

Abstract
Viruses have substantial value as vehicles to transport transgenes into neurons. Each virus has its own set of attributes for addressing neuroscience-related questions. Here we review some of the advantages and limitations of herpes, pseudorabies, rabies, adeno-associated, lentivirus, and others to study the brain. We then explore a novel recombinant vesicular stomatitis virus (dG-VSV) with the G-gene deleted and transgenes engineered into the first position of the RNA genome which replicates only in the first brain cell infected, as corroborated with ultrastructural analysis, eliminating spread of virus. Due to its ability to rapidly replicate and express multiple mRNA copies and additional templates for more copies, reporter gene expression is amplified substantially, over 500-fold in 6 hours, allowing detailed imaging of dendrites, dendritic spines, axons, and axon terminal fields within a few hours to a few days after inoculation. GFP expression is first detected within one hour of inoculation. The virus generates a Golgi-like appearance in all neuron or glia of regions of the brain tested. Whole cell patch clamp electrophysiology, calcium digital imaging with fura-2, and time-lapse digital imaging showed that neurons appeared physiologically normal after expressing viral transgenes. The virus has a wide range of species applicability, including mouse, rat, hamster, human, and drosophila cells. Using dG-VSV, we show efferent projections from the suprachiasmatic nucleus terminating in the periventricular region immediately dorsal to the nucleus. DG-VSVs with genes coding for different color reporters allow multicolor visualization of neurons wherever applied.
Keywords: virus, neuroanatomy, reporter gene, rapid gene expression, neurophysiology
INTRODUCTION
Viruses are obligate parasites whose survival depends on efficient penetration into host cells and expression of viral genes. Many viruses have also evolved sophisticated strategies to penetrate into a cell and outwit antiviral responses. By substituting or adding genes of experimental interest, viruses can be used as vectors which can greatly enhance the transport of transgenes into brain cells to achieve gene expression. Some viruses are useful for short-term experimental purposes, whereas others are more suitable for long-term gene expression. Viruses such as herpes, rabies, or pseudorabies show a natural affinity for infecting neurons (van den Pol, 2006), and this property can be harnessed for studies of brain circuitry.
Introduction to viruses used in experimental neuroscience
Below, we review some of the viruses commonly used for experimental neurobiology. The list is not exhaustive, but addresses some of the advantages and limitations of different types of viruses. A schematic gallery of these viruses with a comparison of size and genome is shown in Table 1. After the general overview here, we describe in detail a novel viral vector (dG-VSV) that may prove useful in some experimental paradigms, particularly those requiring rapid and strong gene expression in acute experiments.
Table 1. Characteristics of six viruses used for brain labeling studies.
Viral structure is shown in schematic drawings, viral particle size is indicated. Genome size is based on the viral wildtype genome. Depending on modifications in the viral genome, foreign genes at various sizes can be inserted. PRV, pseudorabies virus; Herpes, herpes simplex virus; VSV, vesicular stomatitis virus; AAV, adeno-associated virus; ds, double stranded; ss, single stranded, kb, kilo base pairs.
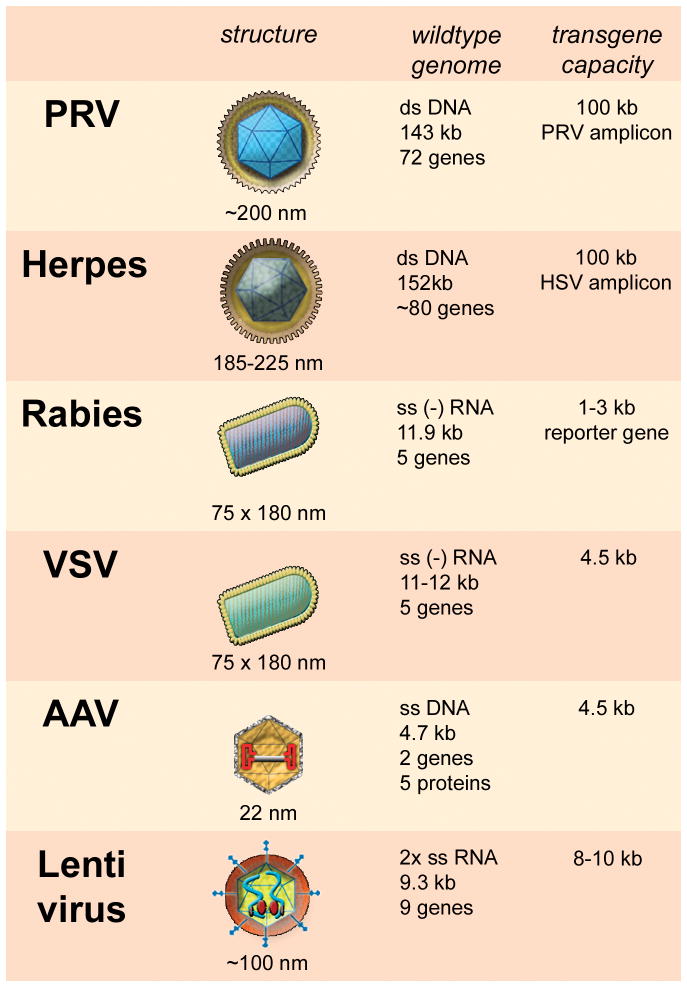 |
Pseudorabies virus (PRV)
PRV is an enveloped (surrounded by membrane) pig alpha herpes virus. The genome is double stranded DNA of about 130,000 base pairs and is proposed to encode 72 genes (Klupp et al., 2004; Ray and Enquist, 2004; Knapp and Enquist, 1997). PRV has no relation to rabies virus except in the initial behavioral dysfunction caused by each of the two viruses; the genomes are completely different.
PRV has been used extensively to trace neuronal circuits (Card et al, 1991; Enquist and Card, 2003; Aston-Jones and Card, 2000). Depending on the strain used, it can travel either in an anterograde and retrograde direction (PRV Becker strain), or primarily in a retrograde direction (PRV Bartha strain; Aston-Jones and Card,2000; Loewy,1998). PRV can be detected by conventional immunostaining with antisera against the virus (Card et al, 1991) or by GFP or RFP reporter genes expressed by recombinant PRVs (Banfield et al, 2003). Because PRV is replication competent, it will continue to infect successive synaptically coupled neurons in a circuit. It has been suggested that PRV Bartha is primarily released from infected cell bodies and dendrites, and selectively infects the presynaptic axon, and is then transported retrogradely back to the afferent neuron cell body where more virus is generated. Glial cells surrounding the infected cell tend to limit diffusion of PRV away from synaptically coupled cells (Aston-Jones and Card, 2000).
In neuronal tract tracing, a substantial advantage of PRV and some of the other viral tracers described below over chemical tracers is that the virus generates a larger signal by making the infected cell synthesize increasing amounts of the viral tracer, and similarly, if the virus continues to replicate in second order infected cells, the viral tracer generates a high amount of tracer in each infected cell; in contrast, the histochemical signal of tracers such as wheat germ agglutinin, horseradish peroxidase, and Phaeseolus vulgaris is reduced by substantial dilution even in the initial cell labeled.
Infection with PRV leads to death of the infected cell, and by successively infecting more neurons in synaptically coupled circuits, eventually leads to death of the host animal. A critical concern therefore in tracing transynaptic pathways with PRV is that of timing. Too short a time may not reveal transported virus, too long a survival interval may result in cell death or morphological shifts related to cell death. Users of PRV employ a systematic analysis at different time intervals to identify synaptically coupled neurons. To differentiate first order synaptically coupled neurons from higher order synaptically coupled neurons, additional non-replicating tracers such as cholera toxin, horseradish peroxidase, or fluorescent tracers can be co-injected with PRV, although these tracers may attenuate the ability of PRV to infect neurons. Although PRV can replicate in human cells in vitro (Wollmann et al, 2005), lethal infections by PRV in humans are rare, and similarly in adult pigs, the natural host in which PRV establishes a latent state after recovery from infection, the virus is generally not lethal. In other species, particularly mice and rats that are commonly used in axonal tract tracing, PRV is ultimately fatal. PRV is particularly valuable for tracing transynaptic circuits both within the brain (Aston-Jones et al, 2001) and from peripheral organs to the brain. It has been a method of choice for many experiments investigating neuronal regulation of peripheral organs including the digestive system, endocrine system, gonads, select muscles, and nociceptive pathways (Jasmin et al, 1997; Lee and Erskine, 2000; Rinaman et al, 2000; Yates et al, 1999). The virus has also been used to study transplant innervation of host brain (Seiler et al, 2008) or the effects of trauma on neural pathways (Lane et al, 2008).
Another interesting permutation of selective neuronal labeling by PRV is the use of a recombinant PRV that replicates conditionally after infecting cells that express cre recombinase. Cre recombinase excises a stop codon in the thymidine kinase viral gene critical for PRV replication, thereby restricting the first round of replication to cells of known phenotype, genetically engineered to express Cre under control of selective promoters. Once the virus replicates, it can continue to infect first and higher order neurons that project to the one from which PRV initially replicated in. This strategy has been used to examine the afferent projections to the NPY or leptin receptor expressing neurons of the hypothalamic arcuate nucleus (DeFalco et al, 2001). A similar strategy has been used in a number of papers to study the afferent input that regulates gonadotropic hormone synthesizing cells : The main olfactory input to the GnRH cell was not confirmed to be the vomeronasal organs, but rather the primary olfactory epithelium projecting through the olfactory cortex (Yoon et al, 2005). Inputs to the GnRH cell included the noradregergnic locus coeruleus and serotonergic raphe (Campbell and Herbison, 2007), and estrogen receptor expressing neurons of the rostral periventricular area (Wintermantel et al, 2006). An added advantage of the PRV approach to labeling the initial GnRH neuron was a clear visualization of somatic and dendritic spines (Campbell, 2007).
Adeno-associated virus (AAV)
AAV is a single strand DNA virus of about 4.7 kb with no membrane. A gene insert of about 4.5 kb can be used in replication deficient AAV; using viral duplexes consisting of two AAVs with partial complementarity, expression of genes of up to 9 kb is possible. In the absence of helper virus, wild type AAV integrates into the human genome in a site specific manner on chromosome 19 (McCarty et al., 2004) though with reportedly very low efficiency. Replication incompetent AAV shows substantially reduced integration into DNA, and instead functions independently from episomal sites in the nucleus (Berns and Parrish, 2007). Wild type AAV is dependent on a co-infection by a second helper virus for replication; as the name indicates, adenovirus is a common helper virus, but a number of other unrelated viruses can substitute for adenovirus, including herpes or cytomegalovirus. Recombinant replication-incompetent AAV can be generated with a helper virus, or by transfection with plasmids coding for adenovirus genes. So far, nine different strains of AAV have been reported, with some suggestions that dozens, if not hundreds, of unique variants may exist (Grieger and Samulski, 2005). Different strains of AAV may have different cellular preferences, and may show different affinities for infecting neurons, glia, or other types of cells (Taymans et al., 2007); some strains (AAV9) are reported to cross the blood brain barrier to infect brain cells (Foust et al, 2009).
Humans are commonly infected by AAV, but in general the virus causes few health problems. In fact, a number of reports have suggested that AAV may be oncoprotective in humans by blocking other oncogenic viruses such as human papilloma virus (Coker et al., 2001). After infection with a replication incompetent AAV, gene expression substantial enough to detect is slow and may take several days to occur, and reaches a maximum after a week or two. Remarkably, gene expression can last for very long periods, with positive tests of several months expression being reported (Michel et al, 2005; McCown et al, 1996; Kaplitt et al, 1994) with minimal insult to the infected or nearby cells (Lo et al., 1999; Taymans et al., 2007). We have found reporter gene expression after AAV infection of mouse brains more than a year post-infection (van den Pol et al, 2004). Due to its low immunogenicity, long term expression, and lack of associated disease, AAV may be the virus of choice for gene therapy in the human brain.
AAV has been used to express the axonal adhesion molecule L1, leading to enhanced recovery from spinal cord damage (Chen et al, 2007). To reduce the probability of seizures, AAV has been used to express neuroprotective peptides such as galanin (Haberman et al,2003) or neuropeptide Y (Noe et al, 2007; Richichi et al, 2004). Studies on GFP-labeled axons in AAV infected brains showed that there is an ongoing plasticity of small axons and boutons even in the mature brain (Stettler et al, 2006). Gene expression can be targeted to specific cells with AAV by using a selective promoter. We used an AAV2 with the promoter from melanin concentrating hormone (MCH) to drive expression selectively in MCH neurons in the lateral hypothalamus; although the recombinant virus infected many cell types, gene expression occurred selectively in the MCH cells, allowing us to study the neurophysiology of cells that would otherwise be difficult to identify (van den Pol et al, 2004).
Rabies virus
Rabies is an enveloped negative-strand RNA virus of the rhabdovirus family. Its genome is about 11.9 kb and codes for 5 genes. Wild type rabies is lethal in animals and to humans, and humans should not work with this virus or with infected animals without prior rabies immunization. World wide, rabies is commonly transmitted from the bite of infected mammals, often infected dogs. Currently in the USA, after a long concerted immunization effort, the primary mode of infection is no longer from dogs; bats, often the silver-haired bat, continue to be a source of concern for rabies transmission. Replication-incompetent rabies is less dangerous. Interestingly, although rabies is the most dangerous of the viruses described here, it is less likely to kill neurons than some of the other viruses described. Instead, after binding to the neuronal plasma membrane and internalization into the cell, the viral RNA genome is released into the cytoplasm (Lyles and Rupprecht,2006). Rabies takes over the synthetic organelles in neurons, and conscripts them to generate viral proteins rather than proteins that neurons would normally make. This results in a loss of normal neuronal function, even though the neuron itself can survive infection. Postmortem examination of rabies-infected brains often reveals relatively little detectable neuropathology; however, with in situ hybridization or immunocytochemistry, wide spread rabies infection can be detected.
The receptor for rabies attachment to a cell has been suggested to be the nicotinic acetylcholine receptor, the low affinity NGF receptor p75, or neural cell adhesion molecule (Lentz et al, 1982). Rabies has been used in the lab for studying neuroanatomical circuits (Taber et al, 2005; Ugolini et al, 1995; Ruigrok et al, 2008). Rabies selectively infects neurons in the brain, and not glia (Taber et al, 2005). Rabies is transported selectively in a retrograde direction from axon terminals to the cell body, and then can be released by the cell body to again be transported in the second order neuron, thereby moving backwards through a synaptically connected chain of neurons. Peripherally, rabies is taken up only at motor neuron endplates, and not at sensory axonal endings or autonomic endings (Ugolini, 2008). Although rabies-infected motor neurons remain alive (Guigoni and Coulon,2002), they may not function normally. For tracing purposes immunohistochemistry can be applied using antisera against one of the rabies virus proteins for detection (Kelly and Strick,2000; Salin et al, 2008). Alternatively, recombinant rabies virus vectors have been designed that express a GFP reporter gene for detection. Other viruses, such as lentiviruses that normally are not transported into the brain, can show enhanced transport by pseudotyping the virus with the rabies surface glycoprotein (Desmaris et al, 2001; Mazarakis et al, 2001); after peripheral infection, these viruses will then be carried into the brain by retrograde axonal transport.
Using a rabies virus lacking the viral gene coding for the rabies G protein, the first order neurons that project to an inoculated site can be identified and studied (Wickersham et al., 2007a; Wickersham et al., 2007b). The recombinant rabies viruses used in these studies have been described to label only those neurons that maintain axon terminals in the brain region into which the virus is injected. Electrophysiological studies of GFP-fluorescently labeled neurons revealed functionally intact neurons.
Lentivirus
Current lentiviral applications are based on recombinant human immunodeficiency virus type 1 (HIV-1) constructed as highly attenuated, replication incompetent vectors (Naldini, 1998) which allow long term gene expression. Other strains currently used for vector applications include feline- and equine- immunodeficiency virus (Jakobsson and Lundberg, 2006). Non-replicating lentivirus vectors have been of considerable use in expressing transgenes in neurons and glia (Adamantidis et al, 2007).
To generate a non-replicating vector, the lentivirus RNA genome of about 10 kb is stripped of all three virus genes gag, pol, env, which allows insertion of an expression cassette of up to 8 kb flanked by viral long terminal repeat (LTR) and cis acting elements required for encapsidation, reverse transcription, and integration. Upon cell entry, the vector RNA strand gets converted into a DNA strand by viral reverse transcriptase and inserts into the host cell genome with the help of a viral integrase. Host cell genome integration is the basis for stable long-term expression of the transgene and the functional DNA state allows the use of cell specific promoters for selective gene expression (Hioki et al., 2007). Lentiviruses are a group of retroviruses that - in contrast to conventional oncoretroviral vectors - have the capacity to transfect both dividing and non-dividing cells; this is due in large part to the ability of lentiviruses to penetrate the nuclear envelope in non-dividing cells. Hence, lentiviral vectors have seen wide application in experimental neuronal gene transfer both in vitro and in vivo (Boyden et al., 2005) and for experimental treatment strategies in animal models for numerous neurological diseases (Wong et al., 2006).
