

Abstract
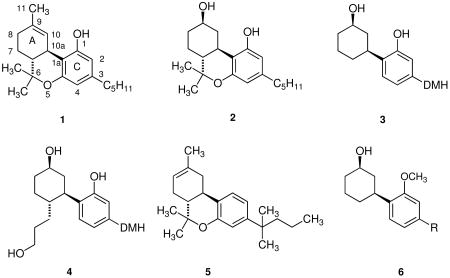
Three 1-methoxy analogs of CP-47,497 (7, 8 and 19) have been synthesized and their affinities for the cannabinoid CB1 and CB2 receptors have been determined. Although these compounds exhibit selectivity for the CB2 receptor none have significant affinity for either receptor. Modeling and receptor docking studies were carried out, which provide a rationalization for the weak affinities of these compounds for either receptor.
Keywords: CB1 receptor, CB2 receptor, nontraditional cannabinoids, 1-methoxy cannabinoids
1. Introduction
The modern era of the study of cannabinoids began with the elucidation of the structure of the principal psychoactive ingredient in marijuana, •9-tetrahydrocannabinol (•9-THC, 1), by Gaoni and Mechoulam.1 Subsequently, a number of analogs of 1 were synthesized and comprehensive structure-activity relationships (SAR) were developed based upon the •9-THC template.2-4 In the course of the development of analgesics derived from the potent synthetic cannabinoid, (-)-9-nor-9β-hydroxyhexahydrocannabinol (HHC, 2), a series of structurally modified analogs of 2 were prepared.5-7 Although useful analgesics were not developed, a series of non-traditional cannabinoids was developed in which the oxygen containing pyran ring of THC was removed to provide a bicyclic system that retained the phenolic hydroxyl group of THC and the 9-hydroxyl of HHC.8 The SAR for traditional cannabinoids specifies maximum potency with a 1,1-dimethylheptyl substituent at the 3-position of the aromatic ring and an equatorial β-orientation of the 9-hydroxyl group.8 These SAR are also valid for these bicyclic non-traditional cannabinoids and the least complex molecule that fulfilled these requirements was CP-47,497 (3, DMH = 1,1-dimethylheptyl), which was found to be more potent than THC in vivo. A hydroxypropyl group at C-4 of the cyclohexanol ring, as in CP-55,940 (4), led to enhanced potency and in 1988 [3H]-CP-55,940 was employed by Howlett's group to identify a cannabinoid receptor in rat brain.9 This G-protein-coupled, transmembrane receptor is now designated as the CB1 receptor and is expressed primarily in the central nervous system.10-12
In 1993 Munro et al. identified a second receptor in macrophages present in the spleen.13 This receptor, designated CB2, is expressed primarily in the in the immune system and it has been suggested that it is responsible for the immunomodulatory effects of cannabinoids,14-19 a conclusion that is supported by the fact that these effects are absent in CB2 receptor knockout mice.19 There is increasing evidence that the endocannabinoid system is of very considerable physiological significance. In particular, CB2 receptors are expressed in C6 glioma cells20 and both CB1 and CB2 receptors are expressed in non-melanoma skin cancer cells.21 There is also a considerable body of evidence that the CB2 receptor is involved in inflammatory pain22-29 Two recent reviews have pointed out the potential of the endocannabinoid system as a therapeutic target and suggest that developing new, selective ligands for the CB2 receptor may lead to the development of new useful drugs for the treatment of a number diseases.30,31
Several years ago we developed a series of 1-deoxy-•8-THC analogs, several of which are highly selective for the CB2 receptor.32 One of these compounds, JWH-133 (5) is a full agonist at the CB2 receptor, has 200-fold selectivity for the receptor and is inactive in the mouse model of cannabinoid activity.33 This series of compounds was developed in analogy with 1-deoxy-3-(1,1-dimethylheptyl)-•8-THC, which is a moderately selective CB2 receptor ligand.32,34 Based upon the observation that 1-deoxy-•8-THC analogs are highly selective ligands for the CB2 receptor, we hypothesized that 1-deoxy analogs of CP-47,497 (3) and CP-55,940 (4) would also exhibit useful selectivity for the CB2 receptor and series of 1-deoxy analogs of both compounds was synthesized and their pharmacology evaluated.35 In contrast to our hypothesis, none of these compounds have more than modest affinity for either the CB1 or CB2 receptor. It appeared possible that the failure of these compounds to bind to either receptor was due to the lack of the oxygen atom at C-1 that was present in the Pfizer compounds and we have now synthesized 1-methoxy analogs of CP-47497 (6) and determined their affinity for the CB1 and CB2 receptors.
2. Results
Typically 1-deoxy and 1-methoxy THC analogs with the highest affinity for both the CB1 and CB2 receptors are those with a 3-dimethylhexyl or dimethylheptyl substituent32,36 and consequently it was determined that 1-methoxy-9β-hydroxy dimethylhexyl (7) and dimethylheptyl (8) CP-47,497 analogs would be the initial synthetic targets (Scheme 1). If either of these compounds exhibited interesting pharmacology, other compounds in this series were to be investigated,
Scheme 1.

(a) NaSPr, DMF, reflux; (b) (EtO)2PO, CCl4, Et3N, 25 °C; (c) Li, NH3, -78 °C; (d) Br2, HOAc, 25 °C; (e) n-BuLi, THF -78 °C; (f) 3-Ethoxy-2-cyclohexen-1-one, THF, reflux, then 10% HCl, 25 °C; (g) Li, NH3, THF, -78 °C; (h) NaBH4, EtOH, 0 °C to 25 °C; (i) Li(sec-butyl)3BH, THF, -78 °C then 25 °C; (j) NaOH, H2O2, EtOH, 25 °C.
The synthesis of the methoxy analogs of CP-47497 as illustrated in Scheme 1 is a modification of the method employed for the synthesis of the 1-deoxy analogs, however using 2-(4-bromo-3-methoxyphenyl)-2-methylheptane (9) and 2-(4-bromo-3-methoxyphenyl)-2-methyloctane (10) as starting materials rather than the dimethylalkylbenzenes used for the deoxy analogs.35 We used aryl bromide 10 a number of years ago in the synthesis of 1-deoxy-11-hydroxy-3-(1,1-dimethylheptyl)-•8-THC37 and employed a modification of that synthesis in the present work.
