

Abstract
The reasons why people smoke are varied, but research has demonstrated that genetic influences on various aspects of nicotine addiction are a major factor. There also is a strong genetic influence on measures of nicotine sensitivity in mice. Despite the established contribution of genetics to nicotine sensitivity in mice and humans, no naturally occurring genetic variation has been identified that demonstrably alters sensitivity to nicotine in either species. However, one genetic variant has been implicated in altering nicotine sensitivity in mice is a T529A polymorphism in Chrna4, the gene that encodes the nicotinic receptor (nAChR) α4 subunit. The Chrna4 T529A polymorphism leads to a threonine to alanine substitution at position 529 of the α4 subunit. To more definitively address whether the Chrna4 T529A polymorphism does, in fact, influence sensitivity to nicotine, knockin mice were generated in which the threonine codon at position 529 was mutated to an alanine codon. Compared to Chrna4 T529 littermate controls, the Chrna4 A529 knockin mice exhibited greater sensitivity to the hypothermic effects of nicotine, reduced oral nicotine consumption and did not develop conditioned place preference to nicotine. The Chrna4 A529 knockin mice also differed from T529 littermates for two parameters of acetylcholine-stimulated 86Rb+ efflux in midbrain: maximal efflux and the percentage of α4β2* receptors with high sensitivity to activation by agonists. Results indicate that the polymorphism affects the function of midbrain α4β2* nAChRs and contributes to individual differences in several behavioral and physiological responses to nicotine thought to be modulated by midbrain α4β2* nAChRs.
Keywords: Nicotinic receptor, genetics, polymorphism, conditioned place preference, oral consumption, addiction
Cigarette smoking is the single most preventable cause of premature death in the United States. It has been estimated that over 440,000 people died annually between the years 1998–2007 from smoking related illnesses [1]. In fact, the number of deaths related to tobacco is more than all deaths related to human immunodeficiency virus (HIV), illegal drug use, motor vehicle injuries, suicides, and murders combined. Despite these morbid statistics, 20% of adults in the United States continue to smoke [1].
Although the specific reasons why people smoke are varied, it is known that genetics plays a vital role. Twin studies, beginning with those of Fisher [2, 3] demonstrated that smoking behavior is heritable with a mean estimate of heritability of 0.53 [4]. Likewise, genetic factors influence the behavioral and physiological effects of nicotine in mice [5]. For example, an examination of 19 inbred mouse strains demonstrated that there is a 2 to 6-fold difference in sensitivity to nicotine across a battery of tests [6, 7]. Moreover, genetic influences on the development of tolerance to nicotine [8–10], nicotine oral consumption [11–13] conditioned place preference [14, 15] and conditioned taste aversion [16] have been reported in mice.
Due to the fact that there are clear genetic influences on nicotine dependence in humans and nicotine sensitivity in rodents, a number of attempts have been made to identify the genetic variants in both species that lead to individual differences in nicotine dependence/sensitivity [17, 18]. Not surprisingly, these studies have uncovered several chromosomal regions and specific polymorphisms that may contribute to variable sensitivity to nicotine. Nonetheless, none of these naturally-occurring polymorphisms have been experimentally demonstrated to contribute to individual differences in nicotine dependence and/or sensitivity. Moreover, it remains to be determined whether any single naturally-occurring polymorphism is actually sufficient to alter individual responsiveness to nicotine in any measurable way.
One genetic variant that has been implicated in altering nicotine sensitivity in mice is the naturally occurring T529A polymorphism in Chrna4, the gene that encodes the nicotinic receptor (nAChR) α4 subunit. Several studies have shown that this polymorphism is associated with individual differences in a variety of measures of nicotine sensitivity in mice [12, 19–21]. The Chrna4 T529A polymorphism leads to a threonine for alanine substitution at amino acid residue 529 of the α4 subunit and is located within a region of the large cytoplasmic loop that is conserved across mammalian species. In mammals, the α4 subunit combines with the nAChR β2 subunit to form the α4β2* nAChR (* indicates the possible inclusion of other nAChR subunits) [22]. The α4β2* nAChR is not only the most abundant nicotinic receptor expressed in the brain [23, 24] but also is critical for self-administration of and positive reinforcement to nicotine [25–28]. Heterologous expression of α4β2 nAChRs possessing the variant forms of the α4 subunit demonstrated that the T529A polymorphism affect the function of α4β2 nAChRs in vitro [29]. Moreover, the Chrna4 T529A polymorphism has been shown to be associated with differences in the function of α4β2* nAChRs in mouse brain synaptosomes [30, 31]. Because the Chrna4 T529A polymorphism is associated with differences in nicotine sensitivity, α4β2* nAChR function, and is located within a gene known to be critical for nicotine reinforcement, it is a strong candidate for a single naturally occurring polymorphism that contributes to individual variability in sensitivity to nicotine. However, all previous studies that have implicated the Chrna4 T529A polymorphism in nicotine sensitivity and nicotinic receptor function in mice were either association or linkage studies. Consequently, it remains to be determined if the Chrna4 T529A polymorphism or a polymorphism that is linked to it is responsible for individual differences in nicotine sensitivity and/or nAChR function. The studies described in this report utilized a knockin mouse for the Chrna4 T529A polymorphism in order to provide a more definitive assessment of the role of the polymorphism in influencing nicotine sensitivity and nAChR function.
Materials and Methods
Animals
Mice were housed in a colony room that was on a 12 h light and 12 h dark cycle (lights on at 0700) and maintained at 21°C ±1. Mice were weaned between 21 and 25 days of age. All studies used male and female adult mice between 60 and 120 days of age. The animals were provided with food (Teklad Rodent Diet (Harlan, Madison, WI)) and water ad libitum. All procedures were approved by the University of Colorado or University of Pittsburgh Institutional Animal Care and Use Committees.