Lentiviral vectors have been used for neuronal circuit tracing in combination with conventional tracing methods. Grinevich and colleagues (2005) used a pyramidal neuron-specific promoter for selective expression of GFP and described an exclusively anterograde labeling pattern. Roberts et al. (2008) used two lentiviral constructs that were driven by a non-specific rous sarcoma virus promoter and were targeted to different brain regions using stereotactic coordinates. In both studies, a synaptophysin-GFP fusion protein construct was used to label pre-synaptic terminals after infection of the cell bodies.
Introduction of regulatory gene switches like the tetracycline on/off system have been successfully applied to lentiviral gene expression in the brain (Kafri et al. 2000; Liu et al., 2008b) allowing some level of external regulation. Since the vector genome does not encode for any viral proteins, local antiviral inflammation is minimal and cells successfully transfected with lentivirus show little to no adverse reactions. Pseudotyping with various envelope glycoproteins, most notably the VSV-G glycoprotein, enhances the range of target cells (Jakobsson and Lundberg, 2006), though titers of vector production vary significantly between different pseudotype variants (Quinonez and Sutton, 2002). Using a neuron-selective promoter, dopamine beta hydroxylase, to drive gene expression, the projections from the brainstem C1 catecholamine system could be studied (Card et al, 2006); the reporter GFP expression was sufficiently strong that C1 axons innervating other regions of the brain could be detected and studied.
Herpes virus
Similar to PRV, human herpes simplex-1 (or -2) is an alpha herpes virus with an enveloped double stranded DNA genome of about 152 kb that encodes around 80 genes, of which nearly half are not essential for viral replication (Mocarski et al, 1980). The majority of adult humans have been infected by herpes, but most show no serious symptoms. After local infection, the virus is carried within the axon to the sensory neuron cell body, where the herpes genome can remain for decades in a latent state as a circular episome in the cell nucleus. During latency, only the latency associated transcript (LAT) is transcribed; upon reactivation, the LAT is involved in regeneration of active infectious virus particles from the latent genome. During latency no virus is present. Because the herpes virus genome can remain latent for the life of its human host, and the virus can be reactivated, it is difficult to completely eliminate from an infected individual.
“Herpes” here is used to refer to human herpes simplex virus. The term herpes is also a family name for a group of 8 viruses that share structural and molecular traits with herpes simplex. Another virus in the herpes family is herpes zoster (also called chicken pox, although it is not a pox virus). After the initial infection in humans, herpes zoster can remain latent for decades, but upon reactivation, can cause a potentially painful condition, shingles. Cytomegalovirus is another member of the herpes family, and although it does not appear to be selectively transported in neurons, cytomegalovirus can cause significant neurological dysfunction if the fetal brain is infected via maternal transmission. Recombinant cytomegalovirus that includes a GFP reporter shows strong expression in neurons and glia (van den Pol et al, 1999; 2002). Most mammalian species are infected by species-specific herpes viruses. When these viruses are transmitted to a foreign species, the new host response ranges between two extremes, from no virus replication (mouse cytomegalovirus in humans) to active replication and death of the host (primate alpha herpes virus in humans).
One of the early uses of herpes in the lab made use of its ability to be axonally transported, with potential transport transynaptically, for instance, to study motor neurons (Ugolini et al, 1987) and other types of cells (Boldogkoi et al, 2004; Norgren and Lehman,1998). Strains of herpes differ in their neuroinvasive potential. Certain herpes simplex virus strains display anterograde transynaptic spread (Zemanick et al., 1991); for instance, the H129 strain of HSV1 is an anterograde trans-neuronal tracer that has been used to study peripheral sensory circuitry after organ infection (Song et al, 2008) and to study the afferent projections of the retina after intraocular administration (Sun et al, 1996).
Another use of herpes is for gene transfer, taking advantage of the ability of the virus to infect neurons, and the potential for replacing many viral genes with foreign DNA (Fraefel et al, 1998). As with AAV, the selectivity of gene expression for herpes can be increased by the use of neuron selective promoters; gene expression can be selective for catecholamine neurons by the use of the tyrosine hydroxylase promoter; alternately, selective virus uptake can be induced by incorporating transgenes coding for glial cell derived neurotrophic factor (GDNF) or brain derived neurotrophic factor (BDNF) into herpes virus vector particles which subsequently bind to cells that express the receptors for GDNF or BDNF on their surfaces (Cao et al, 2008).
A number of permutations of replication-incompetent herpes have been used for gene transfer. One use of replication incompetent herpes has been for the transient amelioration of narcoleptic sleep behavior in mice lacking the hypocretin/orexin gene, by increasing expression of the arousal-enhancing neuropeptide (Liu et al, 2008a). One problem with some herpes vectors is the short-term (days) gene expression, and the potential cell toxicity in the infected cell. Selective removal, or alteration of some of the wild-type herpes genes, together with the use of non-viral promoters such as those driving glutamate decarboxylase or glutamate transporters can allow a more extended gene expression with reduced cytotoxicity (Liu et al, 2005; Smith et al, 2000; Rasmussen et al, 2007). Herpes vectors have also been used to reduce expression of neuronal genes, for instance, herpes coding for the expression of antisense RNA to reduce expression of the NMDA receptor (Cheli et al, 2006).
Adenovirus
Adenovirus (Ad) is a double-stranded DNA virus with no surrounding membrane with a genome size of 30–38 kb, depending on the strain, coding for 30–40 genes. Wild-type virus is commonly associated with colds and respiratory illness. Although there are dozens of variants, a considerable amount of work has been done with Ad-5. Adenovirus has been used in part due to the ease of generating recombinant Ad for gene transfer (van Doren et al,1984; Brody and Crystal,1994; Peltekian et al, 1997; Lowenstein et al, 2003). Labeling of neurons with GFP-expressing Ad-viral vectors in vivo and in vitro has been described (Moriyoshi et al., 1996). GFP fluorescence was reported to appear within 24 hrs post injection with a peak of signal intensity after 48–72 hrs.
Fusion of GFP to a membrane anchor signal sequence from GAP-43 showed enhanced axonal and dendritic labeling. Replication incompetent adenoviral vectors have been shown to be transported retrogradely from the site of injection to the nucleus (Ridoux et al., 1994; Kuo et al, 1995). Transgene products in turn can be transported anterogradely through axonal transport (Terashima et al., 1997). Long ascending and descending spinal tracks have also been successfully labeled with adenovirus based vectors (Tsukamoto et al., 2003). One interesting application of Ad was the generation of a replication-incompetent Ad vector that produced the neuronal tracer wheat germ agglutinin, used to study olfactory pathways (Kinoshita et al, 2002). Further studies involved adenoviral vectors for labeling the neuro-muscular junction (Jacob et al., 2000). Spinal injection of an Ad vector carrying a GFP-VAMP-2 fusion protein led to presynaptic accumulation of GFP signal at remote muscular innervation sites. Since transgenes are encoded in adenoviral vectors as DNA, selective expression can be achieved by using cell-specific promoters. Reported examples include the catecholaminergic selective promoter PRS (Howorth et al., 2009), neuron-specific enolase (NSE), glial fibrillary acidic protein (GFAP), and lysosomal aspartylglucosaminidase (AGA) (Virta et al., 2006) and others.
Retrovirus
Retroviruses have proven useful for labeling developing brain cells. Retroviruses are enveloped viruses with a negative strand RNA genome of 7 to 10 kb, and can accommodate about 5 kb of transgene. A number of retroviruses selectively infect and express genes in dividing cells; after the RNA genome is reverse transcribed into DNA, the DNA integrates into the host cell genome as a provirus, making these viruses particularly useful for cell lineage studies. In this case, daughter cells of the initially infected cell would express the same reporter gene as the parent cell, allowing fate determination at a later stage of development. Since this is an integrating virus, gene expression can continue for long periods. If the retrovirus integrates into a germ cell, then the viral genome can be passed along to descendents of the initially infected host organism. Some estimates suggest that 5% or more of the human genome is composed of retroviral DNA, sometimes considered junk DNA with no obvious purpose.
Retroviruses have been used for a number of purposes in neurobiology. In one, a retrovirus carrying the proopiomelanocortin cDNA was used to infect a number of neuron-like cell lines, but only some lines were able to achieve proteolytic processing of the precursor to generate the peptides found in the hypothalamus (Noel et al, 1989). A good use of retroviruses has been to track cell lineage and precursor origin, as shown by marking glial progenitors which migrate from the subventricular zone and mature into olfactory neurons (Doetsch et al, 1999; Baker et al, 2006), birth dating hippocampal granule cells and their developing axons (Faulkner et al, 2008), or showing microglia fusion to dendrites during development (Ackman et al, 2006). Replication incompetent retroviruses were used to label developing cortical progenitors, revealing that daughter cells were initially radially distributed, but later adopted a non-radial migration pattern (Walsh and Cepko, 1993). Retroviruses may be involved in some human neurological disorders. A mouse retrovirus, Cas-Br-E, can cause lower motor neuron disease associated with spongiform degenerative changes in brain and spinal cord, possibly caused by abortive replication of the virus in neurons (Sharpe et al, 1990). Retroviruses have also been used to increase expression of genes, for instance epidermal growth factor receptors, which may play a role in cortical maturation and cellular response to local modulatory signals (Burrows et al, 1997).
Other viruses
The examples of the viruses above constitute some commonly used viruses in neuroscience research. There are a large number of other viruses that can infect brain cells. Additional viruses that have proven useful for some questions include measles, Sindbis, Semliki Forest, polio, and others. Specific genes from one virus are often used in other viruses, potentially helpful in altering the cell selectivity, expression level, or subcellular targeting. With the increase in our understanding and cloning of viral genes, many of the limitations of different viruses can be attenuated by substituting one or more genes from other viruses. For example, the CMV ie1 promoter is commonly used in many recombinant viruses for strong gene expression (Mocarski,1996; van den Pol and Ghosh, 1998).
Vesicular stomatitis virus (VSV)
VSV is a negative strand RNA membrane-bound virus with an 11.2 kb genome that includes 5 structural genes (Fig. 1). VSV is an interesting virus in that it can infect and replicate very fast, with the first progeny virus being released from infected cells in as little as 1.5 hours (Kretzschmar et al, 1996; Wagner and Rose, 1996). VSV is rare in North America; in contrast, in some regions of Central America, VSV infections are common, and a high percentage of the human population is seropositive for VSV without serious illness. VSV can be problematic to livestock.
Fig. 1. Schematic of recombinant VSVs genomes.

The top shows the 5 VSV structural genes from wild type virus. The RNA genome is shown on the left, together with the color matched proteins, and a schematic of the virus and the virus encoded proteins is shown on the right. The second genome shows an attenuated replication-competent recombinant virus in which GFP was tethered to an extra VSV-G (Dalton and Rose, 2001; van den Pol et al, 2002). The third and fourth genomes depict the replication- restricted dG-VSVs.
VSV can accommodate an insert up to 4.5 kb. By engineering genes from more problematic viruses into VSV, VSV has shown considerable promise as an immunizing vector against a variety of problematic viruses including human immunodeficiency virus, Ebola virus, influenza (Schnell et al, 1997; Wagner and Rose, 1996). Although its genome is RNA-based, recombinant VSV can be generated de novo from a double stranded DNA plasmid by transfecting cells with combinations of plasmids coding for the VSV genome and various viral proteins (Lawson et al, 1995; Wagner and Rose, 1996), together with co-infection with a modified pox virus, vaccinia, that provides RNA capping and cytoplasmic expression of recombinant T7 polymerase. VSV has not been used very much in the neuroscience arena. We and others have used VSV as an oncolytic virus to target and destroy human glioblastoma brain tumors (Wollmann et al, 2004; Ozduman et al, 2008; Lun et al, 2006). Intravenous injections of VSVrp30 cross the blood brain barrier to infect selectively human glioblastoma cells transplanted to the mouse brain (Ozduman et al, 2008).
Within the brain, the recombinant VSVs that are commonly used appear to spread primarily by diffusion (van den Pol et al, 2002), although there is a report of anterograde and retrograde axonal transport of wild-type VSV in the visual system of rodents (Lundh, 1990). Due to the binding of the VSV surface glycoprotein to many different cell types, VSV can infect neurons and glia. In neurons, the virus is released primarily by the cell body and dendrites, and poorly by the axon, and consistent with release, the VSV glycoprotein is found primarily on the somatodendritic complex (Dotti and Simons,1990).
The neuronal target of viruses can be altered by mutating the receptor binding viral protein, or substituting a specific receptor ligand. For instance, as discussed below, by pseudotyping other viruses with the VSV G-gene, the selectivity of targets can be broadened, and vice versa, using a more selective surface protein can target a particular VSV, or other virus, to a narrower range of cells.
In the current work, we generated a replication-restricted dG-VSV to express green or red reporter genes. Previous work has shown that the transmembrane VSV G protein enhances virus binding and fusion of viral particles to cell membranes (Jeetendra et al, 2002), and that the G-protein gene can be deleted from the negative strand virus genome, and in the absence of the gene, can be provided by complementation (Schnell et al, 1997; Miller et al, 2004). This recombinant virus can replicate only in the first cell that is infected, making many copies of itself and many copies of virally encoded mRNA, and these can greatly amplify the expression of transgenes engineered into the viral RNA genome. The virus includes an inserted transgene coding either for green or red fluorescent reporter proteins, GFP or dsRed, at VSV-position 1. The VSV-G gene was removed from the viral genome, and VSV-G was provided in trans from a G-expressing cell line.
This study introduces a new tool for extremely fast and strong labeling of neurons and neuronal networks. Here we show that dG-VSV can be used in vitro both in cultured neurons as well as in acute brain slices and in vivo through direct injection into the brain. Expression of reporter gene fluorescence was apparent within one hour post inoculation. By 6 hrs post inoculation, transgene expression had increased over 500-fold. The virus worked in a wide range of host species. Infected neurons expressing transgenes could be used for neurophysiological experiments and appeared normal after infection, but die at a later point. Intracerebral injections of dG-VSV do not cause illness. Similar to the lectin Phaseolus, axonal projections can be clearly visualized with dG-VSV without histochemical reaction; hypothalamic suprachiasmatic nucleus (SCN) axons show a strong bouton field in the periventricular hypothalamus immediately dorsal to SCN.
MATERIALS AND METHODS
Generation of recombinant dG-VSV
Recombinant VSVs were generated from DNA plasmids as described in detail elsewhere (Schnell et al, 1997; Lawson et al, 1995). The plasmid pVSVΔG1XN-EGFP was constructed by digesting pVSV1XN-EGFP (Ramsburg et al, 2005) and pVSVΔG (Schnell et al, 1997) with HpaI and XbaI. The ~12-kb vector fragment from pVSV1XN-EGFP was separated from the DNA sequence encoding VSV-G and purified. The ~1.4kb insert that contained the ΔG deletion sequence was separated from pVSVΔG and purified. The ΔG insert was cloned into the pVSV1XN-EGFP vector, resulting in pVSVΔG1XN-EGFP. Recombinant virus was recovered from pVSVΔG1XN-EGFP as previously described (Schnell et al, 1997) using stably transfected BHK cells expressing VSV-G controlled by a tetracycline-regulated system.
Plasmid pVSVΔG1XN-dsRed was constructed by digesting pdsRed1-1 with XhoI and XbaI. The 750bp DNA insert encoding dsRed was separated, purified, and ligated into the pVSV1XN vector (Ramsburg et al., 2005) that had been digested with XhoI and NheI. This plasmid, designated pVSV1XN-dsRed, was then digested with HpaI and XbaI to remove the DNA sequence encoding VSV G. The ~1.4kb insert encoding the ΔG deletion was separated from pVSVΔG by digestion with the same enzymes. The ΔG insert was ligated in between the XbaI and HpaI sites of the pVSV1XN-dsRed vector, resulting in pVSVΔG1XN-dsRed. Recombinant virus was recovered from this vector as previously described (Schnell et al, 1997).
VSV-M51-CT9 contains two attenuating mutation, a codon deletion for methionine at position 51 in the VSV M protein and a truncated cytoplasmic tail of the VSV G protein to 9 amino acid residues and was generated as previously described (Publicover et al., 2004; Publicover et al., 2006). VSV-1′-GFP contains a GFP expression cassette at the 1st genome position. The viral genome is shifted downstream resulting in an attenuated phenotype. Details regarding viral production can be found elsewhere (Ramsburg et al., 2005). All viruses were propagated and titered using BHK-21 cells.
