

Abstract
We report here the construction of phenanthroline (Phen) and terpyridine (Terpy) based hybrid coordination motifs (HCMs), which were installed on the surface of the four-helical bundle hemeprotein, cytochrome cb562. The resulting constructs, termed HPhen1, HPhen2, HPhen3 and HTerpy1, feature HCMs that are composed of a histidine ligand and a Phen or Terpy functionality located two helix turns away, yielding stable tri- or tetradentate coordination platforms. Our characterization of the tridentate HCMs indicates that they accommodate many divalent metal ions (Co2+, Ni2+, Cu2+, Zn2+) with nanomolar to femtomolar affinities, lead to significant stabilization of the α-helical protein scaffold through metal-mediated crosslinking, assert tight control over protein dimerization, and provide stable and high-affinity binding sites for substitution-inert metal probes. Our analyses suggest that such tridentate HCMs may be used modularly on any α-helical protein surface in a sequence-independent fashion.
Introduction
The primary biological roles of metals including catalysis, electron transfer and structural stabilization are generally established once they are firmly placed within a protein scaffold.1 Owing to the stability of the resulting complexes, the interactions between metals and the interiors of proteins are readily characterized and have justifiably formed the focus of Bioinorganic Chemistry. One could argue, on the other hand, that metals spend a good majority of their time interacting with protein surfaces, and that such transient, harder-to-characterize interactions carry in vivo and in vitro consequences that rival those of metal-protein interior interactions. The prevalence of metal-protein surface interactions become especially clear when picturing the behavior of metal ions and complexes within the crowded cellular environment, for example, as they are being passed on from one specific protein (e.g. a metallochaperone)2 to another, or as they crosslink together multiple proteins whose aggregation may have dire consequences.3 Similarly, outside the cellular realm, metal-protein surface interactions form the basis of immobilized metal ion affinity chromatography (IMAC)4 as well as the functionalization of protein surfaces with metal complexes that have served as invaluable spectroscopic and functional probes.5, 6 Given such broad importance and utility of metal-protein surface interactions, we envisioned a need for metal coordinating motifs that would enable a better control of inorganic chemistry on protein surfaces.
Our original interest in metal-protein surface interactions stems from our desire to use metal coordination chemistry to direct protein-protein interactions (PPIs)7 and protein self-assembly more predictably and readily than computational design approaches. One caveat to utilizing metal coordination to control PPIs is the presence of numerous metal binding sidechain functionalities on any given protein surface, which bring about the challenge of controlling metal localization. In one strategy to circumvent this challenge, we introduced a Cys-specific bidentate non-natural chelate (5-iodoacetamido-1,10 phenanthroline, IA-Phen) onto the surface of a four-helix-bundle protein, cytochrome cb562, which led to the Ni2+-driven formation of an unusual triangular protein architecture.8 More recently, to exert more control over metal localization as well as metal-directed protein self-assembly, we used another bidentate chelate (5-iodoacetamido-8-hydroxyquinoline, IA-Quin) attached to a Cys (C70) in combination with a His (H63) located two helix turns away on the cyt cb562 surface, yielding the construct HQuin1 (Figure 1a).9 The resulting i/i+7 hybrid coordination motif (HCM) was shown to coordinate various divalent metal ions in a tridentate fashion, which led to: 1) high affinity divalent metal binding with dissociation constants (Kd’s) ranging from nM to fM, 2) stabilization of the protein scaffold via metal-mediated crosslinking of a two-helix turn segment, and 3) tight control over protein dimerization via an octahedral metal coordination geometry. Several potential applications arise from these advantages including site-selective labeling of proteins with metal probes, improved protein separation with IMAC, stabilization of small helical peptides for pharmaceutical purposes, and manipulation of cellular pathways that depend on protein dimerization.
Figure 1.

Cartoon representations for various HCM-bearing cyt cb562 variants. Functionalities that comprise the HCMs and the heme groups are shown as sticks and highlighted in magenta.
Given such possibilities and the ease of constructing an HCM via iodoacetamide (IA)-Cys coupling, we have sought to examine in the present study whether the advantageous properties of the HQuin1 HCM are generalizable, i.e., whether the i/i+7 HCMs that consist of a His and a non-natural chelating ligand can be utilized in a modular fashion on any α-helical surface. Towards this end, we have created a series of additional cyt cb562-based constructs (Figure 1b-e), which have been functionalized with various non-natural chelates (Figure 2): a) HPhen1, the phenanthroline(Phen)-derivatized counterpart of HQuin1, was constructed to probe the generality of the non-natural component; b) HPhen2, which features the opposite placement of His and the Cys-Phen group as in HPhen1 (Cys63 in the i and His70 in the i+7 position), was constructed to test the sensitivity of the i/i+7 HCM to the relative placement of the natural and non-natural ligands, c) HPhen3, which has the HCM motif located elsewhere on the cyt cb562 surface (His70, Cys77), was generated to test the sensitivity of the HCM to location, and, d) HTerpy1, the terpyridine-derivatized counterpart of HQuin1 and HPhen1, as a tetradentate HCM motif. We present here the characterization of these constructs in terms of their metal binding affinities, metal-dependent stabilization and metal-dependent oligomerization properties (Figure 3). Our results suggest that the i/i+7 HCMs may be modularly utilized on any α-helical protein surface towards a number of applications.
Figure 2.
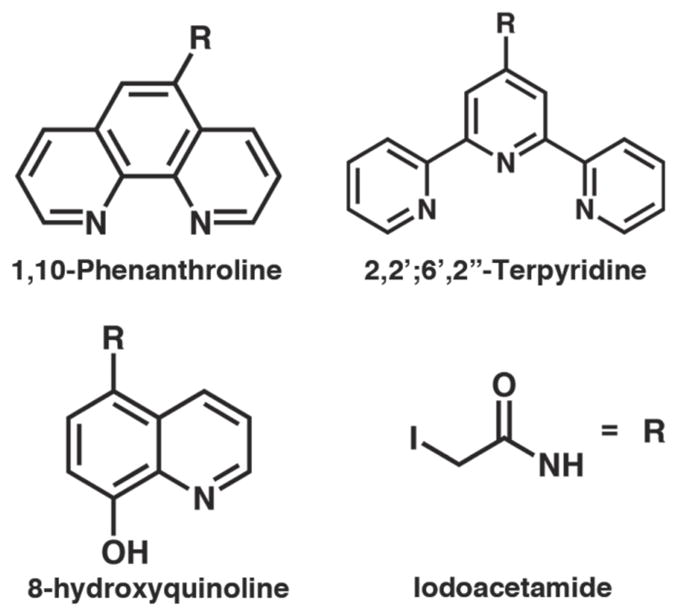
Phenanthroline (Phen), terpyridine (Terpy) and hydroxyquinoline (Quin) derivatives used for the construction of HCMs in this study. The iodoacetamide moiety is attached to the chelating functionalities through the amide nitrogen.