As described previously,37 Selective demethylation of one of the methoxy groups of 2-(3,5-dimethoxyphenyl)-2-methylheptane (11) and 2-(3,5-dimethoxyphenyl)-2-methyloctane (12) using sodium thiopropoxide followed by conversion of the phenol to the corresponding diethyl phosphate and reduction with lithium in ammonia afforded 2-(3-methoxyphenyl)-2-methylheptane (13) and 2-(3-methoxyphenyl)-2-methylheptane (14). Reaction of compounds 13 and 14 with bromine in acetic acid provided aryl bromides 9 and 10.37 The corresponding aryllithiums were prepared via halogen-metal interconversion38 from bromides 9 and 10 using n-butyllithium, which were reacted with 3-ethoxycyclohex-2-en-1-one to provide the corresponding tertiary alcohol. Treatment of the crude reaction products with dilute HCl resulted in hydrolysis of the enol ether, which eliminated the elements of water to provide 3-arylcyclohexenones 15 and 16. Lithium ammonia reduction of these cyclohexenones gave 3-arylcyclohexanones 17 and 18.39 Sodium borohydride reduction40 of 17 and 18 gave respectively (1R*,3S*)3-[2-methoxy-4-(1,1-dimethylhexyl)phenyl]cyclohexanol (7, JWH-440) and (1R*,3S*)3-[2-methoxy-4-(1,1-dimethylheptyl)phenyl]cyclohexanol (8, JWH 441).41 Both of these compounds are racemic as indicated by the notation R* and S*, which indicate relative stereochemistry.41 The equatorial stereochemistry of the hydroxyl group in alcohols 7 and 8 was assigned based upon the method of synthesis40 and the 1H NMR spectrum in which the carbinol proton appears as a multiplet centered about δ 3.79, which is characteristic of an axial carbinol proton.42
The affinities of alcohols 7 (JWH-440) and 8 (JWH-441) for the CB1 receptor were determined by measuring their ability to displace the potent cannabinoid [3H] CP-55,940 from its binding site in a membrane preparation from rat brain as described by Compton et al.44 Affinities for the CB2 receptor were determined by measuring the ability of the compounds to displace [3H] CP-55,940 from a cloned human receptor preparation using the procedure described by Showalter et al.45 The results of these determinations are summarized in Table 1. Also included in Table 1 are the receptor affinities for •8-THC (1) and CP-47,497 (3).
Table 1.
Receptor Affinities (mean ± SEM) of CP-47,497 analogs (7, 8, 19, 20, and 21), •9-THC (1) and CP-47,497 (3).
As shown in Table 1, both JWH-440 (7) and JWH-441 (8) have little affinity for either the CB1 or CB2 receptor. Since it was conceivable that the stereochemistry of the secondary hydroxyl group may have affected the interaction of JWH-440 or JWH-441 with the receptors, ketone 17 was reduced with L-Selectride, a reagent that stereoselectively reduces cyclic ketones to the corresponding axial alcohol,43 to provide racemic (1S*,3S*)3-[2-methoxy-4-(1,1-dimethylhexyl)phenyl]cyclohexanol (19, JWH-442), which has an axial hydroxyl substituent. The 1H NMR spectrum of this compound is consistent with the assigned stereochemistry, with the equatorial carbionol proton as a broadened singlet at δ 4.24.42 This methoxy CP-47,497 analog also has negligible affinity for either the CB1 or CB2 receptor (Table 1).
In order to provide a rationale for the failure of not only compounds 7, 8, and 19 but also the deoxy CP-47,497 analogs we reported previously35 to have appreciable affinity for either receptor, a modeling and receptor docking study was carried out. This study included JWH-441 (8), JWH-324, (1R*,3S*)3-[4-(1,1-dimethylheptyl)phenyl]cyclohexanol (20) and JWH-342, (1S*,3S*)3-[4-(1,1-dimethylheptyl)phenyl]cyclohexanol (21). The affinities of compounds 20 and 21 for the CB1 and CB2 receptors are included in Table 1.
CB2 Docking Studies
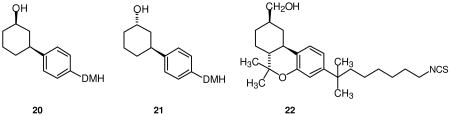
The minimized structure of each ligand was docked in the refined model of the CB2 receptor in its activated state (R*) in the same binding site as described for AM841 (22).46 This CB2 receptor model includes the extracellular and intracellular domains, as well as the transmembrane helix (TMH) bundle. JWH-324 (20), JWH-342 (21) and JWH-441 (8) have low CB2 affinities (JWH-324, Ki = 231 nM; JWH-342, Ki = 178 nM; and JWH-441 Ki = 808 nM). It was found that a significant conformational energy expense is required for each of these ligands to dock in an AM841-like conformation at CB2 R*. For all three ligands, this expense includes a required re-orientation of the dimethylheptyl side chain, as well as a change in the C1-C1a-C10a-C10 torsion angle, which affects the relative orientation of the A and C rings. A reorientation of the methoxy group is also required for JWH-441 (8) and this additional expense may be the reason for the very low CB2 affinity of this compound. Figure 1 (A-C) illustrate the conformational differences between the global minimum energy conformers of JWH-324, JWH-342 and JWH-441 (shown in green) and the final docked conformation of each ligand at CB2 (shown in orange). For JWH-324 (20), the C1-C1a-C10a-C10 torsion angle must shift from its global minimum position of -117° to 15°(see Figure 1A, orange). The total conformational cost for the ligand to dock at CB2 R* is 5.3 kcal/mol. In addition to Van der Waals interactions with residues that line its CB2 R* binding site, JWH-324 (20) can form a hydrogen bond with S7.39 (O-O distance = 2.62 Å; O-H--O angle = 170°) (see Figure 2A). The total interaction energy for JWH-324 (20) at CB2 R* was found to be -36.3 kcal/mol. For JWH-342 (21), the C1-C1a-C10a-C10 torsion angle must shift from its global minimum position of -116° to -48° (see Figure 1B, orange). The total conformational cost for the ligand to dock at CB2 R* is 6.5 kcal/mol. In this conformation, JWH-342 (21) can form a hydrogen bond with S7.39 (O-O distance = 2.62 Å; O-H--O angle = 168°) (see Figure 2B). Although JWH-342 (21) incurs a higher energy expense relative to JWH-324 (20) to dock with CB2 R*, JWH-342 (21) compensates for this higher energy expense with greater Van der Waals interactions, particularly with S6.58 and A7.36, leading to an overall interaction energy (-37.3 kcal/mol) that is just slightly better than that of JWH-324 (20). For JWH-441 (8), it should be noted that the global minimum energy position of the dimethylheptyl side chain differs from that in JWH-324 (20) and JWH-342 (21). In these ligands one of the methyl groups is in the plane of the aromatic ring, pointing up (see Figures 1A-B, green). In JWH-441, steric repulsion from the methoxy group on Ring C, causes the methyl to point down, but the methyl does remain in the plane of the aromatic ring (see Figure 1C, green). For JWH-441 (8), the C1-C1a-C10a-C10 torsion angle must shift from its global minimum position of -117° to 10° and the Ring C methoxy group has to shift 106° out of plane with Ring C (see Figure 1C, orange) to avoid steric interference with L6.54, S6.58, S7.39 or the ligand's A ring. The total conformational cost for the ligand to dock at CB2 is quite high at 10.4 kcal/mol. In addition to Van der Waals interactions with residues that line its binding site, JWH-441 (8) can form a hydrogen bond with S7.39 (O-O distance = 2.67 Å; O-H--O angle = 171°) (see Figure 2C). The total interaction energy for JWH-441 (8) at CB2 was found to be -32.6 kcal/mol. Thus, while each of these ligands can establish some interaction at CB2, the conformational energy expense incurred when each binds to CB2 will significantly impact the affinity of each. These results are consistent with the CB2 affinities for these compounds listed in Table 1.
Figure 1.
