

Abstract
The use of mesoporous silicon particles for drug delivery has been widely explored thanks to their biodegradability and biocompatibility. The ability to tailor the physicochemical properties of porous silicon at the micro and nano scale confers versatility to this material. We present a method for the fabrication of highly reproducible, monodisperse mesoporous silicon particles with controlled physical characteristics through electrochemical etch of patterned silicon trenches. We tailored particle size in the micrometer range and pore size in the nanometer range, shape from tubular to discoidal to hemispherical, and porosity from 46% to over 80%. In addition, we correlated the properties of the porous matrix with the loading of model nanoparticles (Q-dots) and observed their three-dimensional arrangement within the matrix by transmission electron microscopy tomography. The methods developed in this study provide effective means to fabricate mesoporous silicon particles according to the principles of rational design for therapeutic vectors and to characterize the distribution of nanoparticles within the porous matrix
Keywords: Porous Silicon, Microfabrication, Nanoparticles, Drug Delivery, Multi-stage Delivery System
Introduction
Exploratory work demonstrating porous silicon biodegradability[1,2] and biocompatibility[3-7] opened the avenue for its use in implantable devices,[8] drug delivery systems,[9,10] and scaffolds for tissue engineering.[11,12] Porous Silicon Particles (PSPs) have been shown to tune drug solubility,[10] increase bioavailability,[13] and enable sustained release.[9] Our group recently presented a multi-stage drug delivery system (MDS) based on PSPs.[9,14,15] The prototypical MDS consists of different types of therapeutic and diagnostic second stage nanoparticles (NPs) (liposomes, dendrimers, quantum-dots, gold nanoshells, etc) loaded into the pores of an optimally engineered first stage PSP. The use of PSPs for MDS provides the ability to simultaneously carry, protect and release in a controlled fashion a variety of NPs with very different features and functions.
In the last two decades, a vast assortment of NPs has been developed for both therapeutic intervention and imaging.[16-18] They differ significantly in terms of size, composition, and structure, and the NPs loading within the pores of the PSP strictly depends on their relative physicochemical compatibility. Consequently, an efficient MDS requires first stage PSP carriers with variable porous structures capable of hosting and retaining a multitude of different second stage NPs. Furthermore, the interaction of the MDS with target cells[19] and the rates and mechanisms of cellular internalization[15,20] are strongly influenced by the size and shape of the PSPs. Similarly, the PSPs ability to navigate the vasculature proficiently, as dictated by their margination characteristics, depend on their size, geometry and aspect ratio.[21] Finally, the pharmacokinetics of the therapeutic payload can be linked to PSPs biodegradation rates which are determined by their pore size and porosity.[9, 22]
Although some results have been achieved in patterning pSi layers into particles,[23, 24] the necessary control of the features at the micro- and nano-scale was not yet satisfactory. The current standard technique for the fabrication of PSPs consists of sonication or ball milling of porous silicon layers resulting in polydisperse fragments of random size and shape.[10,25,26] In this work, we present a microfabrication technique that enables the fine tailoring of monodisperse PSPs at the sub-micron scale. The technique we describe provides precise control over the key parameters necessary to tune the interaction of PSPs with both biological systems and nanoparticulates: size, shape, and aspect ratio, as well as porosity, pore size, and pore morphology. We furthermore demonstrate the use of this system as a carrier for NPs and describe their loading inside the pores by direct visualization through electron microscopy.
Results and Discussion
A 100mm p++ silicon wafer was patterned by photolitography and etched through a silicon nitride sacrificial layer (figure 1a). The pattern was transferred into the silicon layer by various etch methodologies, determining different shapes of the trench that nucleated PSP with different profiles (Figure 1 b). Subsequently, an electrochemical etch process formed the PSP of desired porosity, pore size and thickness. Following the PSP formation, a high current density was applied to create a highly porous layer at the PSP-wafer interface (release layer) (Figure 1 c). The silicon nitride layer was removed in HF(Figure 1 d), the PSPs were released from the substrate, suspended in isopropanol (IPA) and preserved at 20°C in a controlled temperature environment.