Figure 3.
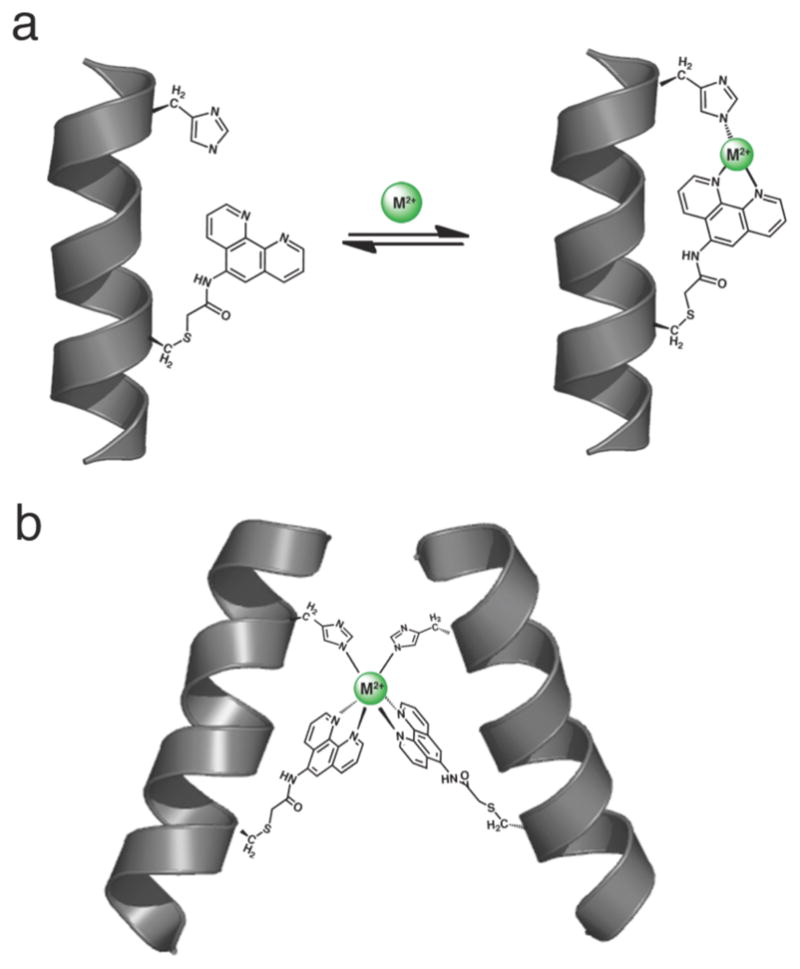
(a) Cartoon depiction for the three coordinate facial binding mode of a His-Phen HCM. (b) Proposed mode of metal-dependent dimerization mediated by a i/i+7 His-Phen HCM.
Results and Discussion
Construction of cyt cb562 variants with Quin, Phen and Terpy-bearing HCMs
One requisite for expanding the biological and chemical utility of HCMs is to demonstrate the modularity of the non-natural metal chelator within the HCM system and the ease of its incorporation. To this end, we site-specifically labeled cyt cb562 variants bearing a single surface Cys residue with IA-derivatized versions of the ubiquitous metal chelators Phen, Terpy and Quin to create the HCM variants shown in Figure 1. These non-natural ligands were chosen because they are commercially available as – or easily converted to – amino-functionalized precursors, their coordination chemistry has been extensively studied, and they represent a small but diverse set if ligands with variations in denticity and overall charge.
The amino-precursors of Phen, Quin and Terpy were converted in a one-pot reaction with iodoacetic anhydride or iodoacetyl chloride into IA-derivatives with 60–75% yield. Although IA-Phen, IA-Quin and IA-Terpy are sparingly soluble in water, they are easily introduced into cyt cb562 solutions after being solubilized in DMF and DMSO; we have found no adverse effects of these organic solvents on cyt cb562 up to a final volume fraction of 50% versus H2O. Cys functionalization reactions proceed rapidly and specifically (provided that the solution pH is kept below 8 to prevent Lys labeling), with overall yields of modification ranging from 60% for IA-Terpy to 95% for IA-Phen after purification. In the case of cyt cb562, the functionalized products are facilely separated from non-functionalized protein using anion-exchange chromatography (Figure S1.1).
Metal Binding Properties of Phen- and Terpy-based HCMs
We had previously examined the divalent metal-binding properties of HQuin1 and confirmed that the i/i+7 His-Quin HCM was able to coordinate metals in a facial, tridentate geometry.9 Here, we have performed similar metal-binding titrations for HPhen1, HPhen2 and HPhen3 using late first-row transition metals (Co2+, Ni2+, Cu2+, Zn2+) to probe if the Phen functionality behaves similarly to Quin in the context of an HCM. It is important to note that the relative positions of the coordinating atoms to the point of protein attachment in the Phen derivative are equivalent to those in the Quin derivative (Figure 2). Metal binding by the His-Phen HCMs were monitored by the distinct 10-nm red-shift in the π-π* absorption band for Phen (metal-free λmax= 272 nm; metal-bound λmax= 282 nm) upon metal binding (Figure 4). It was confirmed through CD spectroscopy that the α-helical fold is not significantly perturbed by metal binding to the HCMs (Figure S1.19).
Figure 4.

Spectral changes that accompany Zn2+ binding to HPhen1 as monitored by UV-vis spectroscopy. Spectra show a typical ferric heme spectrum with a Soret band at 415 nm (ε = 0.148 μM−1 cm−1) along with a transition between metal-free (λmax= 272 nm) and metal-bound (λmax= 282 nm) Phen species. Inset: Close-up view of the UV region showing a clean isobestic point at 274 nm consistent with a 1:1 Zn2+:Phen binding model.