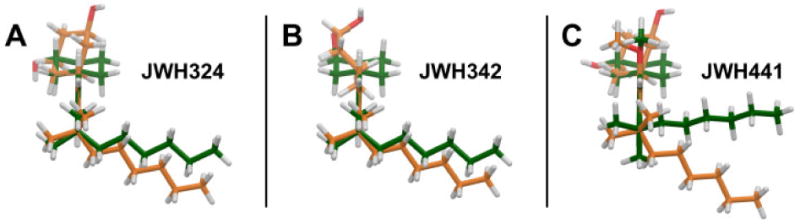
A comparison of the global minimum energy conformation (green) vs. the docked conformation (orange) of JWH-324(20), JWH-342(22) and JWH-441(8) at CB2 R* is illustrated here. The conformers have been overlaid on their phenyl rings (Ring C) and are oriented so that the phenyl ring (Ring C) is perpendicular to the plane of the page, with the dimethylheptyl side chain closest to the viewer and the carbocyclic ring (Ring A) furthest from the viewer (A) For JWH-324(20), the C1-C1a-C10a-C10 torsion angle must shift from its global minimum position of -117° to 15°. The conformational energy difference between the two conformers 5.3 kcal/mol. (B) For JWH-342 (21), the C1-C1a-C10a-C10 torsion angle must shift from its global minimum position of -116° to -48°. The total conformational cost for the ligand to dock at CB2 is 6.5 kcal/mol. (C) For JWH-441 (8), the C1-C1a-C10a-C10 torsion angle must shift from its global minimum position of -117° to 10° and the Ring C methoxy group has to shift 106° out of plane with Ring C to avoid steric interference with L6.54, S6.58, S7.39 or the ligand's A ring. The total conformational cost for the ligand to dock at CB2 R* is therefore quite high at 10.4 kcal/mol.
Figure 2.
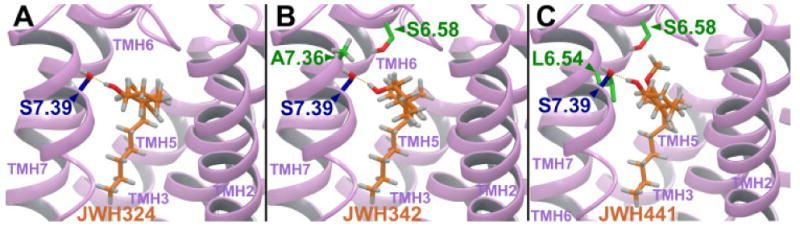
JWH-324 (20), JWH-342 (21) and JWH-441(8) are shown here docked at a model of the CB2 activated (R*) state. The view in each case is from the lipid bilayer, between TMH2 and TMH7 (TMH1 has been omitted for clarity). (A) For JWH-324 (20), in addition to Van der Waals interactions with residues that line its binding site, the ligand can form a hydrogen bond with S7.39 (O-O distance = 2.62 Å; O-H--O angle = 170°). The total interaction energy for JWH-324 (20) at CB2 R* was found to be -36.3 kcal/mol. (b) For JWH-342 (21), the ligand can form a hydrogen bond with S7.39 (O-O distance = 2.62 Å; O-H--O angle = 168°). Although JWH-342 (21) incurs a higher energy expense relative to JWH-324 (20) to dock with CB2 R*, JWH-342 (21) compensates for this higher energy expense with greater Van der Waals interactions, particularly with S6.58 and A7.36, leading to an overall interaction energy (-37.3 kcal/mol) that is just slightly better than that of JWH-324 (20). (c) For JWH-441(8), in addition to Van der Waals interactions with residues that line its binding site, the ligand can form a hydrogen bond with S7.39 (O-O distance = 2.67 Å; O-H--O angle = 171°). The total interaction energy for JWH-441(8) at CB2 R* was found to be -32.6 kcal/mol.
CB1 Docking Studies
The minimized structure of each ligand was docked in the refined model of the CB1 receptor activated (R*) state in the same binding site as described for HU-210.47 This CB1 receptor model includes a truncated N-terminus, all extracellular loops and intracellular domains, as well as the transmembrane helix bundle. JWH-324 (20), JWH-342 (21) and JWH-441 (8) have low/essentially no CB1 affinity (JWH-324, Ki = 2954 nM; JWH-342, Ki = 645 nM; and JWH-441 Ki > 10,000 nM). Docking studies showed that the A ring in all three ligands has severe Van der Waals overlaps with S7.39 or K3.28 when docked in the HU-210 binding site at CB1. In addition, the C ring methoxy group of JWH-441 has severe Van der Waals overlaps with V3.32, L6.51 and C7.42 or, if the C ring is rotated 180°, the methoxy group has severe Van der Waals overlaps with F2.57 and L7.43.
3. Discussion
These methoxy CP-47,497 analogs were designed in analogy to 1-methoxy-3-(dimethylhexyl and dimethylheptyl)-•8-THC, which exhibit respectively 174 and 12-fold selectivity for the CB2 receptor.36 Both these compounds have from good to high affinity for the CB2 receptor (Ki = 18 and 57 nM, respectively). Also, both epimers of the secondary hydroxyl groups of the corresponding 11-nor-9-hydroxy-hexahydrocannabinols, which have from 4.5 to 33-fold selectivity for the CB2 receptor, served as templates for the methoxy CP-47,497 analogs.42 These compounds have from 12 nM to 38 nM affinity for the CB2, In earlier work, we synthesized 1-deoxy analogs of CP-47,497.35 Although all of these compounds exhibit varying degrees of selectivity for the CB2 receptor, only the dimethylheptyl and the dimethyloctyl analogs with axial hydroxyl groups have affinity for the CB2 receptor greater than 200 nM. In that publication it was suggested that an oxygen substituent on the aromatic ring was probably necessary for CB2 receptor binding. This hypothesis led us to the preparation of the methoxy CP-47,497 analogs described above.
It is apparent from the experimentally determined receptor binding data that, although the hypothesis that an oxygen substituent on the aromatic ring was necessary for CB2 binding appeared to be eminently reasonable, the lack of affinity of methoxy CP-47,497 analogs JWH-440 (7), JWH-441 (8) and JWH-442 (19) for the CB2 receptor indicates that this hypothesis is not correct. In order to provide a rationale for the lack of affinity of not only the methoxy CP-47,497 analogs as well as the deoxy CP-47,497 analogs reported previously,35 molecular modeling and docking studies were carried out for deoxy analogs JWH-324 (20), JWH-342 (21) and methoxy analog JWH-441 (8). These three compounds have negligible affinity for the CB1 receptor and the docking studies indicate that the saturated carbocyclic ring of these compounds has severe steric clashes with serine 7.39 and lysine 3.28 of the CB1 receptor. In addition the methoxy group of JWH-441 (8) has severe clashes with valine 3.32, leucine 6.51 and cysteine 7.42. Rotation of the aromatic ring leads to alternative steric clashes with phenylalanine 2.57 and leucine 7.43. These severe steric clashes of these compounds with the amino acids of the CB1 receptor are consistent with the very weak CB1 receptor affinities of JWH-441 (8), JWH-324 (20) and JWH-342 (21).
The two 1-deoxy CP-47,497 analogs, JWH-324 (20) and JWH-342 (21) both have modest affinities for the CB2 receptor (231 ± 48 and 178 ± 15 nM, respectively). For docking of these two compounds with the CB2 receptor model, rotation about the C1a-C10a bond between the aromatic and saturated rings is necessary as well as a reorientation of the dimethylheptyl side chain. The conformational energy expense is 5.3 kcal/mol for JWH-324 (20) and 6.5 kcal/mol for JWH-342 (21). In the case of methoxy analog JWH-441 (8) in addition to reorientation of the dimethylheptyl side chain and rotation of the C1a-C10a bond, the methoxy group must rotate out of the plane of the aromatic ring. This results in a conformational energy expense of 10.4 kcal/mol. The overall interaction energies of JWH-324 (20), JWH-342 (21) and JWH-441 (8) at CB2 were found to be -36.3 kcal/mol, -37.3 kcal/mol and -32.6 kcal/mol, respectively. The trend in interaction energies, JWH-342 (21) < JWH-324 (20) <JWH-441 (8) is consistent with the trend in average Ki vallues for these ligands at CB2. However, the interaction energy of JWH-342 (21) is only 1.0 kcal/mol better than that of JWH-324 (20). This small difference is also consistent with the fact that the Ki values of these two compounds have slightly overlapping ranges when experimental error is taken into account.