Generation of Chrna4 T529A knockin mice
The DNA construct used for homologous recombination was generously provided by Dr. Henry Lester (CalTech) and has been described previously [32]. The mutation introduced by the Lester laboratory [32] was reverted and a new mutation changing the threonine codon to an alanine codon at amino acid position 529 was introduced using site directed mutagenesis. Reversion of the mutation introduced by the Lester laboratory and introduction of the threonine to alanine codon change at residue 529 were verified by sequencing. Sequencing also confirmed that there were no unwanted mutations introduced during the mutagenesis. The construct was introduced into the Strain 129SvJ Go Germline ES cell line (Genome Systems, Inc., St. Louis, MO) and homologous recombinants were identified by Southern blot using the exact same restriction digest and probes as described in Labarca et al. [32]. ES cells containing the targeted construct were introduced into C57BL/6J blastocysts and one chimeric mouse was identified as carrying the mutation germline. An intronic neomycin cassette was removed from the knockin allele of Chrna4 by breeding the Chrna4 T529A knockin progenitor mice with C57BL/6J-TgN(Zp3-Cre)93Knw mice (Jackson Laboratories, Bar Harbor, ME). The ZP3-Cre transgene was subsequently removed by backcrossing the Chrna4 T529A knockin mice with C57BL/6J mice. Chrna4 T529A knockin mice were then backcrossed to C57BL/6J mice for at least 8–9 generations prior to testing.
All animals used in the experiments were produced by matings between mice heterozygous for the T529A polymorphism. For all experiments, Chrna4 A529 knockin mice refer to mice that possess an alanine at amino acid position 529 where as T529 control mice refer to mice that have a threonine at amino acid position 529. Genotyping of the animals was done exactly as described previously by Dobelis et al. [31].
Materials
All materials were purchased (unless otherwise noted) from Sigma Aldrich (St. Louis, MO). Nicotine used for all experiments was free base.
Y-maze and Body Temperature
Using a counterbalanced design, animals received either a 0.5 mg/kg nicotine or saline injection (i.p.) on day one. Three minutes following injection, the animals were transferred to a darkened Y-maze where the number of horizontal and vertical beam breaks was recorded for 3 minutes. Fifteen minutes after the nicotine injection, body temperature was recorded by a rectal thermometer. On the following day, animals were tested under the exact same conditions but received the opposite injection.. Approximately half of the animals per genotype received nicotine on day one and the other half received nicotine on day two.
Nicotine Oral Consumption
A two bottle free choice paradigm was used as previously described [13]. The day before the experiment started, the mice were weighed, singly housed, and given free access to food and water. At the start of the experiment, mice were given two bottles: one containing tap water and the other containing 25 μg/ml nicotine in tap water. Both bottles were made from glass culture tubes fitted with standard stainless steel drinking spouts. The bottles were rotated each day to account for a side bias. After four days, the nicotine concentration was increased to 50 μg/ml and animals received fresh water. Following the four days of 50 μg/ml nicotine solution, the nicotine concentration was increased to 100 μg/ml for four days and animals received fresh water. Bottles were weighed at the start and finish of each new concentration and the volume consumed per bottle was recorded. Animals were reweighed on the final day in order to determine the average weight of each animal throughout the study. In addition, cages without mice were fitted with two bottles to determine volume lost due to spillage and evaporation during the experiment.
Non-biased Nicotine Induced Conditioned Place Preference (CPP)
The place preference apparatus consisted of 3 distinct compartments, two conditioning chambers and a smaller central chamber. Preliminary experiments were done to establish chamber conditions that prevented a bias towards one of the two conditioning chambers. As a result of these preliminary experiments, one conditioning compartments had vertical black and white striped walls with a mesh floor and the other conditioning compartment had black and white checkered walls with a rod floor. The middle chamber consisted of smooth black and white walls with smooth flooring. CPP was conducted in three phases: preconditioning, conditioning, and testing. In the preconditioning phase, mice were allowed to freely roam between the three chambers for 15 minutes. The time spent in each chamber was recorded and then animals were separated into counterbalanced groups. On day two, conditioning began with an injection (i.p.) of 0.09 mg/kg nicotine or saline and immediate isolation in one of the conditioning compartments for 30 minutes. Five hours later the same animal was injected with the opposite drug condition and confined to the other conditioning chamber for 30 minutes. Conditioning was continued for two more days with animals being paired in the same conditioning chamber with the same drug treatment. The experiment was counterbalanced in order to assure that some animals were drug paired in the checkered compartment and some in the striped compartment. On day five, animals were placed in the middle chamber with access to all three chambers for 15 minutes. The preference score for each compartment was determined by subtracting the preconditioning score from the test day score (in seconds).
86Rb+ Efflux
Crude synaptosomes were prepared from hippocampus, striatum, thalamus, and midbrain and loaded with 86Rb+ purchased from Perkin-Elmer Life and Analytical Sciences, Inc. (Waltham, MA, USA) using a technique that has been previously described [33]. Perfusion of synaptosomes with buffer (135 mM NaCl, 5 mM CsCl, 1.5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 20 mM glucose, 50 nM tetrodotoxin, 1 μM atropine, 25 mM HEPES hemisodium, 0.1% bovine serum albumin, pH 7.5) occurred for 5 minutes prior to the initiation of data collection. Data were then collected for 90 seconds to determine basal efflux and immediately thereafter samples were stimulated with acetylcholine for 5 seconds followed by 2 minutes of buffer perfusion. Data were collected on a β-RAM Radioactivity HPLC detector (IN/US Systems, Inc., Tampa, FL). The magnitude of efflux due to stimulation was determined relative to the basal efflux before and after the agonist peak. An exponential decay function was used to fit the basal efflux and subtracted from the efflux resulting from agonist stimulation as previously described [33]. Data was analyzed by determining the magnitude of agonist stimulated 86Rb+ release as the counts per minutes (cpm) exceeding basal efflux during exposure to the agonist. A nonlinear curve fitting algorithm in SigmaPlot 5.0 (Jandel Scientific, San Rafael, CA) was used to fit the data. Michaelis-Menten equations were used to generate concentration-response curves.