As in the case of HQuin1, it was quickly established that Phen-based HCMs bind all tested divalent metals very tightly, which required all titrations to be performed in the presence of ethylene glycol tetraacetic acid (EGTA) as a competing ligand. Due to the inherent ability of Phen-based HCMs to undergo metal-mediated dimerization (Figure 3b), protein concentrations were kept sufficiently low (< 5 μM) to minimize dimer formation. In all cases (HPhen1-3 and all metals), the metal binding isotherms were satisfactorily described by a 1:1 binding model (Figures 5, S1.2 and S1.3).
Figure 5.

Metal-binding titration data and fits for HPhen1 as monitored by UV-visible spectroscopy. A typical titration sample contained 2–5 μM of HPhen1, 50 mM MOPS buffer (pH 7) and 20–100 μM of the competing ligand EGTA. All data were described satisfactorily by a 1:1 binding model. Binding isotherms for HPhen2, HPhen3 are shown in Figure S1.2 and S1.3. Dissociation constants (Kd) determined are listed in Table 1 and S2.1.
An analysis of the determined dissociation constants (Table 1) reveals that all Phen based HCMs display a significant increase in affinity over free Phen for the late first row transition metals, which strongly suggests the participation of the His component of the HCMs in metal binding. Moreover, the affinities of HPhen1, 2 and 3 for divalent metals are similar and vary at most by six-fold, indicating that metal binding ability is not very sensitive to helix location or relative orientation of the HCM (see below for a discussion on the possible effects of intervening residues).
Table 1.
Dissociation constants for HPhen-metal complexes compared to those for free 1,10-phenanthroline (Phen).
| Dissociation Constants (M) | ||||
|---|---|---|---|---|
| HPhen1a | HPhen2a | HPhen3a | Free Phenb | |
| Co2+ | 3.3 (2) × 10−10 | 7.8 (5) × 10−10 | 2.0 (2) × 10−9 | 8.0 × 10−8 |
| Ni2+ | 7.8 (6) × 10−10 | 1.7 (4) × 10−10 | 1.6 (3) × 10−10 | 3.9 × 10−8 |
| Cu2+ | 1.3 (2) × 10−13 | 1.2 (2) × 10−13 | 2.0 (2) × 10−13 | 2.5 × 10−9 |
| Zn2+ | 6 (1) × 10−9 | 3.8 (3) × 10−9 | 3.7 (3) × 10−8 | 3.9 × 10−7 |
Dissociation constants determined by competition with EGTA in 50 mM MOPS (pH 7).
pH-adjusted values based on reported Kd’s.10
In addition to the HPhen variants, we also investigated whether HTerpy1 (Figure 1e) can engage both Terpy and His in a tetradentate coordination motif. Our metal binding titrations and sedimentation velocity (SV) experiments reveal that HTerpy1 almost exclusively forms a stable dimer with a saturation point reached upon addition of half an equivalent of M2+ (Figure S1.4), which has precluded the determination of the HTerpy1 metal binding affinities. While protein unfolding studies (see below) show evidence for metal coordination by both His and Terpy, the unstrained, facial coordination geometry observed in HQuin1 and HPhen variants cannot be accommodated by the large Terpy group, leading to the formation of the thermodynamically and kinetically stable bis-Terpy adduct involving two proteins.
Metal-mediated protein stabilization through Phen- and Terpy-based HCMs
We next sought to determine if the Phen- and Terpy-based HCMs would have any stabilizing effect on the protein scaffold. Since HCMs crosslink a ~7-Å long, two-helix-turn segment of cyt cb562 through metal coordination, an increase in the global stability of the protein should be expected. Metal crosslinking of both natural and non-natural residues at i/i+4 positions has extensively been shown to induce α-helicity in peptides and significantly stabilize helical protein structures.11, 12 Likewise, covalent cross-linking of sidechain functionalities in i/i+4, i/i+7 or i/i+11 positions can lock small peptides in α-helical conformations,13–15 which in turn have proven to be promising pharmaceutical agents that effectively disrupt protein-protein interactions and exhibit increased resistance to proteases in vivo.16
In order to investigate the cross-linking ability of HCMs in the presence of metals, chemical and thermal unfolding studies were undertaken. In a typical chemical unfolding experiment, a solution of folded HPhen or HTerpy variant was titrated with increasing amounts of unfolded protein solution prepared in 8 M guanidine hydrochloride (GuHCl). The folding/unfolding transition was followed by CD spectroscopy, monitoring changes in ellipticity at 222 nm. Thermal unfolding measurements spanning 298 to 373 K were similarly monitored at 222 nm; because of the high stability of all variants, 1.5 M GuHCl was included in each sample to ensure that complete unfolding took place before 373 K. In both chemical and thermal unfolding experiments, metals were present in large excess over protein to ensure full occupancy of HCMs, thereby preventing metal-induced dimerization.
The stability of all HPhen and HTerpy variants tested was found to increase in the presence of divalent metal ions. Figure 6 shows representative unfolding titrations of the variants, each of which display a particularly enhanced stability in the presence of Ni2+ (for other metals and thermal titrations, see Figures S1.5–9); a complete set of results is given in Table 2. At least in the case of the HPhen variants, we attribute the superior stabilizing effect of Ni2+ over other metals to the formation of an unstrained, facial coordination geometry by the His-Phen HCM, which was previously shown to be the case for the His-Quin HCM.
Figure 6.

Chemical unfolding titrations of (a) HPhen1 (b) HPhen2 (c) HPhen3 and (d) HTerpy1 in the presence and absence of Ni2+ as monitored by CD spectroscopy at 222 nm.
Table 2.