4. Conclusions
A priori it appeared that 1-deoxy and 1-methoxy analogs of the very potent bicyclic Pfizer non-traditional cannabinoids would provide compounds that were selective ligands for the CB2 receptor, just as various similar analogs of •8-THC (1) and 11-nor-9-hydroxyhexahydrocannabinol (2) have good CB2 receptor affinity with little affinity for the CB1 receptor. However, the lack of significant affinity for these analogs of CP-47,497 indicates that this hypothesis is not correct. CP-47,497 and related compounds have a phenolic hydroxyl group, which permits energetically significant hydrogen bonding interactions with the receptors, which are lacking in the deoxy and methoxy analogs. It appears that the rigid structure of the dibenzopyran system of the classical cannabinoids that provided the nucleus of the CB2 selective ligands based upon that template provides a superior platform for the synthesis of structurally modified ligands for the CB2 receptor.
5. Experimental
5.1. General
1H and 13C NMR spectra were recorded on Bruker 300AC and JEOL 500 spectrometers. Mass spectral analyses were performed on a Shimadzu QP2010 capillary gas chromatograph/mass spectrometer equipped with a mass sensitive detector at 1.01 kV. HRMS data were obtained in the Mass Spectrometry Laboratory, School of Chemical Sciences, University of Illinois. Ether and THF were distilled from Na-benzophenone ketyl immediately before use, and other solvents were purified using standard procedures. Column chromatography was carried out on Sorbent Technologies silica gel (32 - 63 μ) using the indicated solvents as eluents. All new compounds were homogeneous to TLC and 13C NMR. All target compounds were homogeneous to GLC or TLC in two different solvent systems and GLC. TLC was carried out using 200 μm silica gel plates with the indicated solvents.
5.2. 2-(3-Methoxy-5-hydroxyphenyl)-2-methylheptane
To a suspension of 1.4 g (34 mmol, 60% dispersion in oil) of NaH in 14 mL of dry DMF under argon, was added dropwise 4.2 mL (46 mmol) of 1-propanethiol, and the reaction was stirred for 30 min at ambient temperature. To this mixture was added 1.32 g (5.28 mmol) of 2-(3,5-dimethoxyphenyl)-2-methylheptane32 (11) in 7 mL of dry DMF, and the resultant solution was heated at reflux for 5 h, cooled to ambient temperature and poured into 40 mL of 1 M HCl. The solution was extracted with three portions of ether and the ethereal extracts were washed with successive portions of aqueous NaHCO3, brine, dried (MgSO4) and concentrated in vacuo. The crude product was purified by flash chromatography (petroleum ether/ether, 8:2) to afford 0.96 g (77%) of 2-(3-methoxy-5-hydroxyphenyl)-2-methylheptane as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.81 (t, J = 7.0 Hz, 3H), 0.97-1.15 (m, 2H), 1.16-1.24 (m, 4H), 1.26 (s, 6H), 1.56 (t, J = 6.0 Hz, 2H), 3.79 (s, 3H), 6.28 (s, 1H), 6.36 (s, 1H), 6.45 (s, 1H); 13C NMR (125.77 MHz, CDCl3) δ 14.1, 22.7, 24.5, 28.7, 32.6, 37.6, 44.5, 55.6, 96.6, 105.2, 105.9, 152.9, 156.1, 160.2; MS (EI) m/z (rel intensity) 121 (37), 149 (100), 236 (60).
5.3 2-(3-Methoxyphenyl)-2-methylheptane (13)
To a solution of 1.56 g (6.61 mmol) of 2-(3-hydroxyphenyl-5-methoxyphenyl)-2-methylheptane and 1.08 mL (8.42 mmol) of diethyl phosphite in 4 mL of CCl4 at 0 °C was added dropwise 1 mL (7.17 mmol) of triethylamine. The solution was stirred at 0 °C for 1 h, allowed to warm to ambient temperature and stirred for 7 h at ambient temperature. The mixture was diluted with CH2Cl2 and washed with successive solutions of H2O, 1 M aqueous NaOH, H2O, 1 M HCl, and H2O. The organic layer was dried (MgSO4), concentrated in vacuo and purified by flash chromatography (petroleum ether/ether, 7:3) to give 2.01 g (93%) of 2-(3-hydroxyphenyl-5-methoxyphenyl)-2-methylheptane diethyl phosphate as a red oil, which was used in the next step without further purification:1H NMR (500 MHz, CDCl3) δ 0.82 (t, J = 7.0 Hz, 3H), 0.98-1.14 (m, 2H), 1.15-1.25 (m, 4H), 1.27 (s, 6H), 1.39 (t, J = 7.1 Hz, 6H), 1.56 (t, J = 6.0 Hz, 2H), 3.79 (s, 3H), 4.21-4.26 (m, 4H), 6.64 (s, 1H), 6.71 (s, 1H), 6.78 (s, 1H); 13C NMR (125.77 MHz, CDCl3) δ 13.9, 15.9, 16.8, 22.6, 24.6, 28.6, 31.7, 37.6, 44.3, 55.5, 102.2, 102., 109.2, 109.9,151.3,152.7, 160.1; MS (EI) m/z (rel intensity) 245 (60), 273 (30), 302 (100), 372 (50).
To 49 mL of liquid NH3 at -78 °C was added 0.052 g (7.49 g atom) of lithium shot and the solution was stirred for 15 min. A solution of 0.83 g (2.23 mmol) 2-(3-hydroxyphenyl-5-methoxyphenyl)-2-methylheptane diethyl phosphate in 10 mL of dry THF was added dropwise, and the reaction was stirred at -78°C for 3 h. The reaction was quenched by the addition of solid NH4Cl and the NH3 was evaporated overnight at ambient temperature. The solid residue was taken up in 15 mL of H2O and extracted with two portions of ether. The ethereal extracts were washed with 10 % aqueous HCl and brine, dried (MgSO4), and the solvent removed in vacuo. The yellow oil was purified by flash chromatography (petroleum ether/ether, 9:1) to give 0.35 g (78%) of 13 as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 0.88 (t, J = 7.0 Hz, 3H), 0.97-1.11 (m, 2H), 1.12-1.25 (m, 4H), 1.26 (s, 6H), 1.57 (t, J = 6.0 Hz, 2H), 3.81 (s, 3H), 6.69-6.72 (m, 1H), 6.88-6.91 (m, 2H), 7.18-7.23 (m, 1H); 13C NMR (125.77 MHz, CDCl3) δ 14.1, 22.7, 24.6, 28.9, 32.6, 37.5, 44.7, 55.0, 96.6, 104.8, 118.1, 128.6, 151.7, 159.2; MS (EI) m/z (rel intensity) 57 (40), 205 (100), 220(50).