125I-Epibatidine Binding
Measurement of 125I-epibatidine binding in synaptosomes was quantified as previously described [34]. Briefly, incubations were completed in 96 well polystyrene plates in a final volume of 30 μl in 1X binding buffer (144 mM NaCl, 1.5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 20 mM HEPES, pH = 7.5) at 22°C for 2 h in the presence of 200 pM 125I-epibatidine purchased from Perkin-Elmer Life and Analytical Sciences, Inc. (Waltham, MA, USA). Total and non-specific binding were measured in the presence of binding buffer or 100 μM cytisine, respectively. Samples were counted on a Packard Cobra counter. Specific binding was determined by subtracting the non-specific binding from the total binding. Protein content was detemined by the method of Lowry [35].
Statistical Analyses
Most comparisons between genotypes (Chrna4 A529 and Chrna4 T529) were analyzed using the Student’s t-test. All data are reported as an average ± SEM. Oral nicotine consumption was calculated by determining the μg of nicotine consumed divided by the weight of the mouse (g) ± SEM. Student’s t-test was used to determine genotypic differences for average nicotine consumption and a two-way ANOVA was used to calculate an effect of genotype and nicotine concentration on nicotine consumption. The ACh elicited 86Rb+ efflux was calculated using a nonlinear two site regression with constrained EC50 values in GraphPad Prism 5 (GraphPad Software, La Jolla, CA). A two site regression analysis of the concentration response curves was used to establish EC50 and Emax (maximal efflux) for agonist stimulated 86Rb+ efflux. The goodness of fit (R2) of the data was highest using a two site regression analysis when fitting the data. No significant difference in EC50 values were detected within brain regions between genotype for the high and low sensitivity components. Therefore, for curve-fitting, EC50 values were constrained to be equal between genotypes for each brain region. The constrained EC50 for the high sensitivity component and low sensitivity component were as follows in hippocampus, striatum, midbrain, and thalamus: 2.45 ± 1.24 and 110.50 ± 86.42, 1.24 ± 1.02 and 83.59 ± 43.92, 2.21 ± 1.16 and 198.30 ± 79.77, and 2.31 ± 1.64 and 43.46 ± 45.12 μM ACh, respectively.
Results
Verification of the Chrna4 T529A knockin mutation
Chrna4 T529A knockin mice, hereafter referred to as A529 knockin mice, were generated by converting a threonine codon at position 529 of Chrna4 to an alanine codon as described in the methods. To confirm that the RNA for Chrna4 from the knockin mice differed from wild-type Chrna4 allele for only codon 529, RT-PCR was performed on RNA isolated from the knockin mice and their littermate controls. Sequencing results from the PCR products established that the alanine for threonine codon substitution was the only sequence difference in the RNAs between the knockin animals and controls (data not shown). RT-PCR products from primers designed to span intron-exon boundaries also indicated that splicing of the nAChR α4 subunit RNA was not affected by the engineered mutation (data not shown).
Influence of the Chrna4 T529A polymorphism on measures of nicotine sensitivity
The mouse Chrna4 T529A polymorphism originally was found to be associated with strain differences in nicotine induced changes in body temperature and locomotor activity [20]. To more directly assess whether the Chrna4 T529A polymorphism was responsible for these observed associations the Chrna4 A529 knockin mice and T529 littermate controls were tested for these measures of the acute effects of nicotine. Consistent with previous data, a significant effect of the polymorphism (p < 0.05) was observed for nicotine induced hypothermia (figure 1b) with Chrna4 A529 animals exhibiting greater sensitivity to the body temperature depressant effects of nicotine than Chrna4 T529 animals. In contrast to previous data, no effect of the polymorphism was observed on Y-maze activity (figure 2b). These results indicate that the Chrna4 T529A polymorphism measurably affects sensitivity to the hypothermic effects of nicotine but not sensitivity to the locomotor depressant affects of nicotine.
Figure 1.

Effect of Chrna4 T529A polymorphism on saline (a) and 0.5 mg/kg nicotine induced hypothermia (b). A529 knockin animals (n = 22) were more susceptible to the hypothermic effects of nicotine compared to T529 littermates (n = 25). The mean decrease in body for A529 knockin animals compared to control animals was 1.49 ± 0.29 and 0.70 ± 0.22 degrees Celsius, respectively. Asterisk (*) indicates data values are significantly different (p < 0.05) between genotype. In some instances, the error bars are too small to be seen.
Figure 2.

Effect of saline (a) and 0.5 mg/kg nicotine (b) on Y-maze activity in Chrna4 T529A knockin mice. A529 animals (n = 22) and T529 littermates (n = 25) displayed no difference in nicotine-induced hypolocomotion. The average number of beam breaks for A529 knockin and T529 animals were 48.86 ± 15.28 and 48.32 ± 12.20, respectively.
Another measure that consistently has been associated with the Chrna4 T529A polymorphism is free choice oral nicotine consumption [12, 13]. Consequently, the Chrna4 A529 knockin mice and T529 littermates were evaluated for an effect of the T529A polymorphism on nicotine consumption. The animals were given access to 25, 50 and 100 μg/ml nicotine incrementally for 12 days in a standard two-bottle choice paradigm. Results confirmed that the T529A polymorphism influences oral nicotine consumption (figure 3). Two-way ANOVA indicated that there was an effect of Chrna4 genotype (p < 0.05) and concentration (p < 0.005) on nicotine consumption but not a significant interaction between the variables (p = 0.47). Over the twelve day test period, Chrna4 A529 knockin mice drank less nicotine and consumed a lower average dose (mg/kg) of nicotine than their T529 wildtype littermates.
Figure 3.