Calcium digital imaging and time lapse imaging
Neurons were harvested from Swiss Albino mice on embryonic day 17 and plated on 22×22 mm coverslips. After 3–4 days in serum-free neurobasal medium (Gibco), cultures were inoculated with dG-VSV virus at a multiplicity of infection (MOI) of 0.2. Cultures were used within the first day of infection. 5 μM fura-2 acetoxymethyl ester was incubated with cells for 30 mins at 37°C in standard HEPES buffer solution (10 HEPES, 137 mM NaCl, 25 mM glucose, 5 mM KCl, 1 mM MgCl2, 3 mM CaCl2, pH 7.4). Coverslips were then loaded into a laminar flow perfusion chamber for image acquisition. Two images, one excited by 340 nm and the other by 380 nm light, were taken every 2 seconds. A Sutter filter wheel controlled by a Sutter Lambda 10-2 microprocessor was used to achieve quick alternating excitation filters. An inverted Olympus IX70 microscope was used during image acquisition. The ratiometric images, which correlate with intracellular calcium levels, were processed for each time point using the 340 and 380 images. A QImaging Retiga EX digital camera was used for image acquisition and Openlab and IgorPro software running on an Apple G5 computer were used to analyze the data.
The cells in the perfusion chamber were challenged with a number of stimuli in order to study physiological responses. Electrical stimulation was used to detect synaptic activity of the neurons as described previously (Obrietan and van den Pol,1995; Ho and van den Pol, 2007). A current was evoked in the chamber at 0.06 V/mm2 and duration of 3 ms at 7 Hz using long electrodes on opposite edges of the chamber. Glutamate (50 uM), high K+ (55 mM), and GABA (50 uM) were also used to study the effects of the physiological responses of infected cells.
Time lapse recordings of the appearance of fluorescence over time, and of migrating neurons extending processes were made in a Liebowitz buffer. Cells were maintained at 37° C. Light was provided by mercury or tungsten bulbs that were shutter controlled. Except during image acquisition, light was blocked. Focus was maintained using Slidebook software (Intelligent Imaging, Denver, CO). At each time point over the day, both fluorescent and phase contrast images were taken.
Whole-Cell Patch-Clamp Recording
Brain slices were prepared from 10–16 day old mice in which dG-VSV had been injected into the brain as described below. Coronal slices (200–250 μm) were gassed with ACSF (95% O2, 5% CO2) that contained 124 mM NaCl, 3 mM KCl, 2 mM MgCl2, 2 mM CaCl2,1.23 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose (pH 7.4 with NaOH). The recording chamber (Warner Instruments) was maintained at 36–37 and mounted on an Olympus BX51WI upright microscope equipped with video-enhanced infrared-differential interference contrast (DIC) and fluorescence. Further details of slice preparation and recording are found elsewhere (van den Pol et al, 2004; Fu and van den Pol, 2008).
Whole-cell voltage- and current-clamp recording was done with whole-cell pipettes containing 145 mM KMeSO4, 1 mM MgCl2, 10 mM HEPES, 1.1 mM EGTA, 2 mM Mg-ATP, and 0.5 mM Na2-GTP (pH 7.3 with KOH). DG-VSV virus infected neurons were detected by direct visual observation of GFP fluorescence. Slow and fast capacitance compensation was done using Pulse software (Heka Elektronik, Lambrecht, Germany). An EPC9 amplifier and Pulse software were used for data acquisition (Heka Elektronik). PulseFit (Heka Elektronik), Axograph (Axon Instruments), and Igor Pro (WaveMetrics, Lake Oswego, OR) software were used for analysis. Postsynaptic currents ≥ 5 pA were detected in Axograph 4.8, using a double exponential function sensitive to the kinetics of rise and decay. Data in the Results section are expressed as mean±SEM.
Quantitative real-time PCR
BHK cells were grown to confluency in 24-well dishes and infected with dG-VSV at an MOI of 1. Fifteen minutes later cultures were washed repeatedly with phosphate buffered saline to remove any unbound virus. Experiments were run in quadruplicate. Cells were lysed at indicated time points and RNA was extracted and isolated using TRIzol reagent (Invitrogen) following the manufacturer’s instructions. Total RNA was reverse transcribed using a SuperScriptIII RT kit (Invitrogen) and random hexamer primers (Promega). Gene expression quantitative PCR was performed in 96-well dishes on an iCycler iQ Multicolor Real-time Detection System (Bio-Rad, Hercules, CA) using Custom Taqman Gene Expression Assays (ABI, Part Number 4331348). Primer sequences were as follows: VSV-N gene: negative strand Forward Primer GCCTCGTTCAGATACGGAACT, Reverse Primer CGGTTATTTTGCAGAGGTGTCCAA, FAM Dye Probe CAATGCAGCACAATCT. eGFP positive strand Forward Primer GAGCGCACCATCTTCTTCAAG, Reverse Primer TGTCGCCCTCGAACTTCAC, FAM Dye Probe ACGACGGCAACTACA. The results were normalized to beta-actin expression (Gene Symbol: Actb, RefSeq: NM_007393.1) using Endogenous Control Assay (ABI, Part Number 4352933E), Mouse ACTB FAM Dye/MGB Probe, Non-Primer Limited, Amplicon Size 115 bp.
Tissue culture
Embryonic day 18–20 mice and neonatal mice were used to generate primary brain cultures. Cells were plated on plastic dishes, on glass coverslips, or on plastic dishes with the bottom replaced with a glass coverslip. Substrates were pretreated with poly-L-lysine to enhance cell adhesion during plating. For mixed neuron and glia cultures, Dulbecco’s minimal essential medium with 10% fetal bovine serum was used. For cultures with enriched numbers of neurons, cells were grown in Neurobasal media supplemented with B27.
In vivo brain inoculation
Animals were anesthetized with a combination of ketamine and xylazine. Using a Hamilton microsyringe, 100 nl to 500 nl of dG-VSV was injected into the brain through steel syringe needles, or through narrow glass pipettes. Viral titer ranged from 108 to 106/ml. Animals were later given an overdose of anesthetic (Nembutal or ketamine/xylazine) and perfused transcardially with physiological saline followed by 4% paraformaldehyde. Brains were subsequently removed, immersed in 15% and then 30% sucrose overnight, and sectioned on a cryostat in 15–30 um thick sections. Sections were studied in an Olympus IX70 inverted microscope fitted with a Spot digital camera (Diagnostic Imaging). Contrast and brightness were corrected in Adobe Photoshop. Experiments involving animals were approved by the Yale University Institutional Animal Care and Use Committee.
RESULTS
In vitro neuron infection with dG-VSV
In the first set of in vitro experiments, we infected brain cultures with different recombinant VSVs that all expressed reporter genes to determine which worked best for cell labeling. All VSVs tested infected all cells in vitro, including neurons, astrocytes, ependymal cells, and oligodendrocytes. The primary focus of our study was the replication restricted dG-VSV. Due to the removal of the viral glycoprotein, coded by the VSV G gene (Fig. 1), the replication of this virus is limited to the first cell it infects. However, viral replication and generation of additional RNA transcripts within that cell leads to substantial amplification of any gene engineered into the viral genome.
Figure 2A,B shows a culture of mouse brain with a large number of interconnected cells. Using an MOI of 0.05, only a small number of neurons expressed the GFP reporter, allowing clear visualization of cell body, dendrites and axons by one day after inoculation. Non-infected cells show no fluorescence. Strong GFP fluorescence reaching into distal dendrites could be found within 8–12 hours of infection. Axonal labeling took longer, primarily due to the need for time for diffusion of the GFP from the cell body. By two days after infection, a number of cells showed some signs of a deterioration of cell health, as evidenced by abnormal constrictions and dilations in the neuronal processes. Within 4 days of inoculation, most infected cells showed clear signs of cell death, including loss of truncation of dendritic processes, and reduced attachment of the substrate.
Fig. 2. Selective labeling of cultured cells and processes of a wide variety of species.

A. In culture, 4 mouse neurons show strong GFP expression a day after inoculation. B. In the phase contrast image, dozens of unlabeled cells are seen in the same field. Scale bar, 12 um. C. Drosophila S2 cells are labeled 2 days after inoculation. Bar, 25 um. D. The same field as in C, but shown in phase contrast. E. A control culture of S2 cells plated at the same density shows no fluorescence, indicating that the fluoresc ence in C is due to infection and expression of the GFP reporter. F. Rat neurons and (G) astrocytes express GFP within one day of inoculation. Bar, 12 um in F, 8 um in G. H. Human glioma cells show GFP expression 6 hrs after infection with the dG-VSV. Bar, 15 um.
Other attenuated VSVs
Since wild type VSV in the brain can spread rapidly (Reiss et al, 1998; van den Pol et al, 2002), we tested several recombinant attenuated VSVs to determine their experimental utility. We compared dG-VSV with several other recombinant VSVs with attenuated virulence and fluorescent reporter genes for their ability to infect cultured neurons. These included 1) VSV-G-GFP (Fig. 1) (Dalton and Rose,2001; van den Pol et al,2002; Wollmann et al, 2005), 2) a M51-CT9 VSV (M-51 mutation and the VSV G-protein cytoplasmic tail reduced to 9 amino acids, both mutations attenuate virulence (Clarke et al, 2007; Publicover et al., 2004), and 3) two VSVs in which the N,P,M,G,L genes were all moved one position downward, and the first position was replaced with either the GFP reporter gene or the dsRed reporter gene. These 4 viruses all gave good fluorescence expression. The VSV variant with GFP in the first position gave the strongest fluorescent signal, as expected, based on the principle of attenuated sequential gene expression of VSV (Wagner and Rose, 1996). Each of these four viruses infected brain cultures, and all replicated leading to reporter gene expression in all cells.
The fact that these four replication-competent but attenuated viruses continued to spread and infect more cells complicated their use for studying neuronal morphology. Using these viruses at a multiplicity of infection of 0.1 (MOI 0.1) resulted in infection of about 10% of the cells in the culture dish within 6–8 hours. By 24 hours post inoculation, almost all cells in the culture dish expressed the reporter gene. This indicates that the first set of cells gave rise to a productive infection that resulted in release of progeny virus that infected the remaining cells in the dish. We also tested a CT-1 VSV (G protein cytoplasmic tail reduced to one amino acid) that could potentially show reduced toxicity. However, as it showed a level of toxicity not substantially different from the other 4 replication-competent VSVs, we did not use it further. Given that a primary mode of movement within the brain is random diffusion (van den Pol et al, 2002), these experiments support the view that dG-VSV is the best of this group of recombinant VSVs for generating high levels of gene expression without leading to subsequent infection or expression in other cells.
dG-VSV infects broad range of species
Some viruses are very selective in the species that can be infected. To examine whether dG-VSV showed a wide or restricted species compatibility, we tested cells from different species. dG-VSV infected and expressed transgenes in a wide variety of species. The primary species used in this paper was mouse, which showed robust infection and expression of the reporter gene. In addition, Fig. 2C-H shows expression of the reporter gene in rat, human, and even in invertebrate cells from Drosophila; hamster cells also show strong infection. Although we did not test more species, it is probable that VSV would infect cells from any mammal.
Very rapid expression of reporter transgene in time lapse imaging
To examine the speed with which the GFP reporter gene was expressed, we used time lapse digital imaging studies of hamster cells infected in vitro with dG-VSV. Remarkably, within as little as one hour, GFP expression could be detected in inoculated cultures. Fig 3A shows photomicrographs of a single cell infected at time 0 at intervals after infection by dG-VSV. Fluorescence begins to increase within an hour after the initial infection (Fig. 3A); the graph in Fig. 3B shows the change in fluorescence over time. The focus of this experiment was on early expression of the reporter gene. The intensity of GFP fluorescence increased dramatically over the next few hours.
Fig. 3. Rapid increase in fluorescence after viral infection.
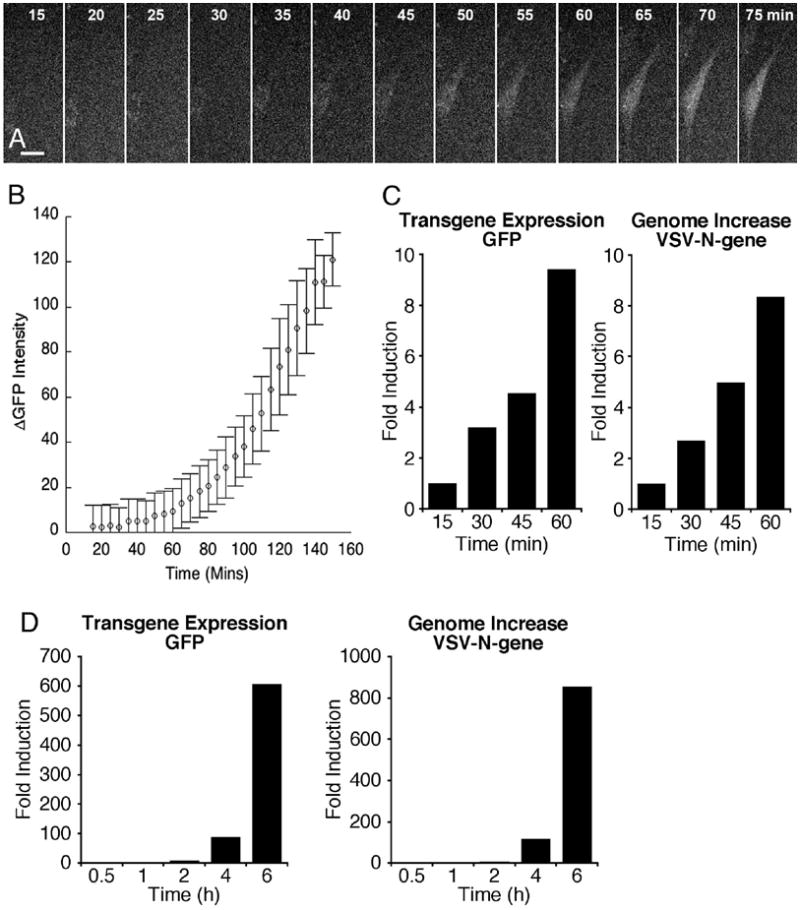
A. After infecting hamster cells in vitro with dG-VSV, digital micrographs were taken at 5 min intervals using a preset exposure. Within one hour after infection, fluorescence of this hamster cell (BHK) begins to show an increase. Bar, 7 um. B. This graph show the increase in fluorescence over baseline in 4 cells imaged simultaneously. Flags are SEM. C. RT-PCR was used to determine the increase in GFP mRNA and VSV-N gene within the first hour of infection. By 1 hr, both had increase by almost 10-fold, compared with the baseline determined by standard from 15 min post-inoculation. D. In a related experiment, the expression of GFP and VSV-N was examined over the time course of 6 hr. By 6 hr, both had increased by between 500 and a thousand fold.
Rapid increase in VSV genomes and mRNA
To examine the early time course of reporter gene mRNA increase after infection, cultured cells (500,000 cells/culture; 2 cultures per time point) were inoculated with an MOI of 1. Cells were harvested, and genomes and mRNA quantified with RT-PCR. At 15 min post-inoculation, the cultures were washed, and the cells harvested; this time period served as the control baseline, and was restricted to parent viral genomes that had adhered or internalized into cells. Already at early time intervals of one hr after adding the virus to the culture solution, a ten-fold increase in the negative sense viral N-gene and in the GFP transgene mRNA could be detected in infected cultures (Fig. 3C). The expression of these genes continued to rise over time, and remarkably by 6 hrs, had increased by more than 500-fold (Fig. 3D).
Release of defective dG-VSV- ultrastructural analysis with electron microscopy
Infection of dG-VSV with an MOI <1 resulted in a limited number of cells that expressed the viral reporter gene; this number did not increase with time, suggesting either that progeny virus was not released from infected cells, or that it did not bind to second order cells after release. To test the hypothesis that dG-VSV was released from infected cells, cultured brain cells were infected with a relatively low MOI of 0.5. After 3 hours, the culture medium was changed 5X in order to wash out any free virus. 48 hours after inoculation, cultures were fixed with 3% glutaraldehyde in a 0.1 M cacodylate buffer, treated with osmium tetroxide, and embedded in plastic. Silver ultrathin sections were cut parallel to the bottom of the culture dish, and examined in a Philips electron microscope. Based on 50 different fields in the electron microscope, a number of cells, or about 20% of the cells examined, were found with a typical morphology showing virus release (Fig. 4A-D); other cells showed no indication of viral infection. In addition, particularly near cells showing virus particles arising from the cellular membrane, free virus was also found between cells. Virus release was, however, attenuated compared to G-protein-containing VSV (van den Pol et al., 2002) reflecting the enhancing role of VSV-G-protein in viral assembly and budding. We found no evidence of virus attaching to the plasma membrane as found with replication competent VSV (van den Pol et al, 2002), and no suggestion of uptake of these daughter virions into endosomes, although presumptive clathrin coated pits were seen on the plasma membrane. (Fig. 4E). Together, these data suggest that the VSV glycoprotein is not necessary for viral budding from the plasma membrane, and that progeny dG-VSV are released from infected cells, but due to the lack of synthesized G protein, cannot bind to or infect additional cells.