Observed changes in the midpoint for the unfolding transition (Δ[GuHCl]m) for HPhen and HTerpy variants upon metal binding.
| Δ[GuHCl]m (M) | ||||
|---|---|---|---|---|
| Co2+ | Ni2+ | Cu2+ | Zn2+ | |
| HPhen1 | 0.84 | 1.15 | 0.41 | 0.78 |
| HPhen2 | 0.25 | 0.6 | 0.22 | 0.4 |
| HPhen3 | 0.24 | 0.6 | −0.02 | 0.29 |
| HTerpy1 | 0.21 | 0.47 | 0.01 | 0.32 |
| APhen1 | 0.02 | 0.12 | −0.19 | 0.01 |
In order to establish that the observed protein stabilization is due to metal-mediated, intrahelical crosslinking, we carried out the unfolding titrations of HPhen1 and HTerpy 1 at pH 5.5, where the His component of the HCM should be partially protonated and unable to fully coordinate metals (Figures 7a and 7b). Additional unfolding titrations were performed for variants of HPhen1 and HTerpy1, where either the Phen or the Terpy moiety is replaced by a carboxymethyl group (CM-G70C cyt cb562, Figure S1.10) or the His63 residue is mutated to Ala (APhen1 and ATerpy1, Figures 7c, 7d and S1.11). The results indicate that, in all cases, Ni-induced stabilization is significantly diminished, confirming the involvement of both His and Phen (or Terpy) in metal coordination.
Figure 7.

Chemical unfolding titrations of (a) HPhen1 at pH 5.5, (b) HTerpy1 at pH 5.5, (c) APhen1 at pH 7.5, and (d) HTerpy1 at pH 7.5. The lack of significant protein stabilization in the presence of Ni2+ ions indicates that both His and Phen or Terpy moieties are involved in metal-mediated protein cross-linking.
It is tempting to link the thermodynamics of metal binding by the HCMs (Table 1) to that of metal-induced protein stabilization (Table 2). Nevertheless, such a correlation is complicated by the fact that net protein stabilization is a function of metal binding not only to the folded but also to the unfolded state, which may display multiple modes of metal coordination (thus deviating from a two-state system). We therefore have avoided presenting free energies of unfolding – which assumes a two-state process – for our variants in the presence of metals. A good case in point is Cu2+, which displays by far the highest affinity for any HCM, yet leads to the smallest extent of stabilization (Table 2). Regardless, the protein unfolding titrations clearly indicate that: 1) all Phen-based HCMs lead to a measurable metal-induced increase in protein stability, 2) this stabilization is not specific to a particular HCM location or orientation, and finally, 3) the Terpy-His HCM displays a diminished stabilizing effect due likely to an unfavorable metal coordination geometry.
Metal-Dependent Self-Assembly Properties of HPhen1
The ability to control protein self-assembly, both temporally and spatially, is an intensely pursued goal that is complicated by the necessity to design extensive molecular surfaces.17, 18 Particularly challenging is to direct the self-assembly of proteins into discrete shapes that can recognize biological targets. One of the most exciting findings about HQuin1 was its ability to specifically dimerize upon Ni2+ binding into a rigid architecture that was shaped appropriately to bind major grooves of a double-helical DNA.9 Since HPhen1 is the closest in composition and chemical behavior to HQuin1 which we had already structurally characterized, we chose to explore its metal-dependent self-assembly properties as a representative of all Phen-bearing variants.
Sedimentation velocity (SV) experiments reveal that HPhen1 readily dimerizes in the presence of half an equivalent of Ni2+ with a sedimentation coefficient of 2.6 S similar to that of the Ni:HQuin12 complex (Figure 8a).9 The dissociation constant for the Ni:HPhen12 dimer (Kd (2mer-1mer)) was determined by sedimentation equilibrium (SE) experiments to be ~ 9 μM, which is lower than the Kd (2mer-1mer) of 42 μM for Ni:HQuin12 (Figure 8b Figure S1.12).9 Significantly, the dimeric stability of Ni:Phen12 now closely approximates that of bZIP family transcription factors which use peripheral leucine zippers domains for dimerization, with Kd‘s in the low micromolar range.19
Figure 8.
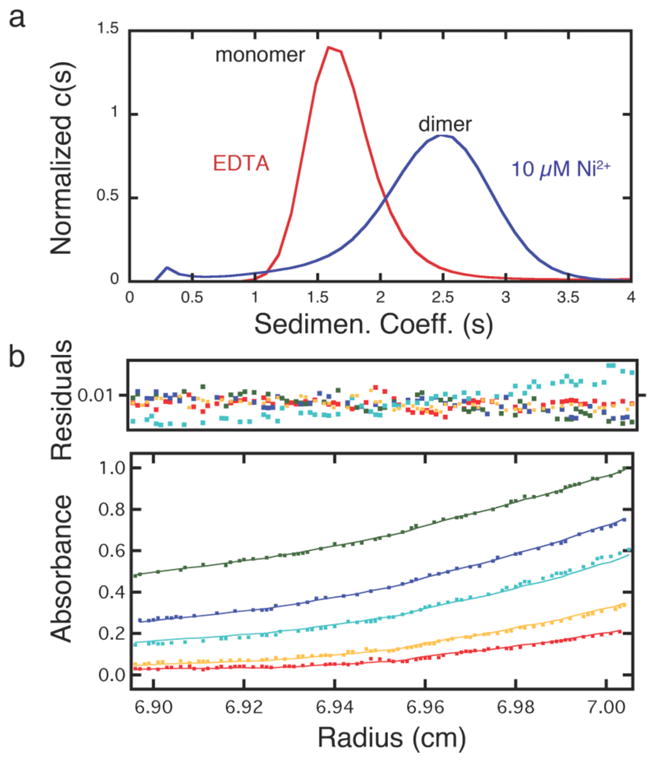
(a) Sedimentation velocity profiles of 20 μM HPhen1 in the absence and presence of Ni2+. (b) Sedimentation equilibrium (SE) profiles for 20 μM HPhen1 in the presence of 10 μM Ni2+, with equilibrium speeds of 20,000 (green), 25,000 (blue), 30,000 (cyan), 35,000 (yellow) and 41,000 rpm (red). Data were fit to a monomer-dimer self-association model with a log K = 5.05 (2) or 8.9 (1) μM.