The influence of the T529A polymorphism on oral nicotine consumption. Overall nicotine consumption in Chrna4 A529 mice (n = 29) was significantly less relative to T529 littermates (n = 26). A529 knockin animals consumed 2.19 ± 0.19, 3.03 ± 0.39, and 3.21 ± 0.56 mg/kg/day nicotine at the 25, 50 and 100 μg/ml concentrations, respectively. In contrast, T529 littermates consumed 2.45 ± .023, 4.16 ± 0.52 and 4.71 ± 0.90 mg/kg/day nicotine at the same nicotine concentrations. In addition, the average daily amount of nicotine consumed (figure 3b) was significantly less for Chrna4 A529 animals (2.82 ± 0.32 mg/kg/day) relative to T529 controls (3.78 ± .047 mg/kg/day). Asterisk (*) indicates p < 0.05.
The observation that Chrna4 T529A genotype affects free choice nicotine consumption suggests that the polymorphism might alter sensitivity to the reinforcing effects of nicotine. In addition, there is evidence that α4* nAChRs are necessary and sufficient for modulating the reinforcing the effects of nicotine [27]. Therefore, to assess whether the Chrna4 T529A polymorphism alters sensitivity to the reinforcing properties of nicotine, Chrna4 T529A knockin animals and wildtype controls were examined for differences in nicotine induced conditioned place preference. Results indicated that the polymorphism influences sensitivity to the reinforcing properties of nicotine (figure 4). Wildtype mice (T529 littermates) exhibited a significant place preference for nicotine while Chrna4 A529 knockin mice did not (p < 0.01). On average, the Chrna4 A529 knockin animals spent no more time in the drug-paired chamber on the test day relative to the preconditioning day whereas T529 animals spent significantly more time in the drug paired chamber on the test day relative to the preconditioning day.
Figure 4.
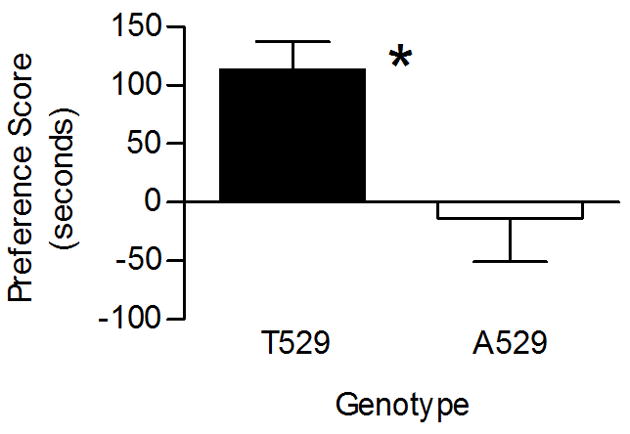
Effect of Chrna4 T529A polymorphism on unbiased nicotine induced conditioned place preference. T529 control animals (n = 12) exhibited significantly increased place preference to nicotine (0.09 mg/kg) relative to A529 knockin animals (n = 9). The average preference score for T529 control animals was 113.17 ± 24.32 seconds compared to −13.56 ± 36.96 seconds for A529 knockin mice. Asterisk (*) indicates p < 0.05.
Influence of the Chrna4 T529A polymorphism on the function of α4β2* nAChRs
Studies have suggested that the Chrna4 T529A polymorphism influences the function of α4β2* nAChRs in both a heterologous expression system and in mouse brain [29–31]. Therefore, the observed effect of the polymorphism on nicotine sensitivity might be explained by an effect of the polymorphism on receptor function. In order to directly test the effect of the Chrna4 T529A polymorphism on receptor function, acetylcholine-stimulated 86Rb+ efflux was measured from synaptosomes prepared from four brain regions of the Chrna4 A529 animals and T529 littermates. The brain regions tested were hippocampus, striatum, midbrain, and thalamus. Results indicate that ACh elicited 86Rb+ efflux in midbrain was different between genotypes. Concentration response curves from midbrain synaptosomes indicated that nAChRs with the T529 variant of α4 produce a greater maximal response to ACh than nAChRs with the A529 variant of α4 (p < 0.05) (Figure 5). In addition, multiple reports over the past decade have shown that α4* containing receptors can exist as two populations differing in sensitivity to activation by nicotinic agonists [36–39]. Therefore, the potential influence of the Chrna4 T529A polymorphism on the proportion of high and low sensitivity α4* nAChRs was assessed (figure 6). Similar to the results of the concentration-response curves, the polymorphism affected this measure of nAChR function in midbrain (p < 0.05) but not in other brain region. In midbrain, the percent of the response to ACh that could be attributed to the high sensitivity component was significantly greater for nAChRs containing the A529 variant of α4 than for nAChRs possessing the T529 variant of α4.
Figure 5.

Effect of Chrna4 T529A polymorphism on ACh elicited 86Rb+ efflux in hippocampus, striatum, midbrain, and thalamus. Genotypic differences in the high sensitivity component were detected in midbrain (c) but not hippocampus (a), striatum (b), and thalamus (d). Additionally, a significant difference in maximal response was observed in midbrain with T529 control animals exhibiting greater function (16.69 ± 1.41 units above baseline, n = 8–9) than A529 knockin animals (11.79 ± 0.55 units above baseline, n = 6–8). Data were fit to a two site curve with the EC50 values constrained for each component (high and low sensitivity). Asterisk (*) indicates data are significantly different (p < 0.05) for a genotypic comparison at the maximal response.
Figure 6.

Influence of the Chrna4 T529A polymorphism on the percentage of high sensitivity α4β2* nAChRs. The contribution of the high sensitivity α4β2* component to the acetylcholine concentration response curve was calculated for hippocampus (a), striatum (b), midbrain (c), and thalamus (d) in A529 knockin animals (n = 6–8) and T529 control animals (n = 8–9). There was no effect of genotype for this measure of receptor function in hippocampus, striatum, and thalamus. However, a significant difference in the percent of high sensitivity was observed in midbrain where 41.4 ± 6.1% of the response to acetylcholine was due to the high sensitivity component in A529 knockin mice and 23.0 ± 2.8% of the acetylcholine response was due to the high sensitivity component in controls. Asterisk (*) indicates p < 0.05.