5.4. 2-(4-Bromo-3-methoxyphenyl)-2-methylheptane (9)
To a solution of 0.281 g (1.27 mmol) of 13 in 0.5 mL of glacial acetic acid was slowly added 0.065 mL (1.27 mmol) of bromine in 0.5 mL of acetic acid at ambient temperature. The solution was stirred for 2 h at ambient temperature, diluted with 5 mL of water and 3 mL of aqueous NaHCO3. The reaction mixture was extracted with two portions of ether and the ethereal extracts were washed with brine, dried (MgSO4), and concentrated in vacuo. The resultant orange oil was purified by flash chromatography (petroleum ether/ether, 9:1) to give 0.201g (79%) of 2-(4-bromo-3-methoxyphenyl)-2-methylheptane (9) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.87 (t, J = 7.0 Hz, 3H), 0.96-1.11 (m, 2H), 1.12-1.24 (m, 4H), 1.25 (s, 6H), 1.60 (t, J = 6, 2H), 3.90 (s, 3H), 6.81 (dd, J = 2.2, 2 Hz, 1H), 6.84 (d, J = 2.2 Hz, 1H); 7.43 (d, J = 8 Hz, 1H); 13C NMR (125.77 MHz, CDCl3) δ 14.1, 22.5, 24.3, 28.9, 29.7, 32.5, 37.9, 44.5, 56.1, 110.1, 119.7, 132.5, 151.1, 155.4; MS (EI) m/z (rel intensity) 91 (80), 148 (90), 199 (70), 227 (94), 298 (60).
5.5. 3-[2-Methoxy-4-(1,1-dimethylhexyl)phenyl]cyclohex-2-en-1-one (15)
To a solution of 0.100g (0.336 mmol) of 2-(4-bromo-3-methoxyphenyl)-2-methylheptane (9) in 3 mL of dry THF at -78 °C under argon was added 0.16 mL (0.403 mmol, 2.5 M solution in cyclohexane) of n-butyllithium and the mixture was stirred for 30 min. A solution of 0.048 g (0.336 mmol) of 3-ethoxycyclohex-en-1-one in 2 mL of dry THF was added dropwise and the solution was heated for 4 h at reflux. After cooling to ambient temperature the reaction was diluted with 15 mL of 10% aqueous HCl, stirred for 30 min and extracted with two portions of ether. The combined ethereal layers were washed with saturated aqueous NaHCO3, brine, dried (MgSO4) and concentrated in vacuo. The residue was chromatographed (petroleum ether/ether, 3:2) to give 0.075 g (71%) of 15 as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 0.82 (t, J = 7.0 Hz, 3H), 0.98-1.26 (m, 6H), 1.27 (s, 6H), 1.59 (t, J = 6.0, 2H), 2.05-2.13 (m, 2H), 2.21-2.29 (m, 2H), 2.74 (t, J = 5.8 Hz, 2H), 3.86 (s, 3H), 6.22 (s, 1H), 6.85 (s, 1H), 6.91 (d, J = 8.0 Hz, 1H), 7.13 (d, J = 8 Hz, 1H); 13C NMR (125.77 MHz, CDCl3) δ 14.1, 22.6, 24.4, 24.6, 28.9, 29.7, 32.4, 34.8, 37.2, 38.6, 44.7, 55.4, 112.9, 118.2, 120.4, 126.2, 133.2.2, 153.7, 155.2, 156.1, 199.6 : MS (EI). m/z (rel intensity) 243 (100), 314 (16).
5.6. 3-[2-Methoxy-4-(1,1-dimethylhexyl)phenyl]cyclohexanone (17)
To 20 ml of liquid ammonia at -78 °C was added 0.004 g (0.579 g atom) of lithium shot and the solution was stirred for 10 min. A solution of 0.072 g (0.23 mmol) of 3-[2-methoxy-4-(1,1-dimethylhexyl)phenyl]cyclohex-2-en-1-one (15) in 25 mL of dry THF was slowly added and the mixture was stirred at -78 °C for 30 min. The reaction was quenched by the addition of NH4Cl and the ammonia was evaporated at ambient temperature. The mixture was diluted with 10 mL of H2O and extracted with two portions of ether. The ethereal extracts were washed with brine, dried (MgSO4) and concentrated in vacuo. The yellow oil was purified by flash chromatography (petroleum ether/ether, 3:2) to give 0.056 g (78%) of 17 as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 0.82 (t, J = 7.1 Hz, 3H), 1.01-1.27 (m, 6H), 1.28 (s, 6H), 1.53-1.60 (m, 2H), 1.71-1.81 (m, 1H), 1.81-1.89 (m, 1H), 2.05-2.14 (m, 1H), 2.12-2.18 (m, 1H), 2.33-2.42(m, 1H), 2.42-2.48 (m, 1H), 2.48-2.56 (m, 1H), 2.57-2.63 (m, 1H), 2.99 (dddd, J =3.8, 3.9, 11.9, 11.9 Hz, 1H), 3.84 (s, 3H) 7.14 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H); 13C NMR (125.77 MHz, CDCl3) δ 14.1, 22.5, 24.3, 25.6, 28.9, 32.5, 32.8, 37.4, 41.2, 44.2, 44.6, 49.0, 55.4, 113.3, 119.1, 126.2, 126.6, 148.2, 159.6, 211.4 : MS (EI). m/z (rel intensity) 60 (25), 245 (100), 316 (17).
5.7. (1R*,3S*)3-[2-Methoxy-4-(1,1-dimethylhexyl)phenyl]cyclohexanol (JWH 440, 7)
To a solution of 0.054 g (0.17 mmol) of 3-[2-methoxy-4-(1,1-dimethylhexyl)phenyl]cyclohexanone (17) in 6 mL of dry ethanol at 0 °C, was slowly added 0.056 g (0.177 mmol) of NaBH4. The mixture was warmed to ambient temperature, stirred for 2 h and quenched by the addition of 10 mL of 10% HCl. The solution was extracted with two portions of ether and the ethereal extracts were washed with saturated NaHCO3, brine, dried (MgSO4) and concentrated in vacuo. The yellow oil was chromatographed (petroleum ether/ether, 1:1) to give 0.038 g (68%) of JWH 440 (7) as a colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.88 (t, J = 7.0 Hz, 3H), 1.01-1.28 (m, 6H), 1.29 (s, 6H), 1.45-1.52 (m, 2H), 1.57-1.63 (m, 4H), 1.78-1.92 (m, 2H), 2.04-2.19 (m, 2H), 2.99 (t, J = 12.0 Hz, 1H), 3.75-3.83 (m, 1H), 3.81 (s, 3H), 7.84 (s, 1H), 6.87 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 8 Hz, 1H); 13C NMR (125.77 MHz, CDCl3) δ 14.1, 22.6, 24.4, 24.6, 28.9, 29.7, 31.9, 32.6, 34.9, 35.6, 37.7, 42.1, 44.7, 55.4, 71.3, 108.4, 117.9, 125.9, 131.2, 148.9, 156.4; MS (EI). m/z (rel intensity) 233 (33), 301 (65), 319 (99); HRMS calcd for C21H35O2 319.2637, found 319.2629.