Figure 1.
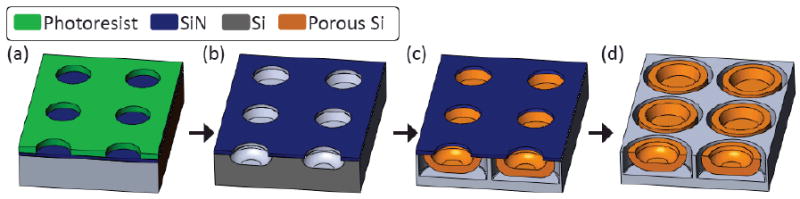
Schematic depiction of the fabrication process. (a) Pattern transfer to the photoresist layer on top of the sacrificial SiN layer. (b) Trench formation in the Si substrate through combination of dry and wet etch. (c) Formation of the PSPs and release layer following anodic etch. (d) PSPs ready to be released by ultrasonication in isopropanol following stripping of the SiN mask
This method, further described in the experimental section, allowed for the microfabrication of a variety of well controlled and reproducible PSPs (Figure 2, Figure 3). The shape of the PSP was determined by the unit shape on the photolithographic mask employed, as the nucleation site retained the exact shape of the lithographic pattern. The shape of the electrochemical etch undercut, constituting the external corona, was determined by the current distribution between the nearest neighbor PSPs and was independent of the profile of the trench. Statistical analysis of Scanning Electron Microscopy (SEM) micrograph of 160 PSPs from independently produced lots confirmed the accuracy and reproducibility of the process. The analysis showed PSPs with a circular nucleation site with 2±0.1 μm, 1±0.1 μm or 0.67±0.08 μm diameter surrounded by a rounded square external corona with 3.2±0.1 μm, 1.6±0.1 μm or 0.97 ±0.08 μm edge length respectively originated from 2 μm, 1 μm or 0.6 μm circular lithographic patterns (Figure 3 a,b). The thickness of the electrochemical etch undercut constituting the external corona was 600 nm, 300 nm or 150 nm for the 2 μm, 1 μm or 0.6 μm patterns respectively. The nucleation site circularity was 0.98±0.05 and the ratio of the particles’ width to its length was 0.98±0.06. The PSP profile was determined by the nucleation trench formed into the silicon prior to its porosification. SEM micrograph analysis of 3.2 μm PSPs showed that wet etch of the masking layer yielded flat disk PSPs (Figure 3 c,d); 4 min CF4 etch formed a 180 nm trench into the silicon that yielded discoidal PSPs (Figure 3 e,f); 4 min SF6 etch formed a 475 nm quasi-isotropic trench that yielded hemispherical PSPs (Figure 3 g,h); 6 min HBr etch followed by 1 min SF6 etch formed a 975 nm anisotropic trench that yielded tubular PSPs (Figure 3 i,j). The combination and timing of different etch procedures allowed a tight control over the PSP final shape and aspect ratio. The undercut for the different shapes was maintained at 600 nm to guarantee a uniform PSP size of 3.2 μm. The resulting porous layer thickness at the bottom of the trench was 605 nm for wet etch, 795 nm for CF4 etch, 720 nm for SF6 etch and 780 nm for HBr+SF6 etch showing a slight dependence of the electrochemical etch isotropy on the trench morphology. The PSP aspect ratio was tuned from 1.8 for tubular PSPs to 5.3 for flat PSPs.
Figure 2.

Scanning electron micrographs of large clusters of PSPs. The PSPs are characterized by size and shape uniformity. (a) HBr6 etched tubular shaped MP3 PSPs still attached to silicon substrate before the removal of the silicon nitride sacrificial layer and the PSPs subsequent release. (b) overview of a large cluster of SF6 etched Bowl shaped MP3 PSPs following release by sonication in IPA. (c) Close-up of a small cluster of CF4 etched discoidal shaped MP3 PSPs following release by sonication in IPA. All scale bars are 1 μm
Figure 3.