The Chrna4 T529A genotype-dependent difference in maximal response to acetylcholine that was detected in midbrain could be the result of differences in receptor function, receptor expression or a combination of the two. Therefore, 125I-Epibatidine binding was measured in the hippocampus, midbrain, striatum and thalamus of Chrna4 A529 knockin and T529 wildtype animals to determine if the polymorphism affects nAChR expression. Results (figure 7) indicated no genotypic difference in 125I-epibatidine binding in hippocampus, striatum, and midbrain, but did detect a genotypic effect in thalamus. However, T529 control mice exhibited significantly (p < 0.05) higher binding in thalamus than did Chrna4 A529 knockin animals.
Figure 7.

125I-Epibatidine binding in hippocampus, striatum, midbrain and thalamus of Chrna4 T529A knockin mice. 125I-Epibatidine (200 pM) binding, which measures high affinity nAChR expression, did not differ between A529 knockin mice and T529 littermates in membrane fractions prepared from hippocampus (a), striatum (b), and midbrain synaptosomes. However, there was a significant difference (p < 0.05) in ligand binding in membrane fractions prepared from thalamic synaptosomes (d). T529 thalamus exhibited higher levels of binding (109.87 ± 7.43 fmol/mg) than thalamus from A529 knockin animals (71.50 ± 7.43 fmol/mg).
Discussion
Previous studies reported that the Chrna4 T529A polymorphism is associated with a wide range of behavioral and physiological responses to nicotine in mice [12, 13, 19–21]. However, it could not be determined whether these reported associations were specifically due to the T529A polymorphism, a different polymorphism in Chrna4 or a polymorphism in a gene linked to Chrna4. In the data reported here, Chrna4 A529 knockin mice were utilized to confirm that the T529A polymorphism does influence nicotine-induced hypothermia and free-choice oral nicotine consumption. Chrna4 A529 knockin mice were found to be more sensitive to the hypothermic effects of nicotine and to consume less nicotine by choice than wildtype controls. The A529 knockin mice also were found to differ from their control littermates for the reinforcing properties of nicotine. Mice possessing the T529 allele exhibited a clear reinforcement to a dose of 0.09 mg/kg nicotine that was absent in animals carrying the A529 allele. The positive reinforcement to nicotine seen in mice possessing the T529 allele of Chrna4 may explain why these animals consume more nicotine by choice than do T529A knockin animals. However, since only a single dose of nicotine was tested in the place preference paradigm, it remains to be determined if the A529 knockin mice completely lack positive reinforcement to nicotine or have a shifted dose-response to nicotine reinforcement.
A529 knockin mice, however, did not differ from controls in sensitivity to the locomotor depressant effects of nicotine across several doses of nicotine (only the data for saline and the 0.5 mg/kg dose are shown in figure 2). This finding suggests that the previous reported association between the T529A polymorphism and nicotine-induced hypolocomotion in the Y-maze may be due to a different polymorphism in either Chrna4 or a linked gene. Alternatively, an effect of the T529A polymorphism on nicotine-induced hypolocomotion may be immeasurable in T529A knockin mice because α4β2* nAChRs are only one of at least three different nAChR subtypes that mediate the effects of nicotine on locomotor activity [40–43]. It should be noted that locomotor activity was only assessed for three minutes in the Y-maze to allow for direct comparisons with previously published data. To more thoroughly assess the role of the Chrna4 T529A polymorphism on locomotor activity a full time course of the effect of nicotine on locomotion is required.
Results of this study demonstrated that the Chrna4 T529A polymorphism alters the function of α4β2* nAChRs in a brain region-specific manner. In the midbrain, two effects of the polymorphism were observed. First, maximal acetylcholine-stimulated 86Rb+ efflux from α4β2* nAChRs was greater in mice possessing the T529 variant of Chrna4 relative to α4β2* nAChRs from mice carrying the A529 allele of Chrna4. Second, the fraction of the total acetylcholine-stimulated 86Rb+ efflux that could be attributed to the high sensitivity population of α4β2* nAChRs was significantly greater in A529 knockin mice. The observation that there is a functional effect of the polymorphism on midbrain α4β2* nAChRs as well as individual differences in oral nicotine consumption, conditioned place preference and nicotine induced hypothermia is consistent with previous studies that have shown that α4β2* nAChRs in the midbrain are essential for nicotine self-administration [25, 26, 44, 45], nicotine place preference [27, 28] and the effects of nicotine on body temperature [40, 41].
Acetylcholine-stimulated 86Rb+ efflux data from midbrain also confirmed previous reports that the Chrna4 T529A polymorphism leads to a shift in the proportion of α4β2* nAChRs with high and low sensitivity to activation by agonists. The phenomena that α4β2* nAChRs can exist in two pharmacologically distinct forms, one activated at low concentrations of agonist and the other activated only at higher agonist concentrations, has been described both in heterologous expression systems [36, 37, 39, 46] and in mouse brain synaptosomes [33, 47]. Moreover, it has been speculated that alterations in the ratio of high sensitivity to low sensitivity α4β2* nAChRs might be important for the addiction process [46]. The data reported here provide the first evidence that innate differences in the ratio of high to low sensitivity α4β2* nAChRs might influence susceptibility to nicotine addiction by altering sensitivity to the reinforcing effects of nicotine.
Although an effect of the Chrna4 T529A polymorphism on α4β2* function was observed in the midbrain, no detectable affect of the polymorphism was observed in the hippocampus, striatum or thalamus. The reason for the region-specific effect of the polymorphism on receptor function is not clear. However, one possible explanation is that these regions, but not the midbrain, express functional α4β2* nAChRs that include α5 [48–50] or other subunits [51]. Inclusion of these other subunits may mask the effect of the polymorphism on receptor function. Studies that examine the effect of the polymorphism on the function of α4β2 nAChRs in mice that lack these other subunits are needed to address this possibility. In addition, assessing the function of subpopulations of α4β2* nAChRs, such as those that contribute to dopamine and GABA release, may uncover functional effects of the Chrna4 T529A polymorphism not detected by the 86Rb+ efflux assay.