Complete structural characterization of the Ni:HPhen12 dimer has remained elusive to this point. However, we project – based on the similarities between HPhen1 and HQuin1 and the fact that dimerization in both cases is entirely dictated by metal coordination – that the structure of Ni:HPhen12 should closely resemble that of the Ni:HQuin12. In the case of Ni:HQuin12, it was determined through density functional theory (DFT) calculations that the most favored inner-sphere coordination arrangement would pose the Quin groups cis to one another in Λ configuration,20 whereby the two phenolate Quin oxygens would lie trans to each other, which is also the crystallographically observed configuration.9
We performed similar DFT (BP86 and OLYP) calculations on Ni:HPhen12, where we investigated the relative energies of two possible inner-sphere arrangements for the His-Phen HCM: one that presents Phen ligands cis to one another (cis-Phen) and one that presents Phen ligands in a trans configuration (trans-Phen) (Figure 9). These calculations suggest that the cis-Phen arrangement of cis- Phen arrangment is ~5.2 kcal/mol more stable than the trans-Phen arrangement. In the case of Ni2+:HQuin12, the higher stability of cis-Quin isomer was attributed to the trans-directing effect of the imine ligands which would render a mutual trans orientation of the weaker-field phenolate ligands the least destabilized configuration. With the N, O groups of Quin now replaced with the N, N groups in the Phen ligands, this argument cannot be made to explain the higher stability of the cis-Phen arrangement. Instead, a close inspection of the calculated structures reveals that in the trans-Phen arrangement, there would be considerable steric clashes between the Phen hydrogens that lie on the Ni equatorial plane, which would be relieved in the cis-Phen arrangement.
Figure 9.
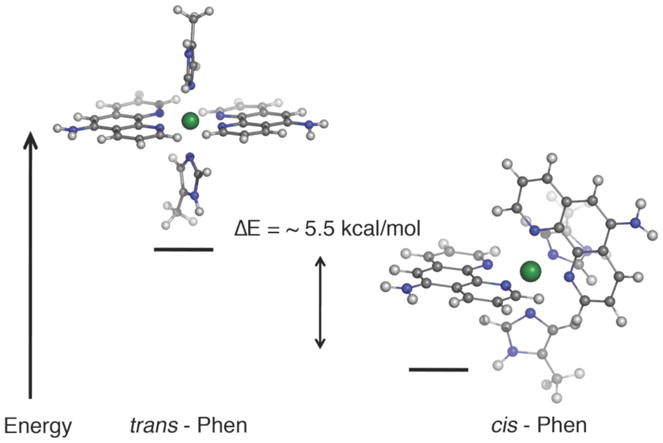
Energy minimized structures (BP86) for the proposed inner-sphere coordination geometry of the Ni:HPhen12 dimer.
Taking together the DFT results and solution studies, we conclude that the i/i+7 His-Phen ICM would yield a Ni2+-induced V-shaped dimer that is equivalent to the crystallographically characterized Ni:HQuin12 architecture (Figure 10).9 Although the reason for the specific formation of this V-shaped structure is different for His-Quin and His-Phen HCMs, both examples demonstrate that self-assembly of proteins can be programmed through a simple consideration of inner-sphere metal coordination, which is far more facile than designing extensive protein interfaces to the same end.
Figure 10.
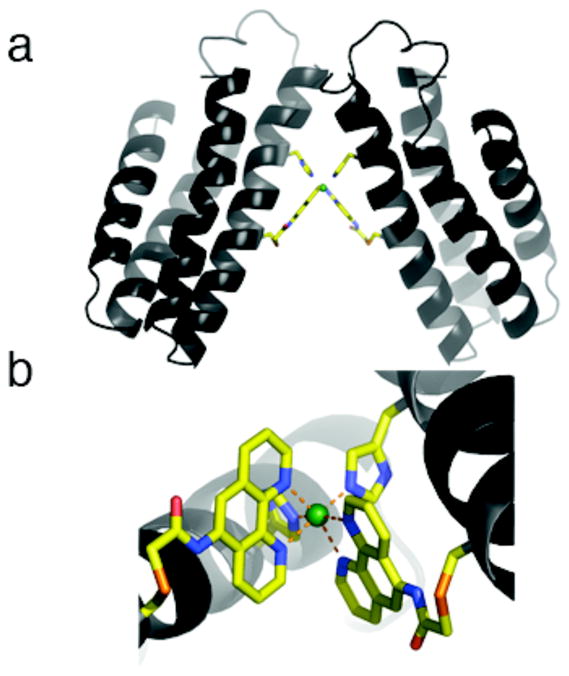
(a) The proposed Ni:HPhen12 architecture modeled after the crystallographically determined Ni:HQuin12 structure. (b) The corresponding inner coordination sphere.
Using Phen-bearing HCMs for protein functionalization
Metal complexes site-specifically attached to protein surfaces have proven to be invaluable functional reporters. Among these, Ru-, Os- and Re-polypyridyl derivatives have been widely used due to their photophysical and photochemical properties.21, 22 Similarly, bifunctional, As-based fluorescent reporters have been designed to specifically bind bis-Cys patterns on proteins and are finding increasing use as target selective in vivo reporters.6 Given the high affinity of Phen-based HCMs for divalent metals and their two-point attachment to the protein scaffold, we envisioned that they could provide stable and specific target sites for functional metal-based probes on α-helical proteins. Moreover, we surmised that if such probes are based on substitution-inert metals, they could result in the improved and irreversible stabilization of α-helical proteins/peptides and may be of value in terms of constructing helical peptide-based pharmaceutical agents. To investigate such possibilities, we explored the interactions of HPhen1 with a p-cymene-capped Ru(II) compound. We chose this particular piano-stool complex as a test case, because the Ru center is capped with an arene group (p-cymene) which should prevent protein dimerization and accommodate facial binding by the His-Phen HCMs. Additionally, it is commercially available in a dimeric, chloro-substituted form (1), and weakly luminescent when bound to a polypyridines.23
In a proof-of-principle study, a solution of HPhen1 was treated with 5-fold molar excess of compound 1 (i.e., 10-fold excess Ru) dissolved in DMSO and stirred at room temperature for ~ 4 days. Reactions were quenched by removing unreacted 1 via gel filtration and subsequently purified by ion exchange chromatography. The FPLC chromatogram and corresponding mass spectra indicate that the only major product of the reaction is HPhen1 bound to a single Ru(p-cymene) adduct (Figure 11), with no discernible unlabeled or multiply labeled species. The absorbance spectra of the isolated Ru(p-cymene)(HPhen1) complex features the expected shift to ~286 nm in the Phen π-π* band due to metal binding as well as a new band at 326 nm (Ru(II) → π* arene MLCT) contributed by Ru-adduct (Figure S1.13 and S1.14). When excited at 326 nm, Ru(p-cymene)- HPhen1 displays a weak emission band centered at 442 nm. Both the absorbance (Figure S1.14) and the emission (Figure S1.15) features of Ru(p-cymene)-HPhen1 are similar to those of a highly analogous model complex, [(p-cymene)Ru(phen)(1-(4-cyanophenyl)imidazole)], in support of the intended mode of Ru coordination to the His-Phen HCM.23
Figure 11.