Fig. 4. Neurons release defective dG-VSV particles- ultrastructure.
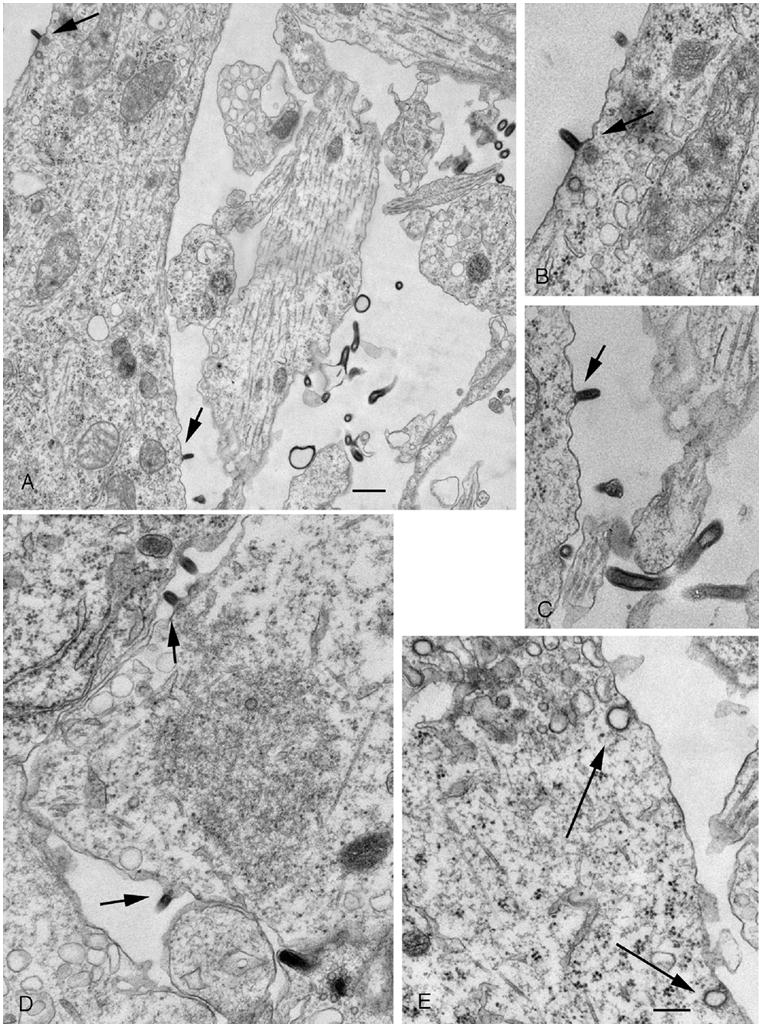
A. In this electron micrograph, several dG-VSV particles are seen budding off the dendritic or somatic membrane. To allow for a higher magnification image, cell body is cut off in the lower left part of the figure. B. Higher magnification of the particle budding from the proximal dendrite in the upper left part of A. C. Higher magnification of a virus budding from the membrane of the cell body, lower part of A. D. VSV buds off a glial cell. E. Although typical clathrin mediated endocytotic figures are seen, none contain newly generated virus particles. Bar, A, 350 nm; B-E, 225 nm.
Inoculation of dG VSV in vivo
dG-VSV-GFP or dsRed was injected into the brains of adult and neonatal mice (n=17). All mice survived. Upon histological analysis, no spread of dG-VSV away from the site of injection was found. The spread of virus was dictated solely by the volume and speed of injection. Unlike wild-type or replication competent recombinant VSVs that can be lethal within days after intracerebral injection, none of the animals given dG-VSV died from the virus, and none showed neurological dysfunction, including mice (n=5) that were maintained for 3 weeks after intracerebral inoculation.
As shown with the in vitro experiments, in vivo injection into the brain resulted in strong expression of the reporter gene in the cell body and dendrites of infected cells. The virus was stereotactically injected into different brain regions to allow an examination of how well cells and their processes were labeled.
When hippocampal granule cells were infected, the mossy fiber axon projections from the dentate gyrus to the CA3 pyramidal field were clearly labeled (Fig. 5A,B). Giant mossy terminals filled with GFP were found to abut on pyramidal dendrites; axons were filled with GFP, and could be followed from the cell body to the terminal fields. Granule cells were labeled from the cell body to distal dendrites and showed numerous GFP-labeled spines (Fig. 5C). Similarly, injections into the substantia nigra labeled nigral neurons, and their projections into the striatum.
Fig. 5. Hippocampal mossy fibers and granule cell spines.
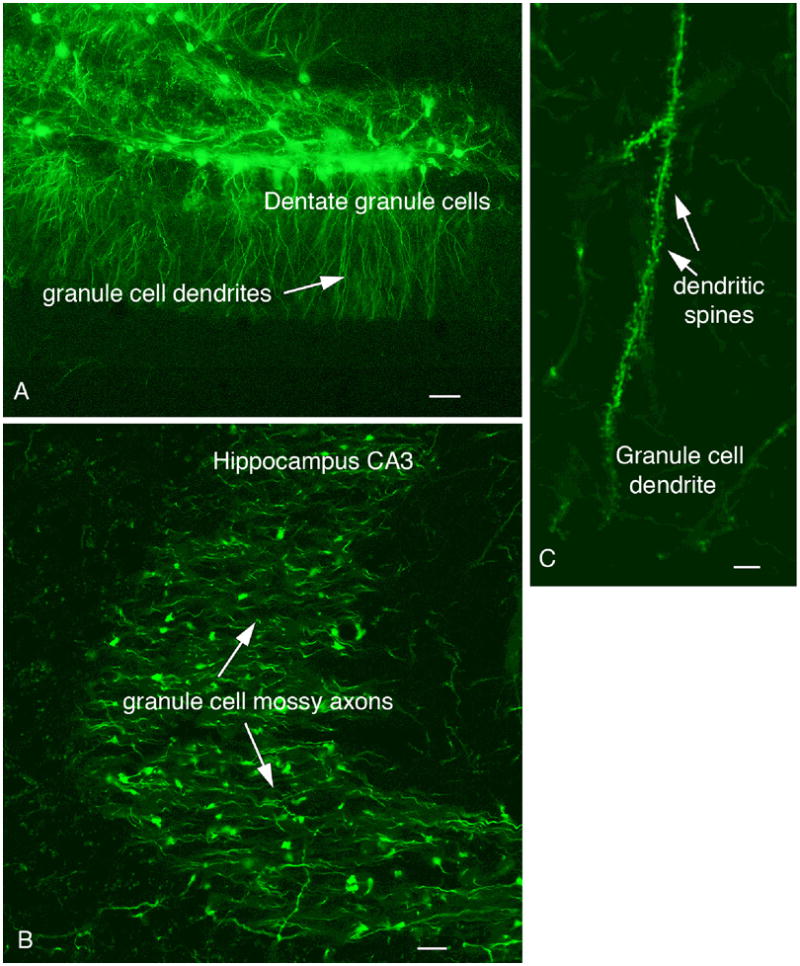
A. An injection of 100 nl of dG-VSV into the hippocampus dentate gyrus labels granule cells and dendrites. Scale bar, 35 um. B. Granule cell axons terminals, the mossy fibers, are labeled in CA3 after an injection of the dentate seen in A. Bar 6 um. C. Granule cell dendrites are covered with GFP-labeled spines. Bar, 5 um.
Injections of dG-VSV into the olfactory bulb labeled the cell body and presynaptic dendrites of different neuron types. In this example, GFP-labeled long dendrites reach from the internal granule cell layer into the external plexiform layer (Fig. 6A,B), where the dendritic boutons release neurotransmitter onto the mitral cell dendrites (Shepherd and Greer, 1998). dG-VSV was also used to inoculate the olfactory mucosa. This labeled olfactory receptor axons in the periphery, and their long axons that reached into the olfactory bulb, and terminated in the glomerular layer (Fig. 6C,D). To identify the glomerular layer, transgenic mice were used that express dsRed in subsets of olfactory axons under control of a beta-actin promoter with a cytomegalovirus enhancer, and individual GFP labeled olfactory axons were found. Since the dG-VSV does not infect second order cells, this suggests that the virus does not require an intermediate cell type, but can diffuse directly from the periphery to infect olfactory receptor neurons in the mucosa.
Fig. 6. Anterograde labeling of axon terminals and presynaptic dendrites.

A. A 500 nl injection into the core of the olfactory bulb labels a large number of granule cells and their presynaptic dendrites. Bar, 35 um. B. Higher magnification shows these presynaptic dendrites and their presynaptic spines. Bar, 5 um. C. After intranasal inoculation and 2 day survival, GFP labeled axons are found innervating glomeruli in the olfactory bulb. Bar, 30 um. D. The same field shows normal glomeruli from a transgenic mouse expressing dsRed in a subset of olfactory axons. Bar, 10 um. E. After injection of dG-VSV into the left cortex, no labeled cell bodies were found in the (F) contralateral cortex. Bar E and F, 20 um. G. After the injection in E, labeled axons were found in the corpus callosum. Bar, 4 um. H. Acute brain slices were cut from postnatal day 7 mice, and inoculated in vitro with dG-VSV. Several hours later, neurons expressing GFP are detected. Bar, 15 um.
When the virus was injected into the cortex on one side of the brain, neurons were strongly labeled near the injection (Fig. 6E). In contrast, no labeled cells were seen in regions of the brain that were connected to the cortex monosynaptically, for example, the thalamus or contralateral cortex (Fig. 6F), however, labeled axons were seen in the corpus callosum (Fig. 6G). In contrast, a positive control injection of pseudorabies virus did label contralateral cortex and ipsilateral thalamus (not shown). These data suggest that dG-VSV is not axonally transported either in a retrograde or anterograde direction. In contrast, the GFP label moves in an anterograde direction down the axon, and clearly labels presynaptic axonal boutons. Similarly, after virus injections into the corpus callosum that contains a large number of axons connecting left and right cortex, no labeled cells were found in the cortex indicating that the virus is not carried by axons of passage.
Acute brain slices are commonly used for neurophysiological analysis of neurons. To determine if dG-VSV could be used to generate rapid transgene expression in an acute slice, brains from postnatal day 7 mice were cut into 350 um thick slices, and dG-VSV was applied to the surface of the slice. GFP labeled neurons could be detected a few hours later, as shown in Figure 6H. This suggests that dG-VSV could be used to generate rapid gene expression of some experimental gene even if used in an acute slice.
Suprachiasmatic nucleus- efferent axonal projections
The neurons of the hypothalamic suprachiasmatic nucleus (SCN) act as a circadian pacemaker. Previous reports have suggested that SCN axons may reach to many regions of the nearby and distal brain (Sofroniew et al, 1981); an alternate suggestion is that the SCN axons project primarily to nearby periventricular regions of the hypothalamus (Watts et al, 1987). To address the question of where in the hypothalamus the SCN axons terminate, microinjections of dG-VSV were made into the SCN in vivo. Of nine mice injected that targeted the SCN, we focus here on brains in which little infection was found outside the SCN. Small diameter axons were found leaving the SCN, particularly in a dorsal direction. Axons and their boutons could be clearly seen. SCN axons could be found in a number of regions of the hypothalamus, but by far the greatest density of boutons was in the periventricular hypothalamus directly dorsal to the SCN. Here large numbers of SCN axons (Fig. 8C) and axonal boutons (Fig. 8B) were found, greater than any other region of the brain (Fig. 7A-D). Projections from the SCN into the periventricular hypothalamus have been previously reported with the lectin Phaseolus vulgaris (Watts et al, 1987).
Fig. 8. Multicolor neuroanatomy with green and red dG-VSV.

A. In a culture from embryonic mouse brain, coinfection with green and red expressing dG-VSVs result in strong expression of green, red, orange, and yellow neurons and glia. This micrograph was recorded by sequential use of GFP and dsRed filter sets on the same field. Bar, 12 um. B. Similarly, when dG-VSV expressing red or green reporter genes are injected into the mouse cortex, within 20 hrs, green, orange, and yellow cells can be seen. Bar, 9 um. C. The dG-VSVdsRed can be coupled with expression of GFP in transgenic mice. Here, mice that express GFP in the hypothalamic neurons that contain melanin concentrating hormone were injected with the dG-VSVdsRed, resulting in one colabeled yellow cell, in addition to green MCH cells and red dG-VSV cells. Bar, 15 um. D. This micrograph shows a VSV-G protein directly labeled with GFP, allowing clear visualization of small and thin membrane bound cellular filopodia. Bar, 2.5 um.
Fig. 7. Axonal projections of suprachiasmatic nucleus.

A. Injection of 50 nl of dG-VSV into the hypothalamic suprachiasmatic nucleus labels a few cells in the SCN. Bar, 50 um. B. In the area immediately dorsal to the SCN, axons and their boutons are labeled with GFP. Bar, 7 um. C. Camera lucida trace of GFP expressing axons within the SCN and in the area immediately dorsal to SCN. Bar, 40 um. D. Representative nissl-labeled section showing the SCN in parallel to C. E. Injections of dG-VSV into the ventromedial nucleus of the hypothalamus (VMH) showed that long dendrites (arrows) reached out of the core of the nucleus into the cell- poor shell. Bar, 12 um.
Ventromedial nucleus
The ventromedial nucleus of the hypothalamus plays a key role in energy homeostasis and modulates a number of behaviors. Lesions of the VMH result in severe obesity and increased appetite (Brobeck, 1946). Anatomical studies have found that many axons tend to remain outside the body of the nucleus and appear to encircle the VMH (Millhouse, 1973b; Broberger et al, 1998). To address the question of whether dendrites from VMH neurons extend outside the nucleus and potentially receive synaptic contact, dG-VSV was injected into the VMH, and 10–20 hours later, the inoculated mice were euthanized. Cell bodies in the VMH consistently extended long dendrites into the cell poor area around the VMH (Fig. 7E), with some of the dendrites leaving the VMH like spokes in a bicycle wheel, and others rolling back to circle the outside of the nucleus. These observations are consistent with detection of dendrites from VMH glutamatergic neurons extending outside the nucleus (Fu and van den Pol, 2008).
A red variant of dG-VSV with multiple color potential
The experiments above focus on the green reporter, GFP. We also generated a dG-VSV that contained the sequence for dsRed (dG-VSVdsRed), a red reporter gene. Similar to the green reporter, neurons infected with dG-VSVdsRed showed bright red fluorescence. When both the green and red dG-VSV were combined and used to inoculate mouse brain cells in culture, some cells showed red fluorescence and others green. Of interest, there were also a number of additional color variants caused by differing proportions of red and green reporter genes (Fig. 8A). These included various shades of red, green, orange, and yellow. The color of the cell body was continued into the processes of the same cell. Similar types of experiments were done by injecting both red and green expressing dG-VSV into the brain. Figure 8B shows green, red, and yellow cells in the cortex.
Viruses can also be used in transgenic animals already expressing reporter genes. For instance, in Figure 8C, dG-VSVdsRed was injected into the brain of a transgenic mouse that expressed GFP selectively in hypothalamic neurons that synthesize the neuropeptide melanin concentrating hormone (MCH). MCH neurons infected with the virus are yellow, whereas other cells in the same brain are green (MCH only) or red (dG-VSVdsRed only). Thus dG-VSV could be used to express genes in subsets of neurons for electrophysiological analysis.
Reporter genes can also be directed to different regions of the cell. In the examples above, reporter genes label the cytoplasmic compartment. Alternately, by generating a GFP fusion with a membrane protein, the reporter can be directed to the plasma membrane, facilitating visualization of the membrane compartment, as shown in Fig. 8D.
Use of infected cells for neurophysiological and imaging experiments
Other viruses have been used for tract tracing or cell identification, followed by electrphysiological testing. For instance, PRV has shown promise for showing the efferent projections of a particular cell, and allowing the labeled cell to be studied with neurophysiological recording (Smith et al, 2000), even though the virus will ultimately kill the recorded cell. To test whether dG-VSV could be used for rapid gene expression in physiological studies, we infected neurons with the virus, and then used fura-2 calcium digital imaging or whole cell patch clamp recording to study the physiological characteristics of infected cells, and to compare the membrane characteristics of the infected cells expressing reporter genes with cells showing no sign of infection. We also used time lapse digital imaging to show that infected cells continue to show mobility and extension of processes.