SEM micrographs of PSPs. (a) Digital composition of three distinct SEM micrographs showing the nucleation side of a 3.2μm, 1.6 μm and 0.97 μm PSP: (1) the external corona and (2) the nucleation site. (b) Digital composition of three distinct SEM micrographs showing the release side of a 3.2μm, 1.6 μm and 0.97 μm PSP. Section along the diameter and the lateral view of: a flat disk PSP obtained by wet etch of the masking layer (c, d respectively); a discoidal PSP obtained by trench formation by CF4 RIE (e, f respectively); a hemispherical PSP obtained by trench formation by SF6 RIE (g, h respectively); a tubular PSP obtained by trench formation by combination of HBr and SF6 RIE (i, j respectively); a XLP1 PSP (k, l respectively). (m) Close-up view of the multilayer structure of an XLP1 particle, from top to bottom: (1) SP layer, (2) transitional layer, (3) XLP layer. (c-m) Nucleation side is at the top of the figure and release side at the bottom. All scale bars are 200nm.
Different PSP types were characterized by N2 desorption isotherms to evaluate their porosity and pore size distribution. The PSPs used for this analysis originated from independent production lots. The analysis proved that each one of the specific pore sizes could be consistently reproduced as demonstrated by the absence of multiple peaks in the pore distribution curves. Figure 4 summarizes the Barrett-Joyner-Halenda (BJH) model analysis of the desorption curves. The average pore size ranged from 5.9±2.1 nm (Small Pores PSPs: SPs) to 51.3 ± 28.7 nm and larger (eXtra Large Pores PSPs: XLPs) depending on the porosification conditions (Figure 4 b). PSPs with pore sizes of 10.1 ± 3.9 nm, 15.2 ± 6.3 nm and 17.9 ± 6.5 nm (Medium Pores PSPs: MPs) as well as PSP with pore sizes of 20.9±7.9 and 26.3±14.6 (Large Pores PSPs: LPs), were also obtained demonstrating the ability to finely control pore size in a PSP (Figure 4 b-d). As previously reported in the literature,[27] the standard deviation increased for larger pore PSPs. Porosity was positively correlated with pore size and ranged from 47.5% for SP to 82.0% for XLP1. Intermediate values were 46.3%, 51.1%, 52.7%, 55.7%, and 66.1% for MP1, MP2, MP3, LP1 and LP2 respectively (Figure 4 b-d). Evaluation of porosity and pore size of XLP1 PSPs was underestimated since the BJH model applies only to the 3-50nm range[27] (Figure 4 c). XLP2 pore size and porosity could not be measured by N2 isotherms and were instead characterized by SEM. Pore morphology varied from randomly oriented densely branched pores for SPs to structures with short branches for MPs to smooth parallel pores for LPs and larger pores (Figure 4 a), as reported in the literature.[28] We also demonstrated the ability to combine multiple porous layers with different pore sizes in a single PSP. This multi-layered structure was crucial to provide the mechanical stability needed to fabricate the XLPs. XLPs were produced through a continuous electrochemical etch process by means of a time varying etch current. The initial SP stabilization layer (approximately 20nm thick), was followed by a transitional layer from SP to XLP (approximately 70nm thick); these two layers guaranteed the PSP structural integrity. The XLP layer of desired thickness and pore size was then formed at the bottom of the transitional layer (Figure 3 k,l,m).
Figure 4.