(a) Anion-exchange FPLC chromatogram of the crude Ru(p-cymene)-HPhen1 reaction mixture. Product eluted at 0.2–0.25 M NaCl using a linear NaCl gradient (0–0.5 M in 10 mM sodium phosphate, pH 8.0). (b) MALDI mass spectra of the major FPLC product identified as the Ru(p-cymene)-HPhen1 complex.
We then examined the chemical unfolding behavior of the Ru(p-cymene)-HPhen1 complex to study the effects of HCM capping by a substitution-inert metal complex on protein stability. As shown in Figure 12, binding of Ru(p-cymene) to HPhen1 leads to a significantly higher stabilization compared to substitution-labile divalent metals (Figure 6 and Table 2), with a corresponding shift in the unfolding midpoint of ~1.5-M GuHCl. Under the reasonable assumption that Ru(p-cymene) is still bound to the His-Phen HCM upon denaturation (which is not necessarily the case for labile metals), the unfolding of Ru(p-cymene)-HPhen1 can now be treated as a two-state process, allowing the determination of the free energy of stabilization (ΔΔGfolding) by Ru(p-cymene) binding to be 4.1 kcal/mol.24 The finding that global protein stability can be raised to such an extent by the metal-mediated crosslinking of a local fragment is particularly significant given that the free energy of unfolding for natural proteins typically ranges from 5 to 15 kcal/mol.25
Figure 12.

Chemical unfolding titrations (monitored by CD spectroscopy) showing the higher stability of the Ru(p-cymene)-HPhen1 complex (blue) with respect to HPhen1 in the absence of metals (red).
Effects of Intervening Residues in i/i+7 HCMs
To probe if tridentate i/i+7 HCMs may be used on any helical protein surface regardless of the amino acid content, we took a closer look at the structural features of our variants with particular focus on the residues that lie between the coordinating His and the functionalized Cys. Figure 13a shows the Ni coordination mode of the His-Quin HCM in the previously determined Ni:HQuin12 structure, and Figure 13b shows the proposed conformation for HPhen1 modeled after the same structure. These structures clearly indicate that the only intervening residues of interest are at the i+3 and i+4 positions, regardless of the relative positions of His and Cys on the helix (i.e., i/i+7 or i/i-7). Importantly, for both i+3 and i+4 positions (Asp66 and Ile67 for HPhen1 and HQuin1), the Cbackbone-Cα vectors that largely dictate the orientation of the sidechains are directed away from the coordinating groups. It could then be expected that the i/i+7 His-Phen or His-Quin HCMs may be universally installed on any regular α-helical surface to coordinate metals without significant interference by the intervening amino acids. This expectation is supported by the finding that HPhen1, HPhen2 and HPhen3 display more or less similar binding affinities for several divalent metal ions (Table 1) despite different sets of intervening residues: Asp66/Ile67 for HPhen1, Ile67/Asp66 for HPhen2 (inverse of HPhen1) and Asp73/Asp74 for HPhen3 (Figures 13a and b).
Figure 13.

(a) Cartoon representation of the HQuin1 structure showing the Ni coordination mode by the i/i+7 His-Quin HCM. Other residues on Helix3 that are important either for the construction of HPhen3 HCM (positions 70 and 77) or those corresponding to the i+3 and i+4 positions for all variants are shown also as sticks. (b) The analogous representation of HPhen1 modeled after the HQuin1 structure/(c) Closeup view of the model for His-Phen HCM coordinated to a Ni2+.
At the same time, a close inspection of the HQuin1 structure and the HPhen1 model (Figure 13c) shows that the side chain of Ile67 forms van der Waals contacts (d ~ 3.0 Å) with the Quin (or Phen) aromatic ring. These favorable interactions would be absent in the case of HPhen2 or HPhen3, which would respectively present Asp66 or Asp74 near the vicinity of Quin or Phen. Such differential interactions are likely culprits for the lack of any obvious trend in the metal binding affinities of HPhen1, HPhen2 and HPhen3 (Table 1). Nevertheless, we envision that the i+3 and i+4 positions within i/i+7 HCMs may be exploited as an additional handles to fine-tune metal coordination by HCMs.
Conclusion
Cumulatively, our studies establish that i/i+7 HCMs that include a single His and a non-natural bidentate ligand like Phen and Quin can form tridentate chelating platforms on α-helices, extending the scope of coordination chemistry on protein surfaces. Such tridentate HCMs not only provide unprecedented metal binding affinities, but are also able to stabilize α-helical structures, lead to the formation of discrete oligomers and provide high-affinity attachment sites for metal-based probes. Our findings and analyses suggest that these HCMs may be utilized as modular units on any α-helical protein surface in a sequence independent fashion.
Experimental Section
Materials and Methods
Unless otherwise noted, all solvents, buffers were purchased from Fisher Scientific or VWR and used without further purification. ACS reagent grade metal salts (CoCl2, NiSO4, CuSO4 and ZnCl2) were purchased from Sigma-Aldrich and used without further purification.
Mass Spectrometry
Protein mass spectrometry was carried out at Biomolecular/Proteomics Mass Spectrometry Facility at UCSD using a Voyager DE-STR MALDI-TOF mass spectrometer. Protein samples (100 μL) were first washed with 3× with 400 μL of nano-pure water (Millipore) using a centrifugal spin column (Millipore) equipped with a 10 KDa cutoff filter. In a typical experiment, 5 μL of a protein sample was mixed in a 1:1 ratio with sinapinic acid (Aligent) as a matrix. 1 μL of the resulting protein/matrix samples was plated on a standard 100 well plate and dried completely before use.
Mass spectrometry (MS) of small molecules was carried out at the Molecular Mass Spectrometry Facility at UCSD using either electrospray ionization (ESI) or an atmospheric pressure chemical ionization (APCI) source on a ThermoFinnigan LCQDECA mass spectrometer equipped with a quadrupole ion trap mass analyzer and Xcalibur data system. The MS detector was operated under both positive and negative ion modes with a mass resolution range of 100 ppm.