5.8. (1S*,3S*)3-[2-Methoxy-4-(1,1-dimethylhexyl)phenyl]cyclohexanol (JWH 442, 19)
To a solution of 0.020 g (0.063 mmol) of 3-[2-methoxy-4-(1,1-dimethylhexyl)phenyl]cyclohexanone (17) in 1 mL of dry THF at -78 °C was added 0.27 mL (0.266 mmol, 1.0 M solution in THF) of L-Selectride and the mixture was stirred for 3 h, warmed to ambient temperature and stirred for 4 h. To the reaction mixture was added 4 mL of H2O, 3 mL of ethanol, 1 ml of 1 M aqueous NaOH and 1 mL of 30% aqueous H2O2. The reaction mixture was stirred for 30 min, extracted with two portions of ether and the ethereal layers were washed with brine, dried (MgSO4), and concentrated in vacuo. The yellow oil was purified by flash chromatography (petroleum ether/ether, 1:1) to give 0.011 g (55%) of JWH 442 (19); 1H NMR (500 MHz) δ 0.88 (t, J = 7.0 Hz, 3H), 1.01-1.28 (m, 6H), 1.29 (s, 6H), 1.45-1.52 (m, 2H), 1.57-1.63 (m, 4H), 1.78-1.92 (m, 2H), 2.04-2.19 (m, 2H), 3.37 (t, J = 12.0 Hz, 1H), 4.24 (bs, 1H), 3.81 (s, 3H), 6.83 (s, 1H), 6.87 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 8.0 Hz, 1H); 13C NMR (125.77 MHz, CDCl3) δ 14.1, 22.6, 24.4, 24.6, 28.9, 29.7, 31.9, 32.4, 34.9, 35.6, 37.7, 42.1, 44.7, 55.4, 67.2, 108.4, 117.9, 125.8, 131.9, 148.7, 156.4; MS (EI) m/z (rel intensity) 233 (33), 301 (65), 319 (99); HRMS calcd for C21H35O2 319.2637, found 319.2630.
5.9. (1R*,3S*)3-[2-Methoxy-4-(1,1-dimethylheptyl)phenyl]cyclohexanol (JWH-441, 8)
2-(3,5-dihydroxyphenyl)-2-methyl octane, provided by Norac Pharma, was converted to 1R*,3S*)3-[2-methoxy-4-(1,1-dimethylheptyl)phenyl]cyclohexanol (8) by the procedure described for the synthesis of JWH-440. From 2.2 g (9.31 mmol) of 2-(3,5-dihydroxyphenyl)-2-methyl octane there was obtained 0.126 g (5.7% for eight steps) of pure JWH-441 after flash chromatography (petroleum ether/ether, 1:1); 1H NMR (500 MHz) δ 0.89 (t, J = 7.0 Hz, 3H), 1.01-1.28 (m, 8H), 1.29 (s, 6H), 1.45-1.52 (m, 4H), 1.57-1.63 (m, 2H), 1.79-1.94 (m, 2H), 2.03-2.17 (m, 2H), 2.99 (t, J = 12.0 Hz, 1H), 3.74-3.83 (m, 1H), 3.82 (s, 3H), 6.83 (s, 1H), 6.87 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 8.0 Hz, 1H), 13C NMR (125.77 MHz, CDCl3) δ 14.1, 22.7, 24.6, 24.7, 28.9, 29.7, 30.0, 31.9, 34.9, 35.6, 37.7, 42.1, 44.7, 55.4, 71.3, 108.4, 118.0, 125.8, 131.6, 148.9, 156.8; MS (EI). m/z (rel intensity) 247 (18), 315 (43), 333 (99); HRMS calcd for C22H37O2 333.2794, found 333.2687.
5.10. Receptor Binding Experiments
5.10.1. Materials
Frozen whole brains of male Sprague-Dawley rats were obtained from Harlan (Dublin, VA). CP-55,940 was provided by Pfizer (Groton, CT). [3H]CP-55,940 was purchased from NEN Life Science Products, Inc. (Boston, MA). Lipofectamine reagent was purchased from Life Technologies (Gaithersburg, MD). Human CB2 cDNA was provided by Dr. Sean Munro (MRC Lab, Cambridge, UK). DMEM and geneticin was purchased from (GIBCO BRL, Grand Island, NY). Fetal clone II was purchased from Hyclone Laboratories, Inc. (Logan, UT). Aquasil was purchased from Pierce (Rockford, IL). GF/C glass-fiber filters (2.4 cm) were purchased from Baxter (McGaw Park, IL). Polyethylenimine and bovine serum albumin were purchased from Sigma Chemical Co. (St. Louis, MO). Scintillation vials and Budget Solve scintillation fluid were purchased from RPI Corp. (Mount Prospect, IL).
5.10.2. Development of hCB2-CHO Cell Line
Chinese hamster ovary cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal clone II and 5% CO2 at 37 °C in a Forma incubator. Cell lines were created by transfection of CB2pcDNA3 into CHO cells by the Lipofectamine reagent. Stable transformants were selected in growth medium containing geneticin (1 mg/mL, reagent). Colonies of about 500 cells were picked (about 2 weeks post transfection) and allowed to expand, then tested for expression of receptor mRNA by northern blot analysis. Cell lines containing moderate to high levels of receptor mRNA were tested for receptor binding properties. Transfected cell lines were maintained in DMEM with 10% fetal clone II plus 0.3-0.5 mg/mL geneticin and 5% CO2 at 37 °C in a Forma incubator.
5.10.3. Membrane Preparation
hCB2-CHO cells were harvested in phosphate-buffered saline containing 1 mM EDTA and centrifuged at 500g. Cell pellets (for CB2) or whole rat brains (for CB1) were homogenized in 10 mL of solution A (50 mM Tris-HCl, 320 mM sucrose, 2 mM EDTA, 5 mM MgCl2, pH 7.4). The homogenate was centrifuged at 1,600 × g (10 min), the supernatant saved, and the pellet washed three times in solution A with subsequent centrifugation. The combined supernatants were centrifuged at 100,000 × g (60 min). The (P2 membrane) pellet was resuspended in 3 mL of buffer B (50 mM Tris-HCl, 1 mM EDTA, 3 mM MgCl2, pH 7.4) to yield a protein concentration of approximately 1 mg/mL. The tissue preparation was divided into equal aliquots, frozen on dry ice, and stored at -70 °C.
5.11. Competition Binding Assays
5.11.1. CB1 Assay
[3H]CP-55,940 binding to P2 membranes was conducted as described elsewhere,49 except whole brain (rather than cortex only) was used. CP-55,940 and all cannabinoid analogs were prepared by suspension in assay buffer from a 1 mg/mL ethanolic stock without evaporation of the ethanol (final concentration of no more than 0.4%). Displacement curves were generated by incubating drugs with 1 nM of [3H]CP-55,940. [3H]CP-55,940 bound to rat brain membranes with a KD value of 0.68 ± 0.07 nM and a Bmax value of 1.7 ± 0.11 pmol/mg. The assays were performed in triplicate, and the results represent the combined data from three individual experiments.