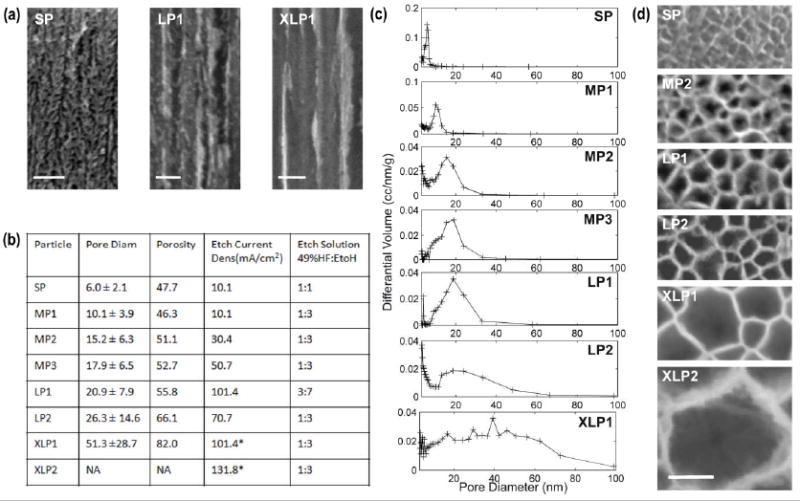
Analysis of the porous structure of the PSPs. (a) SEM micrographs of the PSP porous structure cross section along the pore axis, perpendicular to the Si (100) surface for the most representative pore structures investigated. (b) Summary of the BJH analysis and the electrochemical etch parameters employed in the porosification of the PSPs. The etch current density reported for XLPs refers to the XLP layer. (c) BJH model estimate of the differential volume of pores in the 3nm to 100nm range for all the different types of PSPs investigated. BJH analysis of XLP2 PSPs is not shown due to the inadequacy of the model. (d) SEM micrographs of the central bottom region of the PSP for the most representative pore sizes investigated (all scale bars in the figure are100nm).
We furthermore exploited the positioning of a SP layer within a multi-layer PSP structure to obtain functionally different porous structures. During the standard electrochemical etch, a thin layer of nanopores (nucleation layer) was formed in the initial phase of the etch due to transient currents, effectively capping the pores on the nucleation site. The capping nucleation layer was removed by means of a short CF4 etch following the electrochemical process to expose the underlying porous structure and obtain PSPs with pass-through pores of constant diameter (Figure 5). Inverted PSPs with the nucleation layer removed and a capping layer on the opposite side were obtained combining the short CF4 etch with a two layer PSP structure terminated by an SP layer. The three alternative porous structures obtained (opening on back side, pass through, opening on front side) allow controlling the direction of the loading of NPs within the porous matrix and their subsequent release.
Figure 5.

A short CF4 plasma etch following the formation of the PSP results in the removal of the nucleation layer and formation of pass-through pores. (a) Schematic depiction of a PSP as anodically etched; the small pore nucleation layer is indicated in red. (b) Schematic depticion of a PSP following 15s CF4 plasma etch. (c-f) 45° tilt SEM cross section of a PSP with (a,c) and without (b,d) nucleation layer. No damage to the PSP or the remaining pore structure due to the plasma etch is observed. All scale bars are 200 nm.
To demonstrate the key role of pore size and morphology on the control of the loading of NPs within the porous silicon matrix, we mixed carboxyl terminated quantum dots (Q-dots) with different PSPs functionalized with (3-Aminopropyl)Triethoxysilane to obtain and amine-terminated surface. After optimization of the loading protocols[9] we generated cross sections of the PSPs and analyzed them by High Angle Annular Dark Field Scanning Transmission Electron Microscopy (HAADF-STEM) coupled with mapping Energy Dispersive X-ray Spectroscopy (EDX). Figure 6 shows how differently the Q-dots diffuse within the pores or adhered on the surface of the PSPs as a function of pore size. HAADF-STEM imaging showed surface functionalized CdSe Q-dots (15nm hydrodynamic diameter, 5 nm metal core diameter) as electron dense spots of approximately 5 nm diameter within the Si matrix. Q-dots accumulated on the external surfaces of MPs (and SPs, data not shown) but did not penetrate into the porous matrix due to size exclusion (Figure 6 a). Q-dots distributed uniformly, in a close packed structure, within the porous matrix of LPs while they formed aggregate structures within the matrix of XLPs (Figure 6 a). While the electrostatic interaction between Q-dot and the PSP played an important role in the adhesion of the Q-dots on the surface of the PSP, the ability of the Q-dots to access or not the MP porous matrix was explained by size exclusion. Similarly, the formation of Q-dot aggregates observed within the matrix of XLPs suggested an important role of the electrostatic interaction between the Q-dots and the pore walls. Furthermore the interaction between the carboxyl and amine groups provided a stable bond that allowed loading and retention of the Q-dots within the porous matrix. Upon exposure of the PSP to physiological conditions the biodegradation of the PSP walls will determine the shedding of the silane allowing the free diffusion of Q-dots from the matrix that results in their sustained release.[9]
Figure 6.