One unexpected finding is that the T529A polymorphism affected the expression of α4β2* nAChRs in the thalamus. Previous studies provided little evidence that the T529A polymorphism affects receptor expression or α4 protein levels in mouse brain [31, 52] or transiently transfected cells [29]. Although the basis for the apparent effect of the T529A polymorphism on the expression of thalamic α4β2* nAChRs is not known, one possible explanation is that the level of α4β2* function in the thalamus is under tight homeostatic control. This hypothesis is based upon the observation that, despite the differences in expression of thalamic α4β2* nAChRs, there is no difference in the functional response of these receptors between T529 littermates and A529 knockin animals. Because the T529 variant is the normal allele expressed in C57BL/6J mice, these data suggest that α4β2* nAChRs made from the A529 allele of Chrna4 are down regulated in expression in the thalamus in order to maintain a functional response indistinguishable from the normally expressed α4β2 nAChRs in this mouse strain. If this is the case, then the effect of the T529A polymorphism on receptor expression in the thalamus is not a direct effect of the polymorphism, but rather an indirect effect via homeostatic regulation of receptor function.
The finding that the T529A polymorphism affects the function of α4β2* nAChRs in the A529 knockin mice confirms that the polymorphism is within a novel functional domain of the receptor that influences the ratio of high to low sensitivity α4β2* nAChRs. As described previously [29, 31], the polymorphism is within a region of the large cytoplasmic loop of the α4 subunit that is highly conserved in mammals. Understanding the role of this region in regulating the function of α4β2* nAChRs and how the T529A polymorphism alters this activity provides unique insight into the molecular regulation of α4β2* nAChR function.
Although the Chrna4 T529A polymorphism has not been found in human populations to date, the results of this study are, nonetheless, relevant for understanding the genetics of nicotine dependence in humans. For example, several groups have used genetic approaches to establish that α4β2* nAChRs are critical for nicotine self-administration, conditioned place preference and nicotine-induced hypothermia [26–28, 40, 41, 44, 45]. However, these studies utilized knockout mice in which expression of α4β2* nAChRs was eliminated or hypersensitive knockin mice that express α4β2* nAChRs that exhibit a nearly twenty-fold increase in sensitivity to nicotine relative to native α4β2* nAChRs. Such dramatic changes in gene expression and function are not likely to be common among human genetic variants that influence nicotine dependence and other complex phenotypes. Results from the T529A knockin mice demonstrate that substantially smaller changes in the function or expression of α4β2* nAChRs, similar to those one would expect to occur with modest frequency in the human population, are sufficient to affect nicotine sensitivity in a meaningful way.
In summary, the data reported here are the first to demonstrate that a naturally-occurring polymorphism in the α4 nAChR subunit contributes to individual variability in sensitivity to several affects of nicotine, including positive reinforcement, oral consumption and hypothermia. The effect of the polymorphism on these behavioral and physiological phenotypes is accompanied by an effect of the polymorphism on the function of α4β2* nAChRs in the midbrain, a brain region that modulates these behavioral and physiological phenotypes in an α4β2* dependent manner. Moreover, results from these studies provide the first evidence that the ratio of high to low sensitivity α4β2* nAChRs may be behaviorally and physiologically relevant. However, the mechanism by which the Chrna4 T529A polymorphism affects the ratio of high to low sensitivity α4β2* nAChRs remains to be elucidated. Finally, these results along with the recently published data from Mague et al., [53] demonstrate the utility of using the knockin mouse strategy in understanding the functional consequence of naturally occurring polymorphisms in complex behaviors related to drug addiction.
Acknowledgments
The author’s would like to acknowledge Carolyn Ferguson and Amy Hua for expert technical assistance. Supported by DA014369, DA015663, DA017637, DA024515 and AA010422
Footnotes
Disclosure/conflicts of interest. The authors have no conflicts of interest.
Reference List
- 1.State-specific prevalence and trends in adult cigarette smoking--United States, 1998–2007. MMWR Morb Mortal Wkly Rep. 2009;58:221–226. [PubMed] [Google Scholar]
- 2.FISHER RA. Cancer and smoking. Nature. 1958;182:596. doi: 10.1038/182596a0. [DOI] [PubMed] [Google Scholar]
- 3.FISHER RA. Lung cancer and cigarettes. Nature. 1958;182:108. doi: 10.1038/182108a0. [DOI] [PubMed] [Google Scholar]
- 4.Li MD. The genetics of nicotine dependence. Curr Psychiatry Rep. 2006;8:158–164. doi: 10.1007/s11920-006-0016-0. [DOI] [PubMed] [Google Scholar]
- 5.Stitzel JA, Leonard SS, Collins AC. Genetic Regulation of Nicotine-Related Behaviors and Brain Nicotinic Receptors. In: Clemente F, Gotti C, Fornasari D, editors. Handbook of Experimental Pharmacology: Neuronal Nicotinic Receptors. Milan, Italy: Springer-Verlag; 2000. pp. 563–585. [Google Scholar]
- 6.Marks MJ, Stitzel JA, Collins AC. Genetic influences on nicotine responses. Pharmacol Biochem Behav. 1989;33:667–678. doi: 10.1016/0091-3057(89)90406-1. [DOI] [PubMed] [Google Scholar]