Site Directed Mutagenesis
Site directed mutagenesis was performed on the pETc-b562 plasmid (denoted as wild-type)26 using the QuikChange kit (Stratagene) and employing primers obtained from Integrated DNA Technologies. The mutant plasmids were transformed into XL-1 Blue E. coli cells and purified using the QIAprep Spin Miniprep kit (Qiagen). Point mutations were introduced to obtain the following cyt cb562 variants: G70C-cyt cb562, G70H/H63C-cyt cb562, K77C/G70H/H63A/W59H-cyt cb562, G70C/H63A-cyt cb562. Sequencing of all mutant plasmids was carried out by Retrogen Inc. (San Diego, CA).
General Protein Expression and Purification Protocol
The mutant plasmids isolated from XL-1 blue cells was transformed into BL21(DE3) E. coli cells along with the ccm heme maturation gene cassette plasmid, pEC86.27 Cells were plated on LB agar, containing 100 μg/mL ampicillin and 34 μg/mL chloramphenicol, and grown overnight. From these colonies LB medium was then inoculated and allowed to incubate for 16 h at 37°C, with rotary shaking at 250 rpm. No induction was necessary.
Mutant-expressing cells were sonicated, brought to pH 5 with the addition of HCl, and centrifuged at 16,000 g, 4° C, for 1 hr. The protein was then purified by ion-exchange chromatography on a CM-Sepharose matrix (Amersham Biosciences) using a NaCl gradient in sodium acetate buffer (pH 5). After exchange into sodium phosphate buffer (pH 8) using 6–8 kDa cutoff dialysis tubing (Fisher), the protein was further purified using an Uno-Q (BioRad) anion exchange column on a DuoFlow chromatography workstation (BioRad) using a NaCl gradient. Protein purity was determined by SDS-PAGE gel electrophoresis. Verification of mutations was made through MALDI mass spectrometry:
| cyt cb562 variant | Calc. MW (amu) | Obs. MW (amu) |
|---|---|---|
| G70C | 12386 | 12385 |
| H63C/G70H | 12386 | 12386 |
| K77C/G70H/H63A/H59H | 12280 | 12282 |
| H63A/G70C | 12320 | 12320 |
Synthesis of Iodoacetic anhydride
As a precursor, iodoacetic acid anhydride was freshly prepared by adding 2.64 g (12.8 mmol) of DCC to a stirred solution of 5.0 g (26.8 mmol) iodoacetic acid in 75 mL of ethyl acetate. Dicyclohexylurea precipitates immediately, but the mixture was allowed to stir for 2 h in the dark. The dicyclohexylurea was removed by filtration and the resulting solution was evaporated to dryness and used immediately.
Synthesis of 5-Iodoaceamido-1,10-phenanthroline (IA-Phen)
0.5 g (2.56 mmol) of 5-amino-1-10-phenanthroline (Polysciences) was dissolved in 90 mL of acetonitrile with slight heating. To this stirred solution, the freshly prepared iodoacetic acid anhydride dissolved in 10 mL of acetonitrile was added. The mixture was allowed to react in the dark overnight. The precipitated product was isolated by filtration and washed with cold 5% sodium bicarbonate, followed by water and dried in vacuo. Both the ESI MS (Figure S1.16) and NMR spectra correspond to previously reported literature values.22 (Yield: 75%)
Synthesis of Iodoacetamido-8-hydroxyquinoline (IA-Quin)
0.5 g (2.14 mmol) of 5-amino-8-hydroxyquinoline dihydrochloride (Sigma) was dissolved in 30 mL of acetonitrile by refluxing overnight with 975 μL (7 mmol) of triethylamine. The resulting solution was filtered, and iodoacetic acid anhydride, dissolved in 5 mL of acetonitrile, was added. The mixture was allowed to react in the dark overnight. The product evaporated to dryness and washed extensively with cold 5% sodium bicarbonate followed by water and dried in vacuo (Yield: 75%). Synthesis of IA-Quin was verified by mass spectrometry (ESI-MS, positive mode, Figure S1.17). Measured MW = 328.96 m/z (exp.: 328.9) (M + H+)
Synthesis of 4-Iodoacetamido-2,2′:6′,2″-terpyridine (IA-Terpy)
4-amino-2,2′:6′,2″-terpyridine (NH2-Terpy) was prepared as previously described.28 0.1 g (0.4 mmol) of NH2-Terpy was dissolved in 20 mL of dry dichloromethane. To this solution, 200 μL (1.4 mmol) of triethylamine was added and the reaction mixture was cooled to 0° C by stirring in an ice bath. Once the temperature equilibrated (approx. 20 min), 54 μL (0.6 mmol) of iodoacetyl chloride was added in a dropwise fashion. The mixture was left to react in the dark at 0° C for 30 min and then slowly brought up to room temperature. After a total period of 1 hour, the reaction volume was doubled with dichloromethane and washed extensively with cold 5% sodium bicarbonate followed by water. The organic fractions were evaporated dried in vacuo and used without further purification (Yield: ~60 %). In addition to IA-Terpy, small amounts of the amino precursor and a chloroacetamide terpyridine adduct were present as impurities. However, since only IA-Terpy can efficiently modify the protein under relevant labeling conditions, the product was used without further purification. Product formation was verified by mass spectrometry. (ESI-MS, positive mode, Figure S1.18). Measured MW = 417.05 m/z (exp.: 417.22 m/z) (IA-Terpy + H+); 325.29 m/z (exp.: 325.08 m/z) (ClA-Terpy + H+); 249.44 m/z (exp.: 249.11 m/z) (NH2-Terpy + H+)
General protocol for functionalization of cyt cb562 variants with Phen and Terpy chelates
A solution of 0.3 mM of cyt cb562 protein solution in degassed 0.1 M Tris buffer (pH 7.75) was treated with a 10-fold excess of dithiothreitol (DTT) (Sigma). The protein was allowed to reduce for a period of 30 min. The protein was then dialyzed against 2 × 1 L of degassed 0.1 M Tris buffer (pH 7.75) under an inert atmosphere to remove DTT. A 10-fold excess of iodoacetamide label was dissolved in 2 mL of degassed DMF and added dropwise to the protein solution over the course of 1 min. The mixture was allowed to react in the dark at 25° C overnight. The reaction mixture was then dialyzed again against 2 × 1 L of 10 mM sodium phosphate buffer (pH 8) and 1 mM EDTA. The crude labeled protein was subsequently purified on an Uno-Q anion-exchange column (BioRad) using a sodium chloride gradient. If further purification was necessary, the labeled fractions were combinded and dialyized against 2 × 1 L of 10 mM sodium acetate buffer (pH 5) and 1 mM EDTA. The protein mixture was then purified on an Uno-S (BioRad) cation-exchange column using a sodium chloride gradient. The final purity of the functionalized protein was determined to be greater than 95% by MALDI mass spectrometry and SDS-PAGE electrophoresis. (Labeling yield: 60–95%).