5.11.2. CB2 Assay
Binding was assayed by a modification of Compton et al.44 CP-55,940 and all cannabinoid analogs were prepared by suspension in assay buffer from a 1 mg/mL ethanolic stock without evaporation of the ethanol (final concentration of no more than 0.4%). The incubation was initiated by the addition of 40-50 μg membrane protein to silanized tubes containing [3H]CP-55,940 (102.9 Ci/mmol) and a sufficient volume of buffer C (50 mM Tris-HCl, 1 mM EDTA, 3 mM MgCl2, and 5 mg/mL fatty acid free BSA, pH 7.4) to bring the total volume to 0.5 mL. The addition of 1 μM unlabelled CP-55,940 was used to assess nonspecific binding. Following incubation (30 °C for 1 hour), binding was terminated by the addition of 2 mL of ice cold buffer D (50 mM Tris-HCl, pH 7.4, plus 1 mg/mL BSA) and rapid vacuum filtration through Whatman GF/C filters (pretreated with polyethyleneimine (0.1%) for at least 2 hours). Tubes were rinsed with 2 mL of ice cold buffer D, which was also filtered, and the filters subsequently rinsed twice with 4 mL of ice cold buffer D. Before radioactivity was quantitated by liquid scintillation spectrometry, filters were shaken for 1 hr in 5 mL of scintillation fluid. [3H]CP-55,940 bound to hCB2-CHO cells membranes with a KD value of 0.45 ± 0.07 nM and a Bmax value of 2.93 ± 0.06 pmol/mg.
5.11.3 Data Analysis
Competition assays were conducted with 1 nM [3H]CP-55,940 and 6 concentrations (0.1 nM to 10 μM displacing ligands). Displacement IC50 values were originally determined by unweighted least-squares linear regression of log concentration-percent displacement data and then converted to Ki values using the method of Cheng and Prusoff.50 All experiments were performed in triplicate and repeated 3-6 times. All data are reported as mean values ± SEM.
5.12. Computational methods
5.12.1. Conformational analysis
A conformational search for each ligand using the MCMM/LCMS method as implemented in Macromodel 9.6 (Schrödinger LLC, NY, USA) was conducted. The OPLS2005 forcefield was used with a distance dependent dielectric and nonbonded cutoffs of 20Å, 8.0 Å, and 4.0 Å respectively for electrostatic, VdW, and H-bond interactions. 50,000 structures were evaluated within a 10 kcal/mol window above the lowest energy structure found. All rotatable bonds were varied during the conformational search. Elimination of duplicate structures was performed by considering structures to be different if the maximum atom deviation for any pair of corresponding atoms exceeded 0.5 Å.
5.12.2. CB2 Receptor Docking Studies
The C ring atoms of each ligand were superimposed on the AM841 C ring in our hCB2 receptor model.44 Modifications to ligand torsions were performed using interactive computer graphics in order to fit the compound into the AM841 binding site. Steric overlaps were eliminated using interactive computer graphics. AM841 was then removed from the complex in preparation for subsequent minimization.
5.12.3. Ligand/CB2 Receptor Minimization
Minimization of the hCB2-receptor bundle-JWH compound complex was performed in Macromodel 9.6 (Schrödinger LLC, NY, USA) using the OPLS2005 forcefield with a distance dependent dielectric and nonbonded cutoffs of 20 Å, 8.0 Å, and 4.0 Å respectively for electrostatic, VdW, and H-bond interactions. All residues except D2.50(80), K3.28(109), and D(275) were neutralized during the minimization. To preserve the TMH backbone conformation, torsional restraints of 100 kcal/mol were applied to the phi, psi, and omega dihedrals of each CB2 TMH. To preserve the loop conformations, non-moving fixed atom restraints as implemented in Macromodel were applied to the loop backbone atoms. The full receptor/ligand complex was energy minimized with the Polak-Ribier conjugate gradient method until an energy gradient of 0.1 kcal/mol·Å2 was reached. Approximately 500 steps were required to reach the specified gradient since the receptor/ligand complex was already very near a minimum in each case.
5.12.4. Energy Expense Assessments for Docked Ligands
To calculate the energy difference between the global minimum energy conformation of each compound and its final docked conformation after energy minimization of the ligand/receptor complex, single point energies of the final docked conformation and the global minimum structure were calculated using the OPLS2005 forcefield. A distance dependent dielectric, 8.0 Å extended nonbonded cutoff, 20.0 Å electrostatic cutoff, and 4.0 Å H-bond cutoff were used in the calculation.
5.12.5. Assessment of Pairwise Interaction Energies
After defining the atoms of a ligand in the final energy minimized CB2 R* complex as one group (group 1) and the atoms corresponding to a residue that lines the binding site in the final ligand/CB2 R* energy minimized complex as another group (group 2), Macromodel (version 9.6) was used to output the pair-wise interaction energy (coulombic and van der Waals) for a given pair of atoms. The pairs corresponding to group 1 (ligand) and group 2 (residue of interest) were then summed to yield the interaction energy between the ligand and that residue. A total interaction energy for each ligand with CB2 R* was calculated by summing the pairwise interaction energies for all residues in the binding site of that ligand and adding to this sum, the conformational energy expense for the ligand (see 5.12.4 above).
5.12.6. CB1 Receptor Docking Studies
The C ring atoms of each ligand were superimposed on the HU-210 C ring in our hCB1 receptor model.47 Modifications to ligand torsions were performed using interactive computer graphics to attempt fitting each ligand into the HU-210 binding site. It was not possible to eliminate steric overlaps for any of these compounds. Therefore, no subsequent energy minimzation was performed.
Acknowledgments
The work at Clemson was supported by grant DA003590 to JWH, that at Virginia Commonwealth University by grant DA003672 to JLW and that at the University of North Carolina at Greensboro by grants DA003934 and DA021358 all from the National Institute on Drug Abuse
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Gaoni Y, Mechoulam R. J Am Chem Soc. 1964;86:1648. [Google Scholar]
- 2.Razdan RK. Pharmacol Rev. 1986;38:75. [PubMed] [Google Scholar]
- 3.Rapaka RS, Makriyannis A. Structure-Activity Relationships of the Cannabinoids. National Institute on Drug Abuse; Rockville, MD: 1987. (NIDA Research Monograph 79). [Google Scholar]
- 4.Mechoulam R, Devane WA, Glaser R. In: Marijuana/Cannabinoids: Neurobiology and Neurophysiology. Murphy L, Bartke A, editors. CRC Press; Boca Raton: 1992. pp. 1–33. [Google Scholar]
- 5.Johnson MR, Melvin LS, Milne GM. Life Sci. 1982;31:1703. doi: 10.1016/0024-3205(82)90190-4. [DOI] [PubMed] [Google Scholar]
- 6.Weissman A, Milne GM, Melvin LS., Jr J Pharmacol Exp Ther. 1982;223:516. [PubMed] [Google Scholar]