- 7.Miner LL, Collins AC. Strain comparison of nicotine-induced seizure sensitivity and nicotinic receptors. Pharmacol Biochem Behav. 1989;33:469–475. doi: 10.1016/0091-3057(89)90532-7. [DOI] [PubMed] [Google Scholar]
- 8.Marks MJ, Campbell SM, Romm E, Collins AC. Genotype influences the development of tolerance to nicotine in the mouse. J Pharmacol Exp Ther. 1991;259:392–402. [PubMed] [Google Scholar]
- 9.Marks MJ, Romm E, Gaffney DK, Collins AC. Nicotine-induced tolerance and receptor changes in four mouse strains. J Pharmacol Exp Ther. 1986;237:809–819. [PubMed] [Google Scholar]
- 10.Marks MJ, Stitzel JA, Collins AC. Dose-response analysis of nicotine tolerance and receptor changes in two inbred mouse strains. J Pharmacol Exp Ther. 1986;239:358–364. [PubMed] [Google Scholar]
- 11.Robinson SF, Marks MJ, Collins AC. Inbred mouse strains vary in oral self-selection of nicotine. Psychopharmacology (Berl) 1996;124:332–339. doi: 10.1007/BF02247438. [DOI] [PubMed] [Google Scholar]
- 12.Butt CM, King NM, Hutton SR, Collins AC, Stitzel JA. Modulation of nicotine but not ethanol preference by the mouse Chrna4 A529T polymorphism. Behav Neurosci. 2005;119:26–37. doi: 10.1037/0735-7044.119.1.26. [DOI] [PubMed] [Google Scholar]
- 13.Li XC, Karadsheh MS, Jenkins PM, Stitzel JA. Genetic correlation between the free-choice oral consumption of nicotine and alcohol in C57BL/6JxC3H/HeJ F2 intercross mice. Behav Brain Res. 2005;157:79–90. doi: 10.1016/j.bbr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Schechter MD, Meehan SM, Schechter JB. Genetic selection for nicotine activity in mice correlates with conditioned place preference. Eur J Pharmacol. 1995;279:59–64. doi: 10.1016/0014-2999(95)00139-c. [DOI] [PubMed] [Google Scholar]
- 15.Grabus SD, Martin BR, Brown SE, Damaj MI. Nicotine place preference in the mouse: influences of prior handling, dose and strain and attenuation by nicotinic receptor antagonists. Psychopharmacology (Berl) 2006;184:456–463. doi: 10.1007/s00213-006-0305-7. [DOI] [PubMed] [Google Scholar]
- 16.Risinger FO, Brown MM. Genetic differences in nicotine-induced conditioned taste aversion. Life Sci. 1996;58:223–229. doi: 10.1016/0024-3205(96)00051-3. [DOI] [PubMed] [Google Scholar]
- 17.Portugal GS, Gould TJ. Genetic variability in nicotinic acetylcholine receptors and nicotine addiction: converging evidence from human and animal research. Behav Brain Res. 2008;193:1–16. doi: 10.1016/j.bbr.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li MD. Identifying susceptibility loci for nicotine dependence: 2008 update based on recent genome-wide linkage analyses. Hum Genet. 2008;123:119–131. doi: 10.1007/s00439-008-0473-0. [DOI] [PubMed] [Google Scholar]
- 19.Stitzel JA, Jimenez M, Marks MJ, Tritto T, Collins AC. Potential role of the alpha4 and alpha6 nicotinic receptor subunits in regulating nicotine-induced seizures. J Pharmacol Exp Ther. 2000;293:67–74. [PubMed] [Google Scholar]
- 20.Tritto T, Stitzel JA, Marks MJ, Romm E, Collins AC. Variability in response to nicotine in the LSxSS RI strains: potential role of polymorphisms in alpha4 and alpha6 nicotinic receptor genes. Pharmacogenetics. 2002;12:197–208. doi: 10.1097/00008571-200204000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Tritto T, Marley RJ, Bastidas D, Stitzel JA, Collins AC. Potential regulation of nicotine and ethanol actions by alpha4-containing nicotinic receptors. Alcohol. 2001;24:69–78. doi: 10.1016/s0741-8329(01)00135-5. [DOI] [PubMed] [Google Scholar]
- 22.Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, et al. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev. 1999;51:397–401. [PubMed] [Google Scholar]
- 23.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]
- 24.Whiting P, Lindstrom J. Purification and characterization of a nicotinic acetylcholine receptor from rat brain. Proc Natl Acad Sci U S A. 1987;84:595–599. doi: 10.1073/pnas.84.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corrigall WA, Coen KM, Adamson KL. Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res. 1994;653:278–284. doi: 10.1016/0006-8993(94)90401-4. [DOI] [PubMed] [Google Scholar]
- 26.Epping-Jordan MP, Picciotto MR, Changeux JP, Pich EM. Assessment of nicotinic acetylcholine receptor subunit contributions to nicotine self-administration in mutant mice. Psychopharmacology (Berl) 1999;147:25–26. doi: 10.1007/s002130051135. [DOI] [PubMed] [Google Scholar]
- 27.Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, et al. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- 28.Walters CL, Brown S, Changeux JP, Martin B, Damaj MI. The beta2 but not alpha7 subunit of the nicotinic acetylcholine receptor is required for nicotine-conditioned place preference in mice. Psychopharmacology (Berl) 2006;184:339–344. doi: 10.1007/s00213-005-0295-x. [DOI] [PubMed] [Google Scholar]
- 29.Kim H, Flanagin BA, Qin C, Macdonald RL, Stitzel JA. The mouse Chrna4 A529T polymorphism alters the ratio of high to low affinity alpha 4 beta 2 nAChRs. Neuropharmacology. 2003;45:345–354. doi: 10.1016/s0028-3908(03)00167-9. [DOI] [PubMed] [Google Scholar]
- 30.Butt CM, Hutton SR, Stitzel JA, Balogh SA, Owens JC, Collins AC. A polymorphism in the alpha4 nicotinic receptor gene (Chrna4) modulates enhancement of nicotinic receptor function by ethanol. Alcohol Clin Exp Res. 2003;27:733–742. doi: 10.1097/01.ALC.0000067973.41153.BC. [DOI] [PubMed] [Google Scholar]