(a) HAADF-STEM micrographs of PSPs loaded with 15-20 nm Q-dots. Top right SEM micrograph indicates the region analyzed by STEM-HAADF highlighted by a red box. From left to right, 15.2 nm pores MP2 PSP, 26.3 nm pores LP2 PSP and 51.3 nm pores XLP1 PSP. Arrowheads indicate Q-dots. (b) STEM-EDX mapping analysis. From left to right, top to bottom: HAADF-STEM micrograph of the region of interest, the red box indicates the region analyzed by mapping EDX; HAADF-STEM micrograph of the analyzed region; EDX intensity map for Si; EDX intensity map for Cd; EDX intensity map for Se; Typical STEM-EDX spectrum in the region of interest. All scale bars in the figure are 50 nm
The mapping EDX analysis of the porous matrix of an XLP1 (Figure 6 b) showed co-localization of Cd and Se signals with the electron dense clusters detected by the HAADF-STEM providing elemental analysis confirmation of Q-dot loading. Furthermore we employed HAADF-STEM tomographic imaging[29,30] of the porous matrix of a XLP1 (Supplementary Information Video 1, 2) to confirm spatial localization of the Q-dots within the porous matrix.
Conclusion
In this study we demonstrated the ability to microfabricate porous silicon particles with tailored features at the nanoscale. We controlled with sub micron precision the size (from 0.97 um to 3.2 um) and shape (rounded square) of the nucleation side of the PSP through the size and shape of the photolithographic pattern. We controlled the PSP profile using different silicon etch protocols, resulting in aspect ratios ranging from 1.8 to 5.3 and overall shapes ranging from discoidal to hemispherical to tubular. Control of the PSP aspect ratio can be exploited to affect their biodistribution following intravenous injection[31] while size and shape control is predicted to affect cell uptake mechanisms and kinetics.[32] Furthermore we demonstrated the ability to tailor the porous structure of a PSP in order to control the loading of second stage NPs. We proved the ability to tailor pore size (from 5 to over 50 nm) and porosity (from 47% to over 80%) while preserving the mechanical stability and integrity of the PSP. We described the effect of pore size on the loading of second stage NPs and identified the existence of an optimal pore size for their uniform, close packed loading. In conclusion, we demonstrated the ability to tune particles and pore features over the range that allows to potentially tailor PSPs as delivery agents for combinations of specific biomedically relevant NPs. The tools and protocols provided by this study constitute the foundation for the successful development and use of PSPs for the delivery of NPs in biomedical application.
Experimental Section
Sacrificial layer patterning
In a class 100 cleanroom a uniform masking layer of silicon rich silicon nitride was deposited in a Low Pressure Chemical Vapor Deposition furnace (90 nm, 835° C, 300mTorr, SiH2Cl2:NH3 80%:20%, 25 min) on a heavily doped 4inch p++ type (100) wafer with resistivity less than 0.005 ohm-cm (Silicon Quest, Inc, Santa Clara, CA). 2 μm or 1 μm diameter circles with 2 μm and 1 μm half-pitch respectively were patterned on the nitride film by means of contact photolithography (AZ-5209 photoresist, EVG-620 aligner, 40J dose). The wafer was Reactive Ion Etched (RIE) in pure CF4 plasma (Plasmatherm 790 series, 15 sccm CF4, 200 mTorr, 250 W RF Power, 2 min) to transfer the pattern into the nitride layer on the front side, avoiding overetch into the silicon.
Silicon Etch
Following the sacrificial layer patterning, a trench was formed into the silicon wafer by a second dry or wet etch step. The etch was chosen as a combination of the following processes depending on the desired final shape. SF6 (Oxford plasmalab 80 plus, 10 sccm SF6, 100 mTorr, 300 W RF Power), CF4 (Plasmatherm 790 series, 15 sccm CF4, 200 mTorr, 250 W RF Power), HBr (Plasmatherm 790 series, 4 sccm Cl2 25.4 sccm HBr, 150 mTorr, 250 W RF Power) and Buffered Oxide Etch (BoE).