- 7.Melvin LS, Johnson MR, Herbert CA, Milne GM, Weissman AA. J Med Chem. 1984;27:67. doi: 10.1021/jm00367a013. [DOI] [PubMed] [Google Scholar]
- 8.Johnson MR, Melvin LS. In: Cannabinoids as Therapeutic Agents. Mechoulam R, editor. CRC Press; Boca Raton, FL: 1986. pp. 121–145. [Google Scholar]
- 9.Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. Mol Pharmacol. 1988;34:605. [PubMed] [Google Scholar]
- 10.Huffman JW, Lainton JAH. Curr Med Chem. 1996;3:101. [Google Scholar]
- 11.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, De Costa DR, Rice KC. Proc Natl Acad Sci USA. 1990;87:1932. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pertwee RG. Curr Med Chem. 1999;6:635. [PubMed] [Google Scholar]
- 13.Munro S, Thomas KL, Abu-Shar M. Nature (London) 1993;365:61. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 14.Klein TW, Friedman H, Specter S. J Neuroimmunol. 1998;83:102. doi: 10.1016/s0165-5728(97)00226-9. [DOI] [PubMed] [Google Scholar]
- 15.Galiègue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P. Eur J Biochem. 1995;232:54. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 16.Condie R, Herring A, Koh WS, Lee M, Kaminski NE. J Biol Chem. 1996;271:13175. doi: 10.1074/jbc.271.22.13175. [DOI] [PubMed] [Google Scholar]
- 17.Pettit DA, Anders DL, Harrison MP, Cabral GA. Adv Exp Med Biol. 1996;402:119–129. doi: 10.1007/978-1-4613-0407-4_17. [DOI] [PubMed] [Google Scholar]
- 18.Schatz AR, Lee M, Condie RB, Pulaski JT, Kaminski NE. Toxicol Appl Pharmacol. 1997;142:278. doi: 10.1006/taap.1996.8034. [DOI] [PubMed] [Google Scholar]
- 19.Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, Glass M, Zimmer A. Eur J Pharmacol. 2000;396:141. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez C, de Ceballos ML, Gómez del Pulgar T, Rueda D, Corbacho C, Velasco G, Galve-Roperh I, Huffman JW, Ramón y Cajal S, Guzmán M. Cancer Res. 2001;61:578. [PubMed] [Google Scholar]
- 21.Casanova ML, Blazquez C, Martinez-Palacio J, Villanueva C, Fernandez-Acenero MJ, Huffman JW, Jorcano JL, Guzman M. J Clin Invest. 2003;111:43. doi: 10.1172/JCI16116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanus L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, Pertwee RG, Ross RA, Mechoulam R, Fride E. Proc Natl Acad Sci USA. 1999;96:1422. doi: 10.1073/pnas.96.25.14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conti S, Costa B, Colleoni M, Parolaro D, Giagnoni G. Br J Pharmacol. 2002;135:181. doi: 10.1038/sj.bjp.0704466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Malan TP, Ibrahim MM, Vanderah TW, Makriyannis A, Porreca F. Chem Phys Lipids. 2002;121:191. doi: 10.1016/s0009-3084(02)00155-x. [DOI] [PubMed] [Google Scholar]; (b) Malan TP, Ibrahim MM, Deng H, Liu Q, Main HP, Vanderah T, Porreca F, Makriyannis A. Pain. 2001;93:239. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- 25.Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, Vanderah TW, Lal J, Porreca F, Makriyannis AM, Malan TP. Proc Natl Acad Sci USA. 2003;100:10529. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O'Donnell D. Eur J Neurosci. 2003:2750. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]
- 27.Kehl LJ, Hamamoto DT, Wacnik PW, Croft DL, Norsted BD, Wilcox GL, Simone DA. Pain. 2003;103:175. doi: 10.1016/s0304-3959(02)00450-5. [DOI] [PubMed] [Google Scholar]
- 28.Clayton N, Marshall FH, Bountra C, O'Shaughnessy CT. Pain. 2002;96:253. doi: 10.1016/S0304-3959(01)00454-7. [DOI] [PubMed] [Google Scholar]
- 29.Giblin GMP, O'Shaughnessy CT, Naylor A, Mitchell WL, Eatherton AJ, Slingsby BP, Rawlings DA, Goldsmith P, Brown AJ, Haslam CP, Clayton NM, Wilson AW, Chessel IP, Wittington AR, Green R. J Med Chem. 2007;50:2597. doi: 10.1021/jm061195+. [DOI] [PubMed] [Google Scholar]
- 30.Mackie K. Annu Rev Pharmacol Toxicol. 2006;46:101. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- 31.Pacher P, Batkai S, Kunos G. Pharmacol Rev. 2006;58:389. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, Martin BR. Bioorg Med Chem. 1999;7:2905. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- 33.WIley JL, Jefferson RG, Griffin G, Liddle J, Yu S, Huffman JW, Martin BR. Pharmacol. 2002;66:89. doi: 10.1159/000065631. [DOI] [PubMed] [Google Scholar]
- 34.Gallant M, Dufresne C, Gareau Y, Guay D, Leblanc Y, Prasit P, Rochette C, Sawyer N, Slipetz DM, Tremblay N, Metters KM, Labelle M. Bioorg Med Chem Lett. 1996;6:2263. [Google Scholar]
- 35.Huffman JW, Thompson ALS, Wiley JL, Martin BR. Bioorg Med Chem. 2008;16:322. doi: 10.1016/j.bmc.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huffman JW, Bushell SM, Miller JRA, Wiley JL, Martin BR. Bioorg Med Chem. 2002;10:4119. doi: 10.1016/s0968-0896(02)00331-0. [DOI] [PubMed] [Google Scholar]
- 37.(a) Huffman JW, Yu S, Showalter V, Abood ME, Wiley JL, Compton DR, Martin BR, Bramblett RD, Reggio PH. J Med Chem. 1996;39:3875. doi: 10.1021/jm960394y. [DOI] [PubMed] [Google Scholar]; (b) Yu S. PhD dissertation. Clemson University; 1997. [Google Scholar]; (c) Itagaki N, Sugahara T, Iwabuchi Y. Org Lett. 2005;7:4181. doi: 10.1021/ol051570c. [DOI] [PubMed] [Google Scholar]
- 38.Jones RG, Gilman H. Org React. 1951;6:339. [Google Scholar]
- 39.Caine D. Org React. 1976;23:1. [Google Scholar]
- 40.House HO. Modern Synthetic Reactions. W. A. Benjamin; Menlo Park, CA: 1972. pp. 62–65. [Google Scholar]
- 41.The notation R* or S* indicates relative configuration in racemic compounds. See Eliel EL, Wilen SH, Mander LN. Stereochemistry of Organic Compounds. John Wiley and Sons; New York: 1994. pp. 1205–1206.
- 42.Bhacca NS, Williams DH. Applications of NMR Spectroscopy in Organic Chemistry. Holden-Day; San Francisco: 1966. pp. 77–83. [Google Scholar]
- 43.Brown HC, Krisnamurthy S. J Am Chem Soc. 1972;94:7159. [Google Scholar]
- 44.Compton DR, Rice KC, De Costa BR, Razdan RK, Melvin LS, Johnson MR, Martin BR. J Pharmacol Exp Ther. 1993;265:218. [PubMed] [Google Scholar]
- 45.Showalter VM, Compton DR, Martin BR, Abood ME. J Pharmacol Exp Ther. 1996;278:989. [PubMed] [Google Scholar]
- 46.Pei Y, Mercier RW, Anday JK, Thakur GA, Zvonok AM, Hurst D, Reggio PH, Janero DR, Makriyannis A. Chem Biol. 2008;15:1207. doi: 10.1016/j.chembiol.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapur A, Hurst DP, Fleischer D, Whitnell R, Thakur GA, Makriyannis A, Reggio PH, Abood ME. Mol Pharmacol. 2007;71:1512. doi: 10.1124/mol.107.034645. [DOI] [PubMed] [Google Scholar]
- 48.Marriott KC, Huffman JW, Wiley JL, Martin BR. Bioorg Med Chem. 2006;14:2386. doi: 10.1016/j.bmc.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 49.Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK, Johnson MR, Melvin LS, Mechoulam R, Ward SJ. Pharmacol Biochem Behav. 1991;40:471. doi: 10.1016/0091-3057(91)90349-7. [DOI] [PubMed] [Google Scholar]
- 50.Cheng YC, Prusoff WH. Biochem Pharmacol. 1973 doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]