- 31.Dobelis P, Marks MJ, Whiteaker P, Balogh SA, Collins AC, Stitzel JA. A polymorphism in the mouse neuronal alpha4 nicotinic receptor subunit results in an alteration in receptor function. Mol Pharmacol. 2002;62:334–342. doi: 10.1124/mol.62.2.334. [DOI] [PubMed] [Google Scholar]
- 32.Labarca C, Schwarz J, Deshpande P, Schwarz S, Nowak MW, Fonck C, et al. Point mutant mice with hypersensitive alpha 4 nicotinic receptors show dopaminergic deficits and increased anxiety. Proc Natl Acad Sci U S A. 2001;98:2786–2791. doi: 10.1073/pnas.041582598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marks MJ, Whiteaker P, Calcaterra J, Stitzel JA, Bullock AE, Grady SR, et al. Two pharmacologically distinct components of nicotinic receptor-mediated rubidium efflux in mouse brain require the beta2 subunit. J Pharmacol Exp Ther. 1999;289:1090–1103. [PubMed] [Google Scholar]
- 34.Whiteaker P, Jimenez M, McIntosh JM, Collins AC, Marks MJ. Identification of a novel nicotinic binding site in mouse brain using [(125)I]-epibatidine. Br J Pharmacol. 2000;131:729–739. doi: 10.1038/sj.bjp.0703616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein Measurement with the Folin Phenol Reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 36.Zwart R, Vijverberg HP. Four pharmacologically distinct subtypes of alpha4beta2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol Pharmacol. 1998;54:1124–1131. [PubMed] [Google Scholar]
- 37.Covernton PJ, Connolly JG. Multiple components in the agonist concentration-response relationships of neuronal nicotinic acetylcholine receptors. J Neurosci Methods. 2000;96:63–70. doi: 10.1016/s0165-0270(99)00185-5. [DOI] [PubMed] [Google Scholar]
- 38.Buisson B, Bertrand D. Chronic exposure to nicotine upregulates the human (alpha)4((beta)2 nicotinic acetylcholine receptor function. J Neurosci. 2001;21:1819–1829. doi: 10.1523/JNEUROSCI.21-06-01819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J. Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol. 2003;63:332–341. doi: 10.1124/mol.63.2.332. [DOI] [PubMed] [Google Scholar]
- 40.McCallum SE, Collins AC, Paylor R, Marks MJ. Deletion of the beta 2 nicotinic acetylcholine receptor subunit alters development of tolerance to nicotine and eliminates receptor upregulation. Psychopharmacology (Berl) 2006;184:314–327. doi: 10.1007/s00213-005-0076-6. [DOI] [PubMed] [Google Scholar]
- 41.Tapper AR, McKinney SL, Marks MJ, Lester HA. Nicotine responses in hypersensitive and knockout alpha 4 mice account for tolerance to both hypothermia and locomotor suppression in wild-type mice. Physiol Genomics. 2007;31:422–428. doi: 10.1152/physiolgenomics.00063.2007. [DOI] [PubMed] [Google Scholar]
- 42.Salas R, Cook KD, Bassetto L, De Biasi M. The alpha3 and beta4 nicotinic acetylcholine receptor subunits are necessary for nicotine-induced seizures and hypolocomotion in mice. Neuropharmacology. 2004;47:401–407. doi: 10.1016/j.neuropharm.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 43.Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, et al. In vivo activation of midbraindopamine neurons via sensitized, high-affinity alpha 6 nicotinic acetylcholine receptors. Neuron. 2008;60:123–136. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, et al. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- 45.Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, et al. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci. 2008;28:12318–12327. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buisson B, Bertrand D. Nicotine addiction: the possible role of functional upregulation. Trends Pharmacol Sci. 2002;23:130–136. doi: 10.1016/S0165-6147(00)01979-9. [DOI] [PubMed] [Google Scholar]
- 47.Gotti C, Moretti M, Meinerz NM, Clementi F, Gaimarri A, Collins AC, et al. Partial deletion of the nicotinic cholinergic receptor alpha 4 or beta 2 subunit genes changes the acetylcholine sensitivity of receptor-mediated 86Rb+ efflux in cortex and thalamus and alters relative expression of alpha 4 and beta 2 subunits. Mol Pharmacol. 2008;73:1796–1807. doi: 10.1124/mol.108.045203. [DOI] [PubMed] [Google Scholar]
- 48.Brown RW, Collins AC, Lindstrom JM, Whiteaker P. Nicotinic alpha5 subunit deletion locally reduces high-affinity agonist activation without altering nicotinic receptor numbers. J Neurochem. 2007;103:204–215. doi: 10.1111/j.1471-4159.2007.04700.x. [DOI] [PubMed] [Google Scholar]
- 49.Mao D, Perry DC, Yasuda RP, Wolfe BB, Kellar KJ. The alpha4beta2alpha5 nicotinic cholinergic receptor in rat brain is resistant to up-regulation by nicotine in vivo. J Neurochem. 2008;104:446–456. doi: 10.1111/j.1471-4159.2007.05011.x. [DOI] [PubMed] [Google Scholar]
- 50.McClure-Begley TD, King NM, Collins AC, Stitzel JA, Wehner JM, Butt CM. Acetylcholine-stimulated [3H]GABA release from mouse brain synaptosomes is modulated by alpha4beta2 and alpha4alpha5beta2 nicotinic receptor subtypes. Mol Pharmacol. 2009;75:918–926. doi: 10.1124/mol.108.052274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- 52.Gahring LC, Persiyanov K, Dunn D, Weiss R, Meyer EL, Rogers SW. Mouse strain-specific nicotinic acetylcholine receptor expression by inhibitory interneurons and astrocytes in the dorsal hippocampus. J Comp Neurol. 2004;468:334–346. doi: 10.1002/cne.10943. [DOI] [PubMed] [Google Scholar]
- 53.Mague SD, Isiegas C, Huang P, Liu-Chen LY, Lerman C, Blendy JA. Mouse model of OPRM1 (A118G) polymorphism has sex-specific effects on drug-mediated behavior. Proc Natl Acad Sci. 2009;106:10847–10852. doi: 10.1073/pnas.0901800106. [DOI] [PMC free article] [PubMed] [Google Scholar]