Electrochemical etch
The wafer back side was exposed to CF4 RIE (Plasmatherm 790 series, 15 sccm CF4, 200 mTorr, 250 W RF Power, 2 min 30 sec) to remove the nitride film, and coated with 200 nm of sputtered aluminum (Varian Sputter) to provide a uniform backside electrical contact. A custom made poly(tetrafluoroethylene) electrochemical etch cell with an aluminum foil anode and a platinum mesh cathode was used for anodic electrochemical etch. The anode was contacted to the aluminum coated backside of the wafer and not exposed to the etchant solution, while the cathode faced the patterned wafer front side at a fixed distance. The wafer was electrochemically etched for the time necessary to obtain the desired PSP thickness applying the current density and HF:Ethanol ratio described in Figure 4 b. Following formation of the PSP, a high current density was applied forming a high porosity layer at the PSP-wafer interface (release layer).
PSPs release
The silicon nitride masking layer was stripped in 49% HF for 30 minutes. The wafer was rinsed and spin dried. The wafer was then placed in a glass crystallization dish filled with 40ml of IPA and sonicated for 1 minute to detach the PSPs from the bulk wafer. Finally the release suspension was transferred to a 50ml low retention tube and stored in a controlled temperature environment at 20°C.
Nitrogen Absorption/Desorption
The surface area and pore size of the PSPs were measured using N2 adsorption-desorption isotherms on a Quantachrome Autosorb-3B Surface Analyzer. To prepare the sample, PSPs (10mg) suspended in IPA from 10 independent processes were mixed and centrifuged, and the supernatant was removed leaving about 3ml of PSPs-IPA suspension. The suspension was transferred to a sample cell in a vacuum oven at 80°C to evaporate the solvent. The sample was degassed at 200°C for 12 hours, and the N2 adsorption-desorption isotherm was measured at 77K over the relative pressure (P/P0) range of 0.015-0.995. Nanopore size distributions and porosities were calculated from the desorption branch of the isotherms using the BJH model.
Removal of the Nucleation layer
Following the electrochemical etch, but preceding the removal of the sacrificial layer, the patterned substrate containing the PSPs was dry etched in CF4 plasma (Plasmatherm 790 series, 10 sccm CF4, 25 mTorr, 200 W RF Power, 18 sec)
Loading
SPs, MPs, LPs, XLPs, were oxidized and conjugated with 3’-aminopropyltriethoxysilane (APTES) resulting in a positively charged amine terminated PSP surface. A 100 ml pH 7.3 Tris(hydroxymethyl)aminomethane-HCl buffered suspension of PSPs and 1mM 15nm diameter carboxyl functionalized CdSe Q-dots (Qdot® 565 ITK™, Invitrogen, Carlsbad, CA) was incubated on a rotating wheel for 15 minutes. The suspension was centrifuged for 10 min at 16,000 r.p.m. in a fixed angle table top microcentrifuge.
TEM sample preparation
The supernatant was removed from the loaded PSP suspension, the pellet resuspended in DI water (20 ml) and brought to 40°C in a water bath. low melt 4% agarose solution (100 ml) kept at 60°C was added to the suspension and immediately centrifuged for 20 min at 16,000 r.p.m. in a fixed angle table top microcentrifuge. The resulting PSP-rich gel was diced in 1mm3 cubes and dehydrated with subsequent rinses in 30%-70%-100% anhydrous ethanol followed by 100% acetone. The gel cubes were transferred to a size 3 embedding capsule and Spurr’s resin was added. The capsule was centrifuged for 20 min at 16000 r.p.m. in a fixed angle table top microcentrifuge. The PSP-rich resin was fully cured in an oven at 60°C for 12h. Ultrathin slices (100nm) of PSP-rich resin were microtomed in a Leica UCT and collected on a 400mesh Cu TEM grid.