Calcium digital imaging with fura-2
Because the excitation spectrum for fura-2 was in the 340–380 nm range, and that for GFP in the 480 nm range, we could study calcium responses in cells expressing the GFP reporter (Fig. 9A-D). DG-VSV infected cells that showed GFP expression in cell bodies and processes responded to a variety of electrical and transmitter stimuli, tested 12 h post infection. Glutamate release from synaptic terminals causes an inward calcium flux. Electrical field stimulation (ES; 7 Hz, 7 msec duration, 4.1V/cm2) was used to activate synaptic release of neurotransmitters. Infected cells showed a calcium rise in response to this stimulation (Fig. 9A-H). The block of the ES-induced calcium rise by tetrodotoxin (0.5 uM) showed that the response was due to synaptic release dependent on action potentials, and not to a direct response to ES (Fig. 9E). That the cells remained healthy after the initial TTX challenge was shown by the recovery of the calcium response to electrical stimulation after TTX washout. In addition, infected cells showed a calcium rise in response to glutamate and to the glutamate (100 uM) agonist, NMDA (20 uM). As glia generally do not show a calcium response to NMDA, the NMDA-generated calcium rise is further confirmation that infected cells with a neuronal morphology also showed physiological responses typical of neurons.
Fig. 9. Calcium digital imaging with fura-2 of cells infected with dG-VSV.

A-D Same field showing cultured neurons; A shows infected cells showing GFP expression. B-D are pseudocolor images of fura-2 ratiometric images of 340/380 nm excitation calibrated to known Ca2+ levels. B,C, and D show baseline. B, electrical stimulation (ES) increases calcium levels (C), and recovery after electrical stimulation is stopped D. E,G. Calcium traces from dG VSV-GFP infected neurons showing GFP expression (E,G) and control cells showing no GFP (F,H) respond similarly to ES, ES in TTX (0.5 uM), NMDA, Glutamate, GABA, GABA in TTX, and High K+. I shows a representative Ca2+ trace of dG-VSV-dsRed infected neurons responding to ES, NMDA, Glutamate and High K+. J shows corresponding trace of dsRed-negative control neurons responding similarly to the same stimuli as (I).
In total, 69 GFP-positive cells (infected with dG-VSV) were imaged, with 62 (90%) showing responses to all four conditions of stimulation, including electrical stimulation (ES), ES after recovery from TTX, NMDA and glutamate. Cells not expressing the viral reporter gene in the same cultures were used as controls. In total 530 GFP-negative cells were imaged, with 498 (93%) showing responses to the same four stimuli. Both the proportion of cells responding and response characteristics on the Ca2+ traces appeared to be similar between infected and uninfected cells. Because the 380 nm – related excitation used for fura-2 had a slight overlap with the GFP excitation spectrum, the amplitudes of the Ca2+ responses could not be reliably compared. Fig. 9A-D show the GFP image of infected neurons and ratiometric images from which Ca2+ levels were derived. Fig. 9E-F shows the representative traces of Ca2+ level changes under the different stimuli.
Mature neurons are inhibited by GABA, but during early development, neurons show an excitatory response to GABA before 7 days in vitro, and GABA increases cytoplasmic Ca2+. Responses to GABA (30 uM), ES (7 Hz, 7 msec duration, 2.0 V/cm2) and high K+ (55 mM) were tested in neurons plated 5 days earlier. GFP-positive cells showed similar responses (n=70 of 250 cells) compared to control (87 of 200 cells; Fig. 9G-H). GABA is excitatory in these experiments due to the immaturity (5 days) of the cells. GABA exerted a depolarizing effect on developing neurons at this early stage of in vitro neuronal development in both GFP expressing infected cells (Fig. 9G) and non-GFP control cells (Fig. 9H).
Finally, dG-VSV-dsRed virus was used in similar fura-2 experiments. Imaging was performed 24 hours post inoculation and cells were stimulated with ES (7 Hz, 7 msec duration, 2.0 V/cm2), NMDA, glutamate and high K+. Again, infected cells appear to respond similarly to non-infected cells. One advantage of the dG-VSVdsRed was that the excitation wavelength was shifted to a much longer wavelength than either the GFP reporter or fura-2, allowing a more quantitative comparison of Ca2+ levels in cells showing the red viral reporter, and in non-red controls. To test the mean Ca2+ response to ES, we compared Ca2+ in virus infected cells showing a red color, and in control cells showing no red color 24 h post inoculation (Fig. 10I). The baseline calcium level with no stimulation was examined; mean Ca2+ in infected cells was 98.5 ± 11 (n=20) and 101.12 ± 4.4 for control cells (t-test, p>0.05). In neurons infected with dG-VSVdsRed, Ca2+ during ES was 131.9 ± 14.2 and in responding controls, the Ca2+ was 141.9± 5.9 (n=162) (p>0.05). These data suggest that the virus-infected cells showed similar baseline Ca2+, and similar calcium rises in response to electrical stimulation.
Fig. 10. Electrophysiological properties of dG-VSV infected neurons.

(A) No statistically significant difference (ns) was found between green neurons infected with dG-VSV and control neurons showing no viral reporter expression. Bars show mean resting membrane potential, frequency of postsynaptic currents, spike frequency, or input resistance. Number of cells tested is shown over the respective bar. (B) Examples of spontaneous spikes under resting conditions in dG-VSV virus infected neurons (RMP, -54.1 mV) compared to non-GFP neurons (RMP, −54.6 mV). C. Examples of isolated spontaneous spike shape from infected and control neurons. D. Post-synaptic currents of dG-VSV virus infected neurons compared to non-GFP neurons in voltage-clamp. E. Voltage responses of dG-VSV virus infected neurons compared to non-GFP neurons to a current step protocol of 200 ms 10-pA step pulses from -100 pA to 0 pA. F. Current-voltage relationship displayed as the mean normalized voltage responses of 13 infected neurons and 13 control neurons. Normalized to maximum response at a 100 pA current pulse.
Whole cell patch clamp recording from brain slices
We next tested neurons that had been infected by intracranial injection of dG-VSV. Twelve to 14 hours after inoculation, brain slices were cut at 300 um thickness, and used for whole cell patch clamp recording. A series of tests were run to compare the active and passive membrane properties of neurons showing green reporter gene expression, and non-green control neurons.
dG-VSV infected neurons were compared with nearby non-GFP neurons (n=79) from thick brain slices from mice (n=13). The general characteristics of virus infected neurons were relatively similar to those of nearby non-green neurons. Mean resting membrane potential was almost identical (−54 to −55 mV) in dG-VSV infected neurons (n=13) and non-green control cells (n=13); as shown in Figure 10A, no significant difference was found between the two cell types. Means and standard errors are shown in Figure 10. The frequency of postsynaptic currents was compared. Virus infected neurons compared to non-GFP neurons (5.1 ±2.2 Hz vs. 7.0 ±2.9 Hz, P >0.05, n=17) (Figure 10A, D), displayed no significant difference between the two groups. The spontaneous frequency of action potentials was also compared, with no significant difference found between the two cell types (Fig. 10A,B). Similarly, although the spike shape differed between individual neurons, similar rise, fall, and after-hyperpolarizations were found among each group (Fig. 10C). Both virus infected and non-green cells had similar rates of spontaneous spikes. GFP positive neurons had an average frequency of 3.3±1.1 Hz (n=5), not significantly different from nearby non-GFP neurons with a mean spike frequency of 3.2±0.8 Hz (n=5) (Figure 10A-C). A few neurons from both groups did not have spontaneous spikes; in these cells spikes could be evoked by injection of 20 pA current indicating they were neurons and not glia.
To assess current-voltage relations and input resistance, a series of constant current pulses ( from −100 to 0 pA for 200 ms, with a 10 pA increment, at 1 sec intervals) were injected into the recorded neurons (Fig. 10E). The voltage responses to current injection were similar in the two cell types. The mean normalized current voltage relationship is shown in Figure 10F, and no difference is seen between green infected cells and non-green control cells. dG-VSV virus infected neurons had an average input resistance of 466.0±85.2 MΩ (n=13), not significantly different from non-GFP neurons nearby 424.4±67.1 MΩ (n=13) (Figure 10A,E,F). One day after infection, it was difficult to obtain a high impedence seal of the plasma membrane of infected cells, suggesting that by this time the virus had caused cellular deterioration which would preclude useful recording.
Time lapse imaging of extending neurites
To determine if virus infection blocked neurite outgrowth, neurons cultured from embryonic mice were studied with time lapse digital recording. Neurons imaged over the succeeding hours continued to migrate, and neuronal processes and their growth cones continued to show considerable motility, extending and searching the cellular terrain (Fig. 11).
Fig. 11. Time lapse imaging of infected neurons.
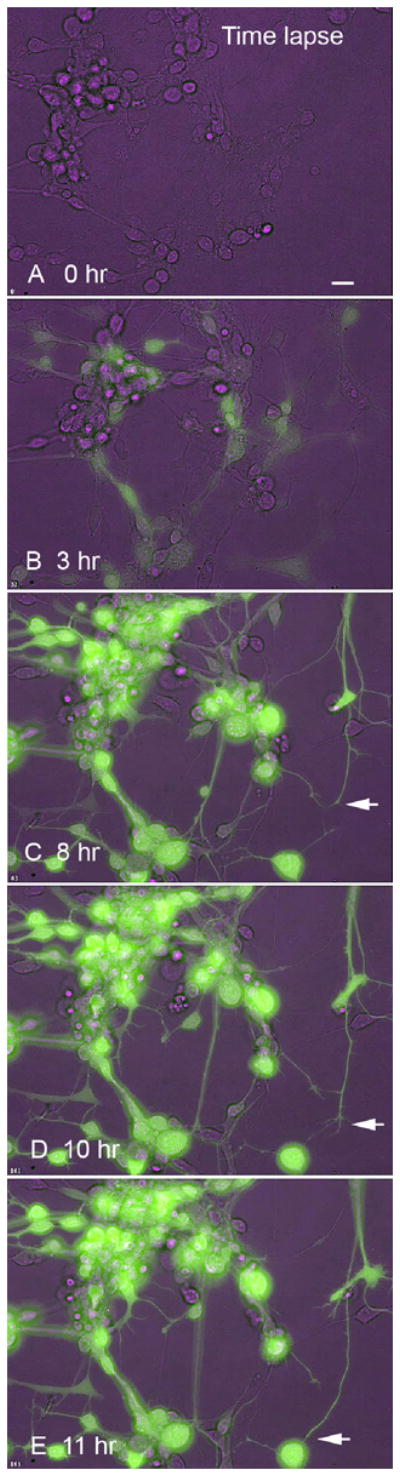
This is a series of time-lapse images after infection with dG-VSV at time 0. Infected cells are noted by their green fluorescence, and other cells are visualized by phase contrast in purple. A – E shows the same microscope field over 11 hrs. Note the increase in fluorescence over time, the mobility of the cells as they change position, the normal morphology, and the extension and exploration of the culture dish by the growing neurite expressing virally expressed GFP denoted by the white arrow. Bar, 15 um.
As with wild type VSV, cells infected with dG-VSV eventually died. Cells looked healthy for a day or two, but then the processes showed a beaded morphology suggestive of dying cells. Within 5 days of infection, most infected cells were dead. Other non-infected cells in the same culture dish survived for weeks and showed no green reporter gene expression, suggesting no virus dissemination after the initial infection, and no substantial release of viral toxins.
The studies above with calcium imaging, whole cell electrophysiology, and time lapse imaging all suggest that dG VSV can be used to label the projections and dendritic arbor of infected cells, to express a transgene, and to allow the study the physiology of these labeled cells. Similar to PRV and herpes viruses, dG-VSV ultimately kills the infected cell, but before the cell deteriorates, the behavior and physiology of the infected neuron appear normal.
DISCUSSION
In the Results of this paper, we explore dG-VSV as a novel tool to study neuronal circuits and morphology, and to generate fast expression of a transgene engineered into the virus. Genes coding for both red and green reporters are rapidly and robustly expressed, with detectable protein levels found as early as 60 minutes after inoculation.
RNA viruses initiate viral gene expression soon after entering a neuron, in part because the nuclear transcription apparatus is not required. The VSV gene expression and replication cycle is confined to the cytoplasmic compartment, shortening the process of gene expression. The genes are expressed sequentially in order from the first position to the fifth position. Genes in the earlier positions are expressed in greater abundance than genes in later positions. The position of the reporter gene in the two dG-VSVs used here was in the first position, giving maximum expression of the fluorescent reporter genes. Conversely, putting a gene of interest further downstream would allow some adjustment of the level of gene expression.
Comparison of dG-VSV with other recombinant VSVs
A number of recombinant VSVs have been used in our lab for brain infections. One of the primary reasons that dG-VSV generates a strong GFP signal is that the coding sequence for GFP is placed in the first position of the VSV genome. Since the intensity of gene expression for VSV is based on the gene order, genes in the first position generate the strongest signal. Other GFP-tagged VSVs, including M51-CT9 VSV, which has a GFP gene in the 5th position, also generate a good signal, but not as strong as the first position-GFP recombinant viruses. First position GFP VSVs that are fully replication competent by virtue of retaining the gene coding for the viral glycoprotein show strong fluorescence similar to that shown by dG-VSV, but due to the rapid diffusion to, and infection of, other nearby cells, these viruses are not as good for studying neuronal morphology. Other VSVs, particularly VSV-G/GFP, have been generated in which the GFP reporter is generated as part of the viral glycoprotein; because the glycoprotein is no longer functional with an attached GFP, a second VSV glycoprotein is included in the VSV sequence (Dalton and Rose, 2001; van den Pol et al, 2002). For visualization of fine membrane structure, for instance filopodia and lamellipodia of axonal and dendritic growth cones, the VSV-G/GFP is beneficial, as it directly labels the neuronal plasma membrane, rather than cytoplasm.
Although dG-VSV is ultimately toxic to the cells it infects, the virus is not lethal to mice, even when injected directly into the brain, as we show above. In contrast, replicating VSVs, including some that are attenuated from recombinant alterations in the virus genome, can still be fatal if injected directly into the brain; in contrast, peripheral inoculation with these recombinant VSVs causes a mild weight loss in rodents, followed by full recovery as the systemic and innate immune system rapidly respond to and eliminate VSV infection.
dG-VSV as a neuronal tracer
Use of dG-VSV for neuronal tracing has some advantages over some other tracers. First, the expression of the reporter gene is very rapid and very strong, and even works in acute brain slices. Even small processes can be clearly labeled, including axonal boutons and dendritic spines. We found nicely labeled axons and terminal boutons after infecting cell bodies in the olfactory mucosa, dentate gyrus, SCN, and a number of other areas. This indicates that sufficient GFP is made in the perikaryon to allow it to diffuse a long distance within the axon to reveal axonal terminal fields. A second advantage is that axons of passage are not labeled. The virus has little affinity for the axon, and injections of virus into axons of passage, for instance in the corpus callosum, does not result in infection or gene expression in the parent cortical cell bodies. We found no evidence of retrograde transport from the injected area to another area, and we also found no evidence of anterograde transport and release of the virus. Whereas dG-VSV works well to detect local or short axons, there is a potential limitation of the use of dG-VSV for anterograde labeling of long axons. Because the axon is labeled by diffusion of fluorescent protein synthesized in the cell body, if the cell were to die before sufficient label has diffused down the entire axon, the distal region of the axon might not be visualized.
Another asset of dG-VSV is that it can be used in a wide variety of species, as we demonstrated with cells from mouse, rat, human, hamster, and drosophila. Furthermore, infection is not restricted to a single cell type, but appears to work with all types of cells tested, including neurons and astrocytes, and also cells from other organs. It works well in both developing brain and mature brain.
A limitation of dG-VSV is that cells that are infected eventually die. The same is true of pseudorabies virus which can be transported and released trans-neuronally and has been used for tract tracing in the brain (Card et al, 1991). PRV has been used to study electrophysiological characteristics of infected cells (Smith et al, 2000; Glatzer et al, 2007: Irraten et al, 2001). PRV is ultimately lethal to cells it infects, and lethal to the animal host. However, between the time of infection and cell death, physiological characteristics of PRV-infected cells can be studied, and appear normal (Smith et al, 2000; Irraten et al, 2001). Unlike PRV, dG-VSV is not lethal to the host animal, and does not spread to second order cells. That cells infected with dG-VSV eventually die could potentially be used as a tool to selectively kill cells at the site of injection. Because the virus is not transported, it would eliminate cell bodies in a region of the brain or spinal cord without damaging axons of passage or nearby uninfected cells. Similar to VSV, the rabies virus G-protein can be deleted; a G-protein defective rabies virus has been found useful for studying cells after retrograde transport and reporter gene expression in the first order neuron (Wickersham et al, 2007a,b).
VSV generates fast and self-amplifying gene expression
Although the dG-VSVs used in the present experiments cannot infect a second round of cells after the first set is infected, the virus does replicate in the first set of infected cells, and as we showed with electron microscopy, infection-defective virions are released. As the replicating viral genomes serve as additional templates for mRNA copies and protein synthesis, even a single virus that infects a cell can lead to strong and robust gene expression.
