI] [PubMed] [Google Scholar]
- 80.Crowley JJ, Treistman SN, Dopico AM. Distinct structural features of phospholipids differentially determine ethanol sensitivity and basal function of BK channels. Mol Pharmacol. 2005;68:4–10. doi: 10.1124/mol.105.012971. [DOI] [PubMed] [Google Scholar]