TEM/EDX Analysis
HAADF-STEM analysis and mapping EDX were performed with a FEI Tecnai G2 F20 X-TWIN TEM (FEI Company, Hillsboro, OR) operating at 200kV with nanoprobe spot 7. EDX analysis was performed with 8000ms dwell time and 12° tilt.
TEM Tomography
HAADF-STEM tilt series were acquired automatically on a FEI Tecnai F20 S-TWIN TEM operating at 200kV using the FEI XPlore3D 2.0 Tomography Suite. The STEM images were collected over an angular range of ±65° in 2° steps below 50° and 1° step above 50°. The STEM probe size was 0.19 nm. The reconstructions of the tilt series were calculated with a FEI Inspect 3D Express tomography pack using a weighted back projection (WBP) method and the visualization was carried out using the FEI Resolve RT/Amira software.
Supplementary Material
Acknowledgments
We thank the Center for Nano and Molecular Science, the Institute for Cellular and Molecular Biology and the Microelectronics Research Center at the University of Texas at Austin for use of instrumentation and facilities. We thank Dwight Romanovicz for help in experimental design and Matthew Landry for excellent graphical support. We thank Michael Strauss, Pal Ove Pedersen, Brandon Van Leer and all the team at the North America Nanoport application team of FEI Company (Hillsboro, OR) for providing the facilities and characterization work. These studies were supported by the following grants: State of Texas’s Emerging Technology Fund, NASA (NNJ06HE06A), DoD (W81XWH-07-2-0 101) and NIH (R01CA128797). The authors would like to recognize the contributions and support from the Alliance for NanoHealth (ANH).
Footnotes
Commercialization rights on intellectual property presented in this paper have been acquired by Leonardo Biosystems Inc. Mauro Ferrari is the founding scientist of Leonardo Biosystems. Ennio Tasciotti and Xuewu Liu are stockholders of Leonardo Biosystems.
Supporting information for this article is available on the WWW under http://www.chemphyschem.org or from the author.
Contributor Information
Xuewu Liu, Email: xuewu.liu@uth.tmc.edu.
Mauro Ferrari, Email: mauro.ferrari@uth.tmc.edu.
References
- 1.Canham LT. Adv Mater. 2005;7:1033–1037. [Google Scholar]
- 2.Anderson SHC, Elliott H, Wallis DJ, Canham LT, Powell JJ. Phys Status Solidi A. 2003;197:331–335. [Google Scholar]
- 3.Martin FJ, Melnik K, West T, Shapiro J, Cohen M, Boiarski AA, Ferrari M. Drugs R D. 2005;6:71–81. doi: 10.2165/00126839-200506020-00002. [DOI] [PubMed] [Google Scholar]
- 4.Low SP, Williams KA, Canham LT, Voelcker NH. Biomaterials. 2006;27:4538–4546. doi: 10.1016/j.biomaterials.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 5.Chin V, Collins BE, Sailor MJ, Bhatia SN. Adv Mater. 2001;13:1877–1880. [Google Scholar]
- 6.Bayliss SC, Heald R, Fletcher DI, Buckberry LD. Adv Mater. 1999;11:318–321. [Google Scholar]
- 7.Goh ASW, Chung AYF, Lo RHG, Lau TN, Yu SWK, Chng M, Satchithanantham S, Loong SLE, Ng DCE, Lim BC. Int J Radiat Oncol Biol Phys. 2007;67:786–792. doi: 10.1016/j.ijrobp.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Canham L, Stewart M, Buriak J, Reeves C, Anderson M, Squire E, Allcock P, Snow P. Phys Status Solidi A. 2000;182:521–525. [Google Scholar]