Another viral vector that can be used for similar purposes is adeno-associated virus (AAV). However, AAV takes much longer to initiate protein synthesis ( a week to ten days to generate substantive expression), and transduction efficiency is low, sometimes requiring administration of a high titer (1012/ml) to achieve strong expression (Kaplitt et al., 1994; Xu et al, 2001). Positive strand RNA viruses such as sindbis and semliki forest virus have been used for rapid transgene expression, but the earliest expression was reported to be in the 4–8 hour range (Ehrengruber et al, 2001; 2002). Adenovirus vectors express GFP in glia in about 8 hours, and in neurons in 16 hours (Sato et al, 2004). The VSV-related rhabdoviral rabies vector shows GFP expression two days after inoculation (Wickersham, 2007b). In contrast, dG-VSV can generate detectable expression in a little over one hour, and virion concentrations down to one virus particle are sufficient to generate large signals. In fact, single particle infections may ultimately result in a greater level of gene expression than multiple copies of the virus, as the cell remains healthy longer, providing a longer time for virus amplification of the gene product. The rapid expression of viral genes is remarkable in that a number of steps must occur first, including virus binding to the host cell, internalization into an endosome, escape from the acidified endosome, transcription into mRNA, translation into protein, and export of the protein through organelles involved in protein synthesis. Expression of the reporter gene is sufficiently fast that the virus can be used to express transgenes in acute brain slices after infection in vitro.
Multiple color potential
In a recent paper, transgenic mice that expressed 3 different color fluorescent proteins in random cells of the brain were proposed for use in studying neural circuits as the combinations of coexpression generated a number of different “Brainbow” colors (Livet et al, 2007). The same strategy could also be used with multiple color dG-VSV infections. For example, simultaneous infection of the brain with dG-VSV expressing either GFP or dsRed gives infected cells a number of color variations ranging from pure green, through various shades of orange or yellow, to pure red. Addition of another color would further extend the potential for multicolor imaging of individual brains within hours after injection. Furthermore, where the site of expression in the Brainbow mouse may be random or predetermined by the transgene insertion site or promoter used, any cell or area of the brain can be selectively labeled with dG-VSV.
The VSV G-gene is often used to pseudotype other viruses to increase their binding and infectivity. But alternately, the G protein could be replaced by a cell-specific protein, for example one that binds specifically to a single cell type, as suggested by experiments replacing the VSV-G gene with one coding for the HIV envelope glycoprotein (Boritz et al, 1999), or providing by protein complementation, the viral glycoprotein that underlies binding of Hanta or Seoul virus to their targets (Ogino et al, 2003). Another possibility is the substitution for the VSV-G gene with a gene coding for a chimeric Sindbis virus glycoprotein coupled with a single chain antibody directed to a specific cell receptor, as used in replicating VSV to target mammary tumors (Gao et al, 2006).
Cells expressing GFP or dsRed had a Golgi-like appearance; dendrites, dendritic spines, and axonal boutons were labeled. As the reporter gene product spreads in the living cell, using dG-VSV to reveal the complete structure of neurons may reduce some of the concern that has been raised about the completeness of Golgi silver chromate labeling that may not fully label the entire dendritic arbor. The hypothalamic ventromedial nucleus has been described as acting as a “satiety” center (Brobeck, 1946; Elmquist et al, 1999); however, many axons, including those involved in energy homeostasis tend to circle around VMH rather than enter it (Broberger et al, 1998; Milhouse, 1973a,b). We show here with dG-VSV that dendrites of the hypothalamic ventromedial nucleus (VMH) commonly leave the borders of the nucleus and radiate outward into the cell-poor region surrounding the nucleus, consistent with possible axodendritic innervation of VMH neurons by axons in the cell poor shell. This provides additional support for the view that arcuate nucleus axons, including those containing agouti related protein that may play an orexigenic role in energy homeostasis, could influence VMH neurons by innervation of the neurons outside the body of the nucleus.
Axonal projections
The dG-VSV GFP reporter strongly labels axons and their terminal boutons after infection of the parent cell bodies, as shown in dentate granule cell terminals in CA3, olfactory receptor axons in the olfactory bulb, and SCN axon terminals in the periventricular hypothalamus. The highest density of axon boutons from the SCN was found in the periventricular area immediately dorsal to the SCN, providing additional support to an earlier suggestion that SCN axons preferentially target the periventricular hypothalamus (Watts et al, 1987). The strong and rapid axonal labeling suggests that the dG-VSV would serve to study anterograde axonal projections, similar to the anterogradely transported lectin, Phaseolis vulgaris. Unlike Phaseolus, dG-VSV requires no histochemical treatment to detect anterogradely labeled axons for light microscopy. Antisera against either the virus, or against reporter genes would enable ultrastructural analysis of brain circuits revealed by dG-VSV.
DG-VSV preserves normal physiology of neurons
We found no significant differences in electrophysiological properties or in calcium responses between GFP-expressing dG-VSV infected neurons and nearby non-GFP control neurons during the periods studied; infected neurons were identified by GFP expression indicating that sufficient time was allowed for transgene expression. At long time intervals, neurons deteriorated. Time lapse imaging of GFP-expressing developing neurons showed that dG-VSV infected cells continued to be mobile, and to extend processes and maintain active growth cones during the initial 24 hours after inoculation. These data are consistent with an earlier report in which a dG-VSV variant was used as an oncolytic agent for glioblastoma cells in vitro; dG-VSV was found to be substantially less toxic to neurons than a wild type VSV (Duntsch et al, 2004). Finally, as spread of dG-VSV to second order cells is blocked, the virus would be relatively safe for the user.
Potential for use of recombinant viruses in a clinical setting
The use and testing of viruses in experimental neuroscience also provides a growing basis for consideration of the use of viruses in the clinic. Although outside the primary focus of this paper, viruses have considerable potential in the treatment of various neurological disorders, particularly as vectors in gene transfer. Viruses that have been considered for gene transfer into the brain include herpes, adeno-, lenti-, retro- and AAV (Glorioso and Fink, 2004; Kaplitt et al, 2007; Martuza, 1992). Given the low likehood of collateral complications, AAV appears to be one of the front-runners in this area.
Another potential use for viruses is in the destruction of brain cancer cells (Chiocca et al., 2003). Some viruses have an affinity for cancer cells, with a number of different mechanisms underlying the selectivity. Viruses have been used in attempts to destroy either peripheral or brain cancer. Some of these viruses have been replication incompetent, but these have not shown great success in directly killing a substantial percent of tumor cells. Replication incompetent viral based vectors have been used to deliver toxic, therapeutic, or disease modulating genes to brain tumor cells (Castro et al., 2003). Replication competent viruses have enjoyed more success at destroying brain tumor cells, although the risk of complications is increased over replication incompetent viruses (Pulkkanen and Yla-Herttuala, 2005; Rainov and Ren, 2003). Potentially oncolytic viruses include herpes simplex (Markert et al., 2009), adenovirus (Alemany et al., 1999), retrovirus (Rainov et al., 2000), measles (Allen et al., 2008), polio (Dobrikova et al., 2008), reovirus (Wilcox et al. 2001), Newcastle disease virus (Csatary et al. 2004), myxoma virus (Barrett et al., 2007), and VSV (Stojdl et al., 2003; Duntsch et al., 2004; Ozduman et al, 2008).
Although viruses have considerable potential in the treatment of brain disease, they are not without hazard. Ad has been used clinically in an attempt to correct a genetic deficiency involving ornithine transcarbamylase; unfortunately a patient died from viral complications, raising concerns about the safety of viral gene therapy (Carmen, 2001). Given the ability to integrate into the host cell genome, retroviruses were thought to have considerable potential for long term expression in clinical use. However, a problem with an integrating virus is that if it integrates into a genomic site that codes or regulates expression of a cancer suppressor gene such as P53, this can increase the oncogenic potential. Retroviruses were used in an attempt to correct a genetic mutation resulting in immune deficiency. Although the primary goal was at least partially achieved in ameliorating the depresssed immunity, an alarming increase in premalignant T-cell proliferation resulting in leukemia was found in a high proportion of treated patients, (Hacein-Bey-Abina et al, 2003; 2008).
The complications notwithstanding, viruses, or viral vectors, are the current method of choice for gene transfer into the brain. Viral-mediated gene transfer holds promise for treatment of many brain diseases, including Parkinson’s disease, epilepsy, Alzheimer’s disease, spinal cord trauma, and stroke.
Overview
A number of viruses have been used to address various questions about neural circuitry or physiology. Each virus has a unique set of characteristics that gives to it a niche for use in neuroscience. No single virus is ideal for all experiments, and each has its own attributes and limitations.
Supplementary Material
Acknowledgments
Grant support provided by NIH NIAID A1/NS48854 and NCI CA124737
We thank Y. Yang, J.N. Davis, and V. Rogulin for excellent technical facilitation.
References
- Ackman JB, Siddiqi F, Walikonis RS, LoTurco JJ. Fusion of microglia with pyramidal neurons after retroviral infection. J Neurosci. 2006;26:11413–11422. doi: 10.1523/JNEUROSCI.3340-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alemany R, Gomez-Manzano C, Balagué C, Yung WK, Curiel DT, Kyritsis AP, Fueyo J. Gene therapy for gliomas: molecular targets, adenoviral vectors, and oncolytic adenoviruses. Exp Cell Res. 1999;252:1–12. doi: 10.1006/excr.1999.4623. [DOI] [PubMed] [Google Scholar]
- Allen C, Paraskevakou G, Liu C, Iankov ID, Msaouel P, Zollman P, Myers R, Peng KW, Russell SJ, Galanis E. Oncolytic measles virus strains in the treatment of gliomas. Expert Opin Biol Ther. 2008;8:213–220. doi: 10.1517/14712598.8.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Card JP. Use of pseudorabies virus to delineate multisynaptic circuits in brain: Opportunities and limitations. J Neurosci Methods. 2000;103:51–61. doi: 10.1016/s0165-0270(00)00295-8. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Chen S, Zhu Y, Oshinsky ML. A neural circuit for circadian regulation of arousal. Nat Neurosci. 2001;4:732–738. doi: 10.1038/89522. [DOI] [PubMed] [Google Scholar]
- Baker KL, Daniels SB, Lennington JB, Lardaro T, Czap A, Notti RQ, Cooper O, Isacson O, Frasca S, Conover JC. Neuroblast protuberances in the subventricular zone of the regenerative MRL/mpj mouse. J Comp Neurol. 2006;498:747–761. doi: 10.1002/cne.21090. [DOI] [PubMed] [Google Scholar]
- Banfield BW, Kaufman JD, Randall JA, Pickard GE. Development of pseudorabies virus strains expressing red fluorescent proteins: New tools for multisynaptic labeling applications. J Virol. 2003;77:10106–10112. doi: 10.1128/JVI.77.18.10106-10112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JW, Alston LR, Wang F, Stanford MM, Gilbert PA, Gao X, Jimenez J, Villeneuve D, Forsyth P, McFadden G. Identification of host range mutants of myxoma virus with altered oncolytic potential in human glioma cells. J Neurovirol. 2007;13:549–560. doi: 10.1080/13550280701591526. [DOI] [PubMed] [Google Scholar]
- Berns KI, Parrish CR. Parvoviridae. In: Griffin DM, Lab RA, Martin MA, Roizman B, Straus SE, editors. Fields Virology. 5. Vol. 2. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2437–2477. [Google Scholar]
- Boldogköi Z, Sík A, Dénes A, Reichart A, Toldi J, Gerendai I, Kovács KJ, Palkovits M. Novel tracing paradigms--genetically engineered herpesviruses as tools for mapping functional circuits within the CNS: present status and future prospects. Prog Neurobiol. 2004;72:417–445. doi: 10.1016/j.pneurobio.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Boritz E, Gerlach J, Johnson JE, Rose JK. Replication-competent rhabdoviruses with human immunodeficiency virus type 1 coats and green fluorescent protein: entry by a pH-independent pathway. J Virol. 1999;73:6937–6945. doi: 10.1128/jvi.73.8.6937-6945.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Brobeck JR. Mechanism of the development of obesity in animals with hypothalamic lesions. Physiol Rev. 1946;26:541–559. doi: 10.1152/physrev.1946.26.4.541. [DOI] [PubMed] [Google Scholar]
- Broberger C, Johansen J, Johansson C, Schalling M, Hokfelt T. The neuropeptide Y/agouti gene-related protein (AgRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci U S A. 1998;95:15043–15048. doi: 10.1073/pnas.95.25.15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody SL, Crystal RG. Adenovirus-mediated in vivo gene transfer. Ann N Y Acad Sci. 1994;716:90–101. doi: 10.1111/j.1749-6632.1994.tb21705.x. [DOI] [PubMed] [Google Scholar]
- Burrows RC, Wancio D, Levitt P, Lillien L. Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron. 1997;19:251–267. doi: 10.1016/s0896-6273(00)80937-x. [DOI] [PubMed] [Google Scholar]
- Campbell RE. Defining the gonadotrophin-releasing hormone neuronal network: Transgenic approaches to understanding neurocircuitry. Journal of Neuroendocrinology. 2007;19:561–573. doi: 10.1111/j.1365-2826.2007.01561.x. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Herbison AE. Definition of brainstem afferents to gonadotropin-releasing hormone neurons in the mouse using conditional viral tract tracing. Endocrinology. 2007;148:5884–5890. doi: 10.1210/en.2007-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Zhang GR, Wang X, Kong L, Geller AI. Enhanced nigrostriatal neuron-specific, long-term expression by using neural-specific promoters in combination with targeted gene transfer by modified helper virus-free HSV-1 vector particles. BMC Neurosci. 2008;9:37. doi: 10.1186/1471-2202-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card JP, Whealy ME, Robbins AK, Moore RY, Enquist LW. Two alpha-herpesvirus strains are transported differentially in the rodent visual system. Neuron. 1991;6:957–969. doi: 10.1016/0896-6273(91)90236-s. [DOI] [PubMed] [Google Scholar]
- Card JP, Sved JC, Craig B, Raizada M, Vazquez J, Sved AF. Efferent projections of rat rostroventrolateral medulla C1 catecholamine neurons: Implications for the central control of cardiovascular regulation. J Comp Neurol. 2006;499:840–859. doi: 10.1002/cne.21140. [DOI] [PubMed] [Google Scholar]
- Carmen IH. A death in the laboratory: the politics of the Gelsinger aftermath. Mol Ther. 2001;3:425–428. doi: 10.1006/mthe.2001.0305. [DOI] [PubMed] [Google Scholar]
- Castro MG, Cowen R, Williamson IK, David A, Jimenez-Dalmaroni MJ, Yuan X, Bigliari A, Williams JC, Hu J, Lowenstein PR. Current and future strategies for the treatment of malignant brain tumors. Pharmacol Ther. 2003;98:71–108. doi: 10.1016/s0163-7258(03)00014-7. [DOI] [PubMed] [Google Scholar]
- Cheli V, Adrover M, Blanco C, Ferrari C, Cornea A, Pitossi F, Epstein AL, Jerusalinsky D. Knocking-down the NMDAR1 subunit in a limited amount of neurons in the rat hippocampus impairs learning. J Neurochem. 2006;97(Suppl 1):68–73. doi: 10.1111/j.1471-4159.2005.03592.x. [DOI] [PubMed] [Google Scholar]