| Cyt cb562 variant | Calc. MW (amu) | Obs. MW (amu) |
|---|---|---|
| G70C-Phen (HPhen1) | 12622 | 12622 |
| G70H/H63C-Phen (HPhen2) | 12622 | 12620 |
| W59H/H63A/G70H/K77C-Phen (HPhen3) | 12516 | 12513 |
| G70C-Quin (HQuin1) | 12589 | 12590 |
| G70C-Terpy (HTerpy1) | 12675 | 12680 |
| H63A/G70C-Phen (APhen1) | 12556 | 12552 |
Metal Binding Titrations
Unless otherwise stated, all metal (M2+) binding titrations were prepared by diluting a concentrated protein stock solution to a final volume of 2 mL with a final protein concentration ranging from 2 to 5 μM. All titrations were performed in 50 mM MOPS buffer (pH 7) previously treated with Chelex resin (BioRad). All pipette tips were rinsed 3× in an analytical grade 5% HNO3 (Fluka) solution before use. All further procedures followed to ensure a metal-free environment have been previously outline by Linse.29
Titration data obtained by monitoring changes in the Phen/Terpy absorption were fit using non-linear regression on Dynafit 3 (BioKin). All absorption spectra were obtained on an HP 8452A spectrophotometer. HPhen and HTerpy concentrations were determined based on the Soret absorption maximum for cyt at 415 nm (ε = 0.148 μM−1 cm−1). All data were baseline-corrected and adjusted cb562 for dilution. Due to the tendency of the HPhen and HTerpy variants to undergo metal-mediated dimerization, data were separately fit to both 1:1 and 1:1/1:2 models. The later model accounts for both metal binding and metal-mediated protein dimerization.
Chemical Unfolding Studies
5 mL of an unfolded protein solution containing 5 μM of HPhen/HTerpy and 1 mM of M2+ or EDTA was freshly prepared in ~ 8 M guanidine HCl (GuHCl) in the appropriate buffer (either 100 mM Tris buffer (pH 7.5) or 100 mM sodium acetate (pH 5.5)). In parallel, 3 mL of a folded protein solution containing 5 μM of the same variant protein in the appropriate buffer and 1 mM M2+ or EDTA was prepared. The unfolded protein stock was titrated into the folded protein stock at 25°C using an autotitrator (Microlab 500 Series), keeping the sample volume constant at 2 mL. Protein unfolding was monitored by CD spectroscopy (222 nm) on an Aviv 215 spectrometer. For every titration point, the solution was allowed to stir for 30 seconds in order to reach equilibrium. This procedure was repeated for a minimum of 30 points covering a GuHCl range of 0.1–6.5 M. GuHCl concentrations were calculated using the refractive indices of the folded and unfolded protein stock solutions.30 Unfolding data were fit using Kaleidagraph (Synergy Software) with an expression that assumes a two-state folding/unfolding equilibrium as described by Pace (eq. 1):31
| (1) |
where m1 represents the slope of the unfolding transition and is defined as (∂ΔGH2O/∂ [GuHCl]), and m 2 represents the midpoint GuHCl concentration where 50% of the protein is unfolded. All HPhen and HTerpy variants remain in their monomeric form at under the conditions (5 μM of protein, 1 mM M2+) used in chemical denaturation experiments.
Thermal Denaturation
A 3 mL solution of 5 μM HPhen/HTerpy in 100 mM Tris buffer (pH 7.5) and 1.5 M GuHCl was prepared. Inclusion of 1.5 M GuHCl was necessary to ensure that the protein fully unfolds below 373 K. To the protein solutions either 1 mM Ni2+ or EDTA was added. The unfolding reaction was monitored over a range of 300–376 K by CD spectroscopy (222 nm). At each temperature point, the solution was allowed to stir for 30 seconds in order to reach equilibrium. Although the thermal unfolding of HCMs is not completely reversible, the curve was fit to a two-state model as described by John and Weeks32 to obtain an apparent ΔTmmetal. Unfolding data were fit using Kaleidagraph (Synergy Software).
Sedimentation Velocity Experiments
SV experiments were performed in order to determine the solution-state oligomerization behavior of each HPhen/HTerpy variant. All SV samples were prepared in 20 mM Tris buffer (pH 7). Measurements were made on a Beckman XL-I Analytical Ultracentrifuge (Beckman-Coulter Instruments) using an An-60 Ti rotor at 41,000 rpm for a total of 250 scans per sample. The following wavelengths were used for detection: 418 nm (5 μM protein), 420 nm (10 μM protein), 425 nm (20 μM protein), 540 nm (40 μM protein), 545 nm (60μM protein) and 560 nm (100 μM protein).
All data were processed using SEDFIT.33 Buffer viscosity, buffer density, and protein partial specific volume values were calculated at 25° C with SEDNTERP (http://www.jphilo.mailway.com). Partial specific volume (Vbar) for each variant was calculated assuming a partial specific volume of heme of 0.82 mg/mL and 0.71 mg/mL, 0.75 mg/mL, 0.87 mg/mL for Phen, Quin and Terpy respectively. All data were processed using fixed values for buffer density (ρ) (0.99764 g/mL) and buffer viscosity (0.0089485 poise).
DFT Calculations
DFT calculations were performed with Amsterdam Density Functional (ADF) program suite,34, 35 version 2007.01,36 on a home-built 72-CPU (1 × 8 master, 8 × 8 slave) Rocks 4.3 Linux cluster featuring Intel Xeon E5335 Quad-Core 2.00GHz processors. Job control was implemented with the