- 9.Tasciotti E, Liu X, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MMC, Decuzzi P, Tour JM, Robertson F. Nature Nanotech. 2008;3:151–157. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 10.Salonen J, Laitinen L, Kaukonen AM, Tuura J, Björkqvist M, Heikkilä T, Vähä-Heikkilä K, Hirvonen J, Lehto VP. J of Controlled Release. 2005;108:362–374. doi: 10.1016/j.jconrel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Sun W, Puzas JE, Sheu TJ, Fauchet PM. Phys Status Solidi A. 2007;204:1429–1433. [Google Scholar]
- 12.Khung Y, Graney SD, Voelcker NH. Biotechnol Prog. 2006;22:1388–1393. doi: 10.1021/bp060115s. [DOI] [PubMed] [Google Scholar]
- 13.Prestidge CA, Barnes TJ, Lau C, Barnett C, Loni A, Canham LT. Expert Opin Drug Delivery. 2007;4:101–110. doi: 10.1517/17425247.4.2.101. [DOI] [PubMed] [Google Scholar]
- 14.Ferrari M. Nat Rev Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 15.Serda RE, Gu J, Bhavane RC, Liu X, Chiappini C, Decuzzi P, Ferrari M. Biomaterials. 2009;30:2440–2448. doi: 10.1016/j.biomaterials.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 16.Cuenca AG, Jiang H, Hochwald SN, Delano M, Cance WG, Grobmyer SR. Cancer. 2006;107:459–466. doi: 10.1002/cncr.22035. [DOI] [PubMed] [Google Scholar]
- 17.Yezhelyev MV, Gao X, Xing Y, Al-Hajj A, Nie S, O’Regan RM. Lancet Oncol. 2006;7:657–667. doi: 10.1016/S1470-2045(06)70793-8. [DOI] [PubMed] [Google Scholar]
- 18.Brannon-Peppas L, Blanchette JO. Adv Drug Deliver Rev. 2004;56:1649–1659. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 19.Decuzzi P, Ferrari M. Biomaterials. 2007;28:2915–2922. doi: 10.1016/j.biomaterials.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Rolland JP, Maynor BW, Euliss LE, Exner AE, Denison GM, DeSimone JM. J Am Chem Soc. 2005;127:10096–10100. doi: 10.1021/ja051977c. [DOI] [PubMed] [Google Scholar]
- 21.Gentile F, Chiappini C, Fine D, Bhavane RC, Peluccio MS, Cheng MM, Liu X, Ferrari M, Decuzzi P. J Biomech. 2008;41:2312–8. doi: 10.1016/j.jbiomech.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 22.Decuzzi P, Ferrari M. Biomaterials. 2008;29:377–384. doi: 10.1016/j.biomaterials.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 23.Cunin F, Schmedake TA, Link JR, Li YY, Koh J, Bhatia SN, Sailor MJ. Nat Mater. 2002;1:39–41. doi: 10.1038/nmat702. [DOI] [PubMed] [Google Scholar]
- 24.Cohen MH, Melnik K, Boiarski AA, Ferrari M, Martin FJ. Biomed Microdevices. 2003;5:253–259. [Google Scholar]
- 25.Dorvee JR, Sailor MJ, Miskelly GM. Dalton Trans. 2008;2008:721–730. doi: 10.1039/b714594b. [DOI] [PubMed] [Google Scholar]
- 26.Cheng L, Anglin E, Cunin F, Kim D, Sailor MJ, Falkenstein I, Tammewar A, Freeman WR. Br J Ophthalmol. 2008;92:705–711. doi: 10.1136/bjo.2007.133587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herino R. In: The Properties of Porous Silicon. Canham LT, editor. Ch 2.2 INSPEC-IEE; London, UK: 2007. [Google Scholar]
- 28.Zhang XG. J Electrochem Soc. 2004;151:C69–C80. [Google Scholar]
- 29.Kübel C, Voigt A, Schoenmakers R, Otten M, Su D, Lee T, Carlsson S, Bradley J. Microsc Microanal. 2005;11:378–400. doi: 10.1017/S1431927605050361. [DOI] [PubMed] [Google Scholar]
- 30.Drummy LF, Wang YC, Schoenmakers R, May K, Jackson M, Koerner H, Farmer BL, Mauryama D, Vaia RA. Macromolecules. 2008;41:2135–2143. [Google Scholar]
- 31.Decuzzi P, Pasqualini R, Arap W, Ferrari M. Pharm Res. 2009;26:235–243. doi: 10.1007/s11095-008-9697-x. [DOI] [PubMed] [Google Scholar]
- 32.Gratton SE, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. Proc Natl Acad Sci U S A. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.