- Chen J, Wu J, Apostolova I, Skup M, Irintchev A, Kügler S, Schachner M. Adeno-associated virus-mediated L1 expression promotes functional recovery after spinal cord injury. Brain. 2007;130:954–969. doi: 10.1093/brain/awm049. [DOI] [PubMed] [Google Scholar]
- Chiocca EA, Aghi M, Fulci G. Viral therapy for glioblastoma. Cancer J. 2003;9:167–179. doi: 10.1097/00130404-200305000-00005. [DOI] [PubMed] [Google Scholar]
- Clarke DK, Nasar F, Lee M, Johnson JE, Wright K, Calderon P, Guo M, Natuk R, Cooper D, Hendry RM, Udem SA. Synergistic attenuation of vesicular stomatitis virus by combination of specific G gene truncations and N gene translocations. J Virol. 2007;81:2056–2064. doi: 10.1128/JVI.01911-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coker AL, Russell RB, Bond SM, Pirisi L, Liu Y, Mane M, Kokorina N, Gerasimova T, Hermonat PL. Adeno-associated virus is associated with a lower risk of high-grade cervical neoplasia. Exp Mol Pathol. 2001;70:83–89. doi: 10.1006/exmp.2000.2347. [DOI] [PubMed] [Google Scholar]
- Csatary LK, Gosztonyi G, Szeberenyi J, Fabian Z, Liszka V, Bodey B, Csatary CM. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J Neurooncol. 2004;67:83–93. doi: 10.1023/b:neon.0000021735.85511.05. [DOI] [PubMed] [Google Scholar]
- Dalton KP, Rose JK. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology. 2001;279:414–421. doi: 10.1006/viro.2000.0736. [DOI] [PubMed] [Google Scholar]
- DeFalco J, Tomishima M, Liu H, Zhao C, Cai X, Marth JD, Enquist L, Friedman JM. Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science. 2001;291:2608–2613. doi: 10.1126/science.1056602. [DOI] [PubMed] [Google Scholar]
- Desmaris N, Bosch A, Salaün C, Petit C, Prévost MC, Tordo N, Perrin P, Schwartz O, de Rocquigny H, Heard JM. Production and neurotropism of lentivirus vectors pseudotyped with lyssavirus envelope glycoproteins. Mol Ther. 2001;4:149–156. doi: 10.1006/mthe.2001.0431. [DOI] [PubMed] [Google Scholar]
- Dobrikova EY, Broadt T, Poiley-Nelson J, Yang X, Soman G, Giardina S, Harris R, Gromeier M. Recombinant oncolytic poliovirus eliminates glioma in vivo without genetic adaptation to a pathogenic phenotype. Mol Ther. 2008;16:1865–1872. doi: 10.1038/mt.2008.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Duntsch CD, Zhou Q, Jayakar HR, Weimar JD, Robertson JH, Pfeffer LM, Wang L, Xiang Z, Whitt MA. Recombinant vesicular stomatitis virus vectors as oncolytic agents in the treatment of high-grade gliomas in an organotypic brain tissue slice-glioma coculture model. J Neurosurgery. 2004;100:1049–1059. doi: 10.3171/jns.2004.100.6.1049. [DOI] [PubMed] [Google Scholar]
- Ehrengruber MU, Hennou S, Büeler H, Naim HY, Déglon N, Lundstrom K. Gene transfer into neurons from hippocampal slices: comparison of recombinant Semliki Forest Virus, adenovirus, adeno-associated virus, lentivirus, and measles virus. Mol Cell Neurosci. 2001;17:855–871. doi: 10.1006/mcne.2001.0982. [DOI] [PubMed] [Google Scholar]
- Ehrengruber MU, Lundstrom K, Schweitzer C, Heuss C, Schlesinger S, Gähwiler BH. Recombinant Semliki Forest virus and Sindbis virus efficiently infect neurons in hippocampal slice cultures. Proc Natl Acad Sci U S A. 2002;96:7041–7046. doi: 10.1073/pnas.96.12.7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Enquist LW, Card JP. Recent advances in the use of neurotropic viruses for circuit analysis. Curr Opin Neurobiol. 2003;13:603–606. doi: 10.1016/j.conb.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Faulkner RL, Jang MH, Liu XB, Duan X, Sailor KA, Kim JY, Ge S, Jones EG, Ming GL, Song H, Cheng HJ. Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain. Proc Natl Acad Sci. 2008;105:14157–14162. doi: 10.1073/pnas.0806658105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraefel C, Breakefield XO, Jacoby DR. HSV-1 Amplicon. In: Chiocca EA, Breakefield XO, editors. Gene Therapy for Neurological Disorders and Brain Tumors. Totowa, NJ: Humana Press; 1998. pp. 63–82. [Google Scholar]
- Fu LY, van den Pol AN. Agouti-related peptide and MC3/4 receptor agonists both inhibit excitatory hypothalamic ventromedial nucleus neurons. J Neurosci. 2008;28:5433–5449. doi: 10.1523/JNEUROSCI.0749-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Whitaker-Dowling P, Watkins SC, Griffin JA, Bergman I. Rapid adaptation of a recombinant vesicular stomatitis virus to a targeted cell line. J Virol. 2006;80:8603–8612. doi: 10.1128/JVI.00142-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieger JC, Samulski RJ. Adeno-associated virus as a gene therapy vector: vector development, production and clinical applications. Adv Biochem Eng Biotechnol. 2005;99:119–145. [PubMed] [Google Scholar]
- Grinevich V, Brecht M, Osten P. Monosynaptic pathway from rat vibrissa motor cortex to facial motor neurons revealed by lentivirus-based axonal tracing. J Neurosci. 2005;25:8250–8258. doi: 10.1523/JNEUROSCI.2235-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatzer NR, Derbenev AV, Banfield BW, Smith BN. Endomorphin-1 modulates intrinsic inhibition in the dorsal vagal complex. J Neurophysiol. 2007;98:1591–1599. doi: 10.1152/jn.00336.2007. [DOI] [PubMed] [Google Scholar]
- Glorioso JC, Fink DJ. Herpes vector-mediated gene transfer in treatment of diseases of the nervous system. Annu Rev Microbiol. 2004;58:253–271. doi: 10.1146/annurev.micro.58.030603.123709. [DOI] [PubMed] [Google Scholar]
- Guigoni C, Coulon P. Rabies virus is not cytolytic for rat spinal motoneurons in vitro. J Neurovirol. 2002;8:306–317. doi: 10.1080/13550280290100761. [DOI] [PubMed] [Google Scholar]
- Haberman RP, Samulski RJ, McCown TJ. Attenuation of seizures and neuronal death by adeno-associated virus vector galanin expression and secretion. Nat Med. 2003;9:1076–1080. doi: 10.1038/nm901. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Hioki H, Kameda H, Nakamura H, Okunomiya T, Ohira K, Nakamura K, Kuroda M, Furuta T, Kaneko T. Efficient gene transduction of neurons by lentivirus with enhanced neuron-specific promoters. Gene Ther. 2007;14:872–882. doi: 10.1038/sj.gt.3302924. [DOI] [PubMed] [Google Scholar]
- Ho WSC, van den Pol AN. Bystander attenuation of neuronal and astrocyte intercellular communication by murine cytomegalovirus infection of glia. J Virol. 2007;81:7286–7292. doi: 10.1128/JVI.02501-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howorth PW, Teschemacher AG, Pickering AE. Retrograde adenoviral vector targeting of nociresponsive pontospinal noradrenergic neurons in the rat in vivo. J Comp Neurol. 2009;512:141–157. doi: 10.1002/cne.21879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob JM, Zhou Q, Liu Y. Novel method for the labeling of distant neuromuscular junctions. J Neurosci Res. 2000;61:61–66. doi: 10.1002/1097-4547(20000701)61:1<61::AID-JNR7>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Jakobsson J, Lundberg C. Lentiviral vectors for use in the central nervous system. Mol Ther. 2006;13:484–493. doi: 10.1016/j.ymthe.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Burkey AR, Card JP, Basbaum AI. Transneuronal labeling of a nociceptive pathway, the spino-(trigemino-)parabrachio-amygdaloid, in the rat. The J Neurosci. 1997;17:3751–3765. doi: 10.1523/JNEUROSCI.17-10-03751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeetendra E, Robison CS, Albritton LM, Whitt MA. The membrane-proximal domain of vesicular stomatitis virus G protein functions as a membrane fusion potentiator and can induce hemifusion. J Virol. 2002;76:12300–12311. doi: 10.1128/JVI.76.23.12300-12311.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafri T, van Praag H, Gage FH, Verma IM. Lentiviral vectors: regulated gene expression. Mol Ther. 2000;1:516–21. doi: 10.1006/mthe.2000.0083. [DOI] [PubMed] [Google Scholar]
- Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, Bland RJ, Young D, Strybing K, Eidelberg D, During MJ. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet. 2007;369:2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- Kaplitt MG, Leone P, Samulski RJ, Xiao X, Pfaff DW, O’Malley KL, During MJ. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet. 1994;8:148–154. doi: 10.1038/ng1094-148. [DOI] [PubMed] [Google Scholar]
- Kelly RM, Strick PL. Rabies as a transneuronal tracer of circuits in the central nervous system. J Neurosci Methods. 2000;103:63–71. doi: 10.1016/s0165-0270(00)00296-x. [DOI] [PubMed] [Google Scholar]
- Kinoshita N, Mizuno T, Yoshihara Y. Adenovirus-mediated WGA gene delivery for transsynaptic labeling of mouse olfactory pathways. Chem Senses. 2002;27:215–223. doi: 10.1093/chemse/27.3.215. [DOI] [PubMed] [Google Scholar]
- Klupp BG, Hengartner CJ, Mettenleiter TC, Enquist LW. Complete, annotated sequence of the pseudorabies virus genome. J Virol. 2004;78:424–440. doi: 10.1128/JVI.78.1.424-440.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp AC, Enquist LW. Pseudorabies virus recombinants expressing functional virulence determinants ge and gi from bovine herpesvirus 1.1. J Virol. 1997;71:2731–2739. doi: 10.1128/jvi.71.4.2731-2739.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar E, Peluso R, Schnell MJ, Whitt MA, Rose JK. Normal replicaiton of vesicular stomatis virus without C proteins. Virology. 1996;216:309–316. doi: 10.1006/viro.1996.0066. [DOI] [PubMed] [Google Scholar]
- Kuo H, Ingram DK, Crystal RG, Mastrangeli A. Retrograde transfer of replication deficient recombinant adenovirus vector in the central nervous system for tracing studies. Brain Res. 1995;705:31–38. doi: 10.1016/0006-8993(95)01065-3. [DOI] [PubMed] [Google Scholar]
- Lane MA, White TE, Coutts MA, Jones AL, Sandhu MS, Bloom DC, Bolser DC, Yates BJ, Fuller DD, Reier PJ. Cervical prephrenic interneurons in the normal and lesioned spinal cord of the adult rat. J Comp Neurol. 2008;511:692–709. doi: 10.1002/cne.21864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson ND, Stillman EA, Whitt MA, Rose JK. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci USA. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Erskine MS. Pseudorabies virus tracing of neural pathways between the uterine cervix and CNS: Effects of survival time, estrogen treatment, rhizotomy, and pelvic nerve transection. J Comp Neurol. 2000;418:484–503. [PubMed] [Google Scholar]
- Lentz TL, Burrage TG, Smith AL, Crick J, Tignor GH. Is the acetylcholine receptor a rabies virus receptor? Science. 1982;215:182–184. doi: 10.1126/science.7053569. [DOI] [PubMed] [Google Scholar]
- Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, Sanes JR, Lichtman JW. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- Liu M, Thankachan S, Kaur S, Begum S, Blanco-Centurion C, Sakurai T, Yanagisawa M, Neve R, Shiromani PJ. Orexin (hypocretin) gene transfer diminishes narcoleptic sleep behavior in mice. Eur J Neurosci. 2008a;28:1382–1393. doi: 10.1111/j.1460-9568.2008.06446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Tang J, Wang X, Yang T, Geller AI. Enhanced long-term expression from helper virus-free HSV-1 vectors packaged in the presence of deletions in genes that modulate the function of VP16, U L 46 and U L 47. J Neurosci Methods. 2005;145:1–9. doi: 10.1016/j.jneumeth.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Liu B, Wang S, Brenner M, Paton JF, Kasparov S. Enhancement of cell-specific transgene expression from a Tet-Off regulatory system using a transcriptional amplification strategy in the rat brain. J Gene Med. 2008b;10:583–592. doi: 10.1002/jgm.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WD, Qu G, Sferra TJ, Clark R, Chen R, Johnson PR. Adeno-associated virus-mediated gene transfer to the brain: duration and modulation of expression. Hum Gene Ther. 1999;10:201–213. doi: 10.1089/10430349950018995. [DOI] [PubMed] [Google Scholar]
- Loewy AD. Viruses as transneuronal tracers for defining neural circuits. Neurosci Biobehav Rev. 1998;22:679–684. doi: 10.1016/s0149-7634(98)00006-2. [DOI] [PubMed] [Google Scholar]
- Lowenstein PR, Suwelack D, Hu J, Yuan X, Jimenez-Dalmaroni M, Goverdhana S, Castro MG. Nonneurotropic adenovirus: a vector for gene transfer to the brain and gene therapy of neurological disorders. Int Rev Neurobiol. 2003;55:3–64. doi: 10.1016/s0074-7742(03)01001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun X, Senger DL, Alain T, Oprea A, Parato K, Stojdl D, Lichty B, Power A, Johnston RN, Hamilton M, Parney I, Bell JC, Forsyth PA. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas. J Natl Cancer Inst. 2006;98:1546–1557. doi: 10.1093/jnci/djj413. [DOI] [PubMed] [Google Scholar]
- Lundh B. Spread of vesicular stomatitis virus along the visual pathways after retinal infection in the mouse. Acta Neuropathol. 1990;79:395–401. doi: 10.1007/BF00308715. [DOI] [PubMed] [Google Scholar]
- Lyles DS, Rupprecht CE. Rhabdoviridae. In: Fields BN, Knipe DM, Howley PM, Griffin DE, editors. Fields Virology. Lippincot, Williams, Wilkins Press; 2006. pp. 1363–1408. [Google Scholar]
- Mao H, Rosenthal KS. Strain-dependent structural variants of herpes simplex virus type 1 ICP34.5 determine viral plaque size, efficiency of glycoprotein processing, and viral release and neuroinvasive disease potential. J Virol. 2003;77:3409–3417. doi: 10.1128/JVI.77.6.3409-3417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, Nabors LB, Markiewicz M, Lakeman AD, Palmer CA, Parker JN, Whitley RJ, Gillespie GY. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009;17:199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martuza RL. Molecular neurosurgery for glial and neuronal disorders. Stereotactic and Functional Neurosurgery. 1992;59:92–99. doi: 10.1159/000098923. [DOI] [PubMed] [Google Scholar]
- Mazarakis ND, Azzouz M, Rohll JB, Ellard FM, Wilkes FJ, Olsen AL, Carter EE, Barber RD, Baban DF, Kingsman SM, Kingsman AJ, O’Malley K, Mitrophanous KA. Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum Mol Genet. 2001;10:2109–2121. doi: 10.1093/hmg/10.19.2109. [DOI] [PubMed] [Google Scholar]
- McCarty DM, Young SM, Jr, Samulski RJ. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu Rev Genet. 2004;38:819–845. doi: 10.1146/annurev.genet.37.110801.143717. [DOI] [PubMed] [Google Scholar]
- McCown TJ, Xiao X, Li J, Breese GR, Samulski RJ. Differential and persistent expression patterns of CNS gene transfer by an adeno-associated virus (AAV) vector. Brain Res. 1996;713:99–107. doi: 10.1016/0006-8993(95)01488-8. [DOI] [PubMed] [Google Scholar]
- Mebatsion T, Konig M, Conzelmann KK. Budding of rabies virus particles in the absence of the spike glycoprotein. Cell. 1996;84:941–951. doi: 10.1016/s0092-8674(00)81072-7. [DOI] [PubMed] [Google Scholar]
- McGettigan JP, Pomerantz RJ, Siler CA, McKenna PM, Foley HD, Dietzschold B, Schnell MJ. Second-generation rabies virus-based vaccine vectors expressing human immunodeficiency virus type 1 gag have greatly reduced pathogenicity but are highly immunogenic. J Virol. 2003;77:237–244. doi: 10.1128/JVI.77.1.237-244.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel U, Malik I, Ebert S, Bähr M, Kügler S. Long-term in vivo and in vitro AAV-2-mediated RNA interference in rat retinal ganglion cells and cultured primary neurons. Biochem Biophys Res Commun. 2005;326:307–312. doi: 10.1016/j.bbrc.2004.11.029. [DOI] [PubMed] [Google Scholar]
- Millhouse OE. The organization of the ventromedial hypothalamic nucleus. Brain Res. 1973a;55:71–87. [PubMed] [Google Scholar]
- Millhouse OE. Certain ventromedial hypothalamic afferents. Brain Res. 1973b;55:89–105. doi: 10.1016/0006-8993(73)90490-3. [DOI] [PubMed] [Google Scholar]
- Miller MA, Lavine CL, Klas SD, Pfeffer LM, Whitt MA. Recombinant replication-restricted VSV as an expression vector for murine cytokines. Protein Expr Purif. 2004;33:92–103. doi: 10.1016/j.pep.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Mocarski ES, Post LE, Roizman B. Molecular engineering of the herpes simplex virus genome: insertion of a second L-S junction into the genome causes additional genome inversions. Cell. 1980;22:243–255. doi: 10.1016/0092-8674(80)90172-5. [DOI] [PubMed] [Google Scholar]
- Mocarski E. Cytomegaloviruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields virology. 3. Lippincott-Raven Publishers; Philadelphia, PA: 1996. pp. 2447–2492. [Google Scholar]
- Moriyoshi K, Richards LJ, Akazawa C, O’Leary DD, Nakanishi S. Labeling neural cells using adenoviral gene transfer of membrane-targeted GFP. Neuron. 1996;16:255–260. doi: 10.1016/s0896-6273(00)80044-6. [DOI] [PubMed] [Google Scholar]
- Naldini L. Lentiviruses as gene transfer agents for delivery to non-dividing cells. Curr Opin Biotechnol. 1998;9:457–463. doi: 10.1016/s0958-1669(98)80029-3. [DOI] [PubMed] [Google Scholar]
- Noe’ F, Nissinen J, Pitkänen A, Gobbi M, Sperk G, During M, Vezzani A. Gene therapy in epilepsy: the focus on NPY. Peptides. 2007;28:377–383. doi: 10.1016/j.peptides.2006.07.025. [DOI] [PubMed] [Google Scholar]
- Noël G, Zollinger L, Laliberté F, Rassart E, Crine P, Boileau G. Targeting and processing of pro-opiomelanocortin in neuronal cell lines. J Neurochem. 1989;52:1050–1057. doi: 10.1111/j.1471-4159.1989.tb01846.x. [DOI] [PubMed] [Google Scholar]
- Norgren RB, Lehman MN. Herpes simplex virus as a transneuronal tracer. Neurosci Biobehav Rev. 1998;22:695–708. doi: 10.1016/s0149-7634(98)00008-6. [DOI] [PubMed] [Google Scholar]
- Obrietan K, van den Pol AN. GABA neurotransmission in the hypothalamus: developmental reversal from Ca2+ elevating to depressing. J Neurosci. 1995;15:5065–5078. doi: 10.1523/JNEUROSCI.15-07-05065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino M, Ebihara H, Lee BH, Araki K, Lundkvist A, Kawaoka Y, Yoshimatsu K, Arikawa J. Use of vesicular stomatitis virus pseudotypes bearing hantaan or seoul virus envelope proteins in a rapid and safe neutralization test. Clin Diagn Lab Immunol. 2003;10:154–60. doi: 10.1128/CDLI.10.1.154-160.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozduman K, Wollmann G, Peipmeier J, van den Pol AN. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcicoma in brain. J Neurosci. 2008;28:1882–1893. doi: 10.1523/JNEUROSCI.4905-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltékian E, Parrish E, Bouchard C, Peschanski M, Lisovoski F. Adenovirus-mediated gene transfer to the brain: methodological assessment. J Neurosci Methods. 1997;71:77–84. doi: 10.1016/s0165-0270(96)00128-8. [DOI] [PubMed] [Google Scholar]
- Publicover J, Ramsburg E, Rose JK. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J Virol. 2004;78:9317–9324. doi: 10.1128/JVI.78.17.9317-9324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Publicover J, Ramsburg E, Robek M, Rose JK. Rapid pathogenesis induced by a vesicular stomatitis virus matrix protein mutant: viral pathogenesis is linked to induction of tumor necrosis factor alpha. J Virol. 2006;80:7028–7036. doi: 10.1128/JVI.00478-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulkkanen KJ, Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585–598. doi: 10.1016/j.ymthe.2005.07.357. [DOI] [PubMed] [Google Scholar]
- Quinonez R, Sutton RE. Lentiviral vectors for gene delivery into cells. DNA Cell Biol. 2002;21:937–951. doi: 10.1089/104454902762053873. [DOI] [PubMed] [Google Scholar]
- Rainov NG, Kramm CM, Banning U, Riemann D, Holzhausen HJ, Heidecke V, Burger KJ, Burkert W, Körholz D. Immune response induced by retrovirus-mediated HSV-tk/GCV pharmacogene therapy in patients with glioblastoma multiforme. Gene Ther. 2000;7:1853–1858. doi: 10.1038/sj.gt.3301311. [DOI] [PubMed] [Google Scholar]
- Rainov NG, Ren H. Oncolytic viruses for treatment of malignant brain tumours. Acta Neurochir Suppl. 2003;88:113–123. doi: 10.1007/978-3-7091-6090-9_17. [DOI] [PubMed] [Google Scholar]
- Ramsburg E, Publicover J, Buonocore L, Poholek A, Robek M, Palin A, Rose JK. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J Virol. 2005;79:15043–15053. doi: 10.1128/JVI.79.24.15043-15053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Kong L, Zhang GR, Liu M, Wang X, Szabo G, Curthoys NP, Geller AI. Glutamatergic or GABAergic neuron-specific, long-term expression in neocortical neurons from helper virus-free HSV-1 vectors containing the phosphate-activated glutaminase, vesicular glutamate transporter-1, or glutamic acid decarboxylase promoter. Brain Res. 2007;1144:19–32. doi: 10.1016/j.brainres.2007.01.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray N, Enquist LW. Transcriptional response of a common permissive cell type to infection by two diverse alphaherpesviruses. J Virol. 2004;78:3489–3501. doi: 10.1128/JVI.78.7.3489-3501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss CS, Plakhov IV, Komatsu T. Viral replication in olfactory receptor neurons and entry into the olfactory bulb and brain. Ann NY Acad Sci. 1998;855:751–761. doi: 10.1111/j.1749-6632.1998.tb10655.x. [DOI] [PubMed] [Google Scholar]
- Richichi C, Lin EJ, Stefanin D, Colella D, Ravizza T, Grignaschi G, Veglianese P, Sperk G, During MJ, Vezzani A. Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci. 2004;24:3051–3059. doi: 10.1523/JNEUROSCI.4056-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridoux V, Robert JJ, Zhang X, Perricaudet M, Mallet J, Le Gal La Salle G. Adenovirus vectors as functional retrograde neuronal tracers. Brain Res. 1994;648:171–175. doi: 10.1016/0006-8993(94)91919-4. [DOI] [PubMed] [Google Scholar]
- Rinaman L, Levitt P, Card JP. Progressive postnatal assembly of limbic-autonomic circuits revealed by central transneuronal transport of pseudorabies virus. J Neurosci. 2000;20:2731–2741. doi: 10.1523/JNEUROSCI.20-07-02731.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TF, Klein ME, Kubke MF, Wild JM, Mooney R. Telencephalic neurons monosynaptically link brainstem and forebrain premotor networks necessary for song. J Neurosci. 2008;28:3479–3489. doi: 10.1523/JNEUROSCI.0177-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok TJ, Pijpers A, Goedknegt-Sabel E, Coulon P. Multiple cerebellar zones are involved in the control of individual muscles: a retrograde transneuronal tracing study with rabies virus in the rat. Eur J Neurosci. 2008;28:181–200. doi: 10.1111/j.1460-9568.2008.06294.x. [DOI] [PubMed] [Google Scholar]
- Salin P, Castle M, Kachidian P, Barroso-Chinea P, López IP, Rico AJ, Kerkerian-Le Goff L, Coulon P, Lanciego JL. High-resolution neuroanatomical tract-tracing for the analysis of striatal microcircuits. Brain Res. 2008;1221:49–58. doi: 10.1016/j.brainres.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Sato Y, Shiraishi Y, Furuichi T. Cell specificity and efficiency of the Semliki forest virus vector- and adenovirus vector-mediated gene expression in mouse cerebellum. J Neurosci Methods. 2004;137:111–121. doi: 10.1016/j.jneumeth.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Schnell MJ, Johnson JE, Buonocore L, Rose JK. Construction of a novel virus that targets HIV-1-infected cells and controls HIV-1 infection. Cell. 1997;90:849–857. doi: 10.1016/s0092-8674(00)80350-5. [DOI] [PubMed] [Google Scholar]
- Seiler MJ, Thomas BB, Chen Z, Wu R, Sadda SR, Aramant RB. Retinal transplants restore visual responses: Trans-Synaptic tracing from visually responsive sites labels transplant neurons. Eur J Neurosci. 2008;28:208–220. doi: 10.1111/j.1460-9568.2008.06279.x. [DOI] [PubMed] [Google Scholar]
- Sharpe AH, Hunter JJ, Chassler P, Jaenisch R. Role of abortive retroviral infection of neurons in spongiform CNS degeneration. Nature. 1990;346:181–183. doi: 10.1038/346181a0. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Greer CA. The olfactory bulb. In: Shepherd G, editor. The synaptic organization of the brain. 4. New York: Oxford University Press; 1998. pp. 159–203. [Google Scholar]
- Smith BN, Banfield BW, Smeraski CA, Wilcox CL, Dudek FE, Enquist LW, Pickard GE. Pseudorabies virus expressing enhanced green fluorescent protein: A tool for in vitro electrophysiologicalanalysis of transsynaptically labeled neurons in identified central nervous system circuits. Proc Natl Acad Sci U S A. 2000;97:9264–9269. doi: 10.1073/pnas.97.16.9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Lachmann RH, Efstathiou S. Expression from the herpes simplex virus type 1 latency-associated promoter in the murine central nervous system. J Gen Virol. 2000;81:649–662. doi: 10.1099/0022-1317-81-3-649. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Weindl A, Schrell U, Wetzstein R. Immunohistochemistry of vasopressin, oxytocin and neurophysin in the hypothalamus and extrahypothalamic regions of the human and primate brain. Acta Histochem Suppl. 1981;24:79–95. [PubMed] [Google Scholar]
- Song CK, Schwartz GJ, Bartness TJ. Anterograde Transneuronal Viral Tract Tracing Reveals Central Sensory Circuits From White Adipose Tissue. Am J Physiol Regul Integr Comp Physiol. 2008 doi: 10.1152/ajpregu.90786.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stettler DD, Yamahachi H, Li W, Denk W, Gilbert CD. Axons and synaptic boutons are highly dynamic in adult visual cortex. Neuron. 2006;49:877–887. doi: 10.1016/j.neuron.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- Sun N, Cassell MD, Perlman S. Anterograde, transneuronal transport of herpes simplex virus type 1 strain H129 in the murine visual system. J Virol. 1996;70:5405–5413. doi: 10.1128/jvi.70.8.5405-5413.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taber KH, Strick PL, Hurley RA. Rabies and the cerebellum: new methods for tracing circuits in the brain. J Neuropsychiatry Clin Neurosci. 2005;17:133–139. doi: 10.1176/jnp.17.2.133. [DOI] [PubMed] [Google Scholar]
- Taymans JM, Vandenberghe LH, Haute CV, Thiry I, Deroose CM, Mortelmans L, Wilson JM, Debyser Z, Baekelandt V. Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Hum Gene Ther. 2007;18:195–206. doi: 10.1089/hum.2006.178. [DOI] [PubMed] [Google Scholar]
- Terashima T, Miwa A, Kanegae Y, Saito I, Okado H. Retrograde and anterograde labeling of cerebellar afferent projection by the injection of recombinant adenoviral vectors into the mouse cerebellar cortex. Anat Embryol (Berl) 1997;196:363–382. doi: 10.1007/s004290050105. [DOI] [PubMed] [Google Scholar]
- Tsukamoto Y, Yamamoto T, Okado H, Nibu K, Terashima T. Retrograde labeling of mouse spinal descending tracts by a recombinant adenovirus. Arch Histol Cytol. 2003;66:209–220. doi: 10.1679/aohc.66.209. [DOI] [PubMed] [Google Scholar]
- Ugolini G, Kuypers HG, Simmons A. Retrograde transneuronal transfer of herpes simplex virus type 1 (HSV 1) from motoneurones. Brain Res. 1987;422:242–256. doi: 10.1016/0006-8993(87)90931-0. [DOI] [PubMed] [Google Scholar]
- Ugolini G. Specificity of rabies virus as a transneuronal tracer of motor networks: transfer from hypoglossal motoneurons to connected second-order and higher order central nervous system cell groups. J Comp Neurol. 1995;356:457–480. doi: 10.1002/cne.903560312. [DOI] [PubMed] [Google Scholar]
- Ugolini G. Use of rabies virus as a transneuronal tracer of neuronal connections: implications for the understanding of rabies pathogenesis. Dev Biol (Basel) 2008;131:493–506. [PubMed] [Google Scholar]
- van den Pol AN, Dalton K, Rose J. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J Virology. 2002;76:1309–1327. doi: 10.1128/JVI.76.3.1309-1327.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Acuna C, Clark R, Ghosh PK. Physiological properties of hypothalamic MCH neurons identified with selective expression of reporter gene after recombinant virus infection. Neuron. 2004;42:635–652. doi: 10.1016/s0896-6273(04)00251-x. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Reuter J, Santarelli J. Enhanced cytomegalovirus infection of the developing brain independent of the adaptive immune system. J Virology. 2002;76:8842–8854. doi: 10.1128/JVI.76.17.8842-8854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN. Viral infections in the developing and mature brain. Trends Neurosci. 2006;29:398–406. doi: 10.1016/j.tins.2006.06.002. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Ghosh P. Selective neuronal expression of green fluorescent protein with cytomegalovirus promoter reveals entire neuronal arbor in transgenic mice. J Neurosci. 1998;18:10640–10651. doi: 10.1523/JNEUROSCI.18-24-10640.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Mocarski E, Saederup N, Vieira J, Meier TJ. Cytomegalovirus cell tropism, replication, and gene transfer in brain. J Neurosci. 1999;19:10948–10965. doi: 10.1523/JNEUROSCI.19-24-10948.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Vieira J, Spencer DD, Santarelli JG. Mouse cytomegalovirus in developing brain tissue - analysis of 11 species with GFP-expressing recombinant virus. J Comp Neurol. 2000;427:559–580. doi: 10.1002/1096-9861(20001127)427:4<559::aid-cne5>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Van Doren K, Hanahan D, Gluzman Y. Infection of eucaryotic cells by helper-independent recombinant adenoviruses: early region 1 is not obligatory for integration of viral DNA. J Virol. 1984;50:606–614. doi: 10.1128/jvi.50.2.606-614.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virta S, Rapola J, Jalanko A, Laine M. Use of nonviral promoters in adenovirus-mediated gene therapy: reduction of lysosomal storage in the aspartylglucosaminuria mouse. J Gene Med. 2006;8:699–706. doi: 10.1002/jgm.892. [DOI] [PubMed] [Google Scholar]
- Wagner RR, Rose JK. Rhabdoviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. 3. New York: Lippincott-Raven; 1996. pp. 1121–1131. [Google Scholar]
- Walsh C, Cepko CL. Clonal dispersion in proliferative layers of developing cerebral cortex. Nature. 1993;362:632–635. doi: 10.1038/362632a0. [DOI] [PubMed] [Google Scholar]
- Watts AG, Swanson LW, Sanchez-Watts G. Efferent projections of the suprachiasmatic nucleus: I. Studies using anterograde transport of Phaseolus vulgaris leucoagglutinin in the rat. J Comp Neurol. 1987;258:204–229. doi: 10.1002/cne.902580204. [DOI] [PubMed] [Google Scholar]
- Wickersham IR, Lyon DC, Barnard RJO, Mori T, Finke S, Conzelmann KK, Young JAT, Callaway EM. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 2007a;53:639–647. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham IR, Finke S, Conzelmann KK, Callaway EM. Retrograde neuronal tracing with a deletion mutant rabies virus. Nature Methods. 2007b;4:47–49. doi: 10.1038/NMETH999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox ME, Yang W, Senger D, Rewcastle NB, Morris DG, Brasher PM, Shi ZQ, Johnston RN, Nishikawa S, Lee PW, Forsyth PA. Reovirus as an oncolytic agent against experimental human malignant gliomas. J Natl Cancer Inst. 2001;93:903–912. doi: 10.1093/jnci/93.12.903. [DOI] [PubMed] [Google Scholar]
- Wintermantel TM, Campbell RE, Porteous R, Bock D, Gröne HJ, Todman MG, Korach KS, Greiner E, Pérez CA, Schütz G, Herbison AE. Definition of estrogen receptor pathway critical for estrogen postive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron. 2006;52:271–280. doi: 10.1016/j.neuron.2006.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmann G, Tattersall P, van den Pol AN. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J Virol. 2005;79:6005–6022. doi: 10.1128/JVI.79.10.6005-6022.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LF, Goodhead L, Prat C, Mitrophanous KA, Kingsman SM, Mazarakis ND. Lentivirus-mediated gene transfer to the central nervous system: therapeutic and research applications. Hum Gene Ther. 2006;17:1–9. doi: 10.1089/hum.2006.17.1. [DOI] [PubMed] [Google Scholar]
- Xu R, Janson CG, Mastakov M, Lawlor P, Young D, Mouravlev A, Fitzsimons H, Choi KL, Ma H, Dragunow M, Leone P, Chen Q, Dicker B, During MJ. Quantitative comparison of expression with adeno-associated virus (AAV-2) brain-specific gene cassettes. Gene Ther. 2001;8:1323–1332. doi: 10.1038/sj.gt.3301529. [DOI] [PubMed] [Google Scholar]
- Yates BJ, Smail JA, Stocker SD, Card JP. Transneuronal tracing of neural pathways controlling activity of diaphragm motoneurons in the ferret. Neuroscience. 1999;90:1501–1513. doi: 10.1016/s0306-4522(98)00554-5. [DOI] [PubMed] [Google Scholar]
- Yoon H, Enquist LW, Dulac C. Olfactory inputs to hypothalamic neurons controlling reproduction and fertility. Cell. 2005;123:669–682. doi: 10.1016/j.cell.2005.08.039. [DOI] [PubMed] [Google Scholar]
- Zemanick MC, Strick PL, Dix RD. Direction of transneuronal transport of herpes simplex virus 1 in the primate motor system is strain-dependent. Proc Natl Acad Sci U S A. 1991;88:8048–8051. doi: 10.1073/pnas.88.18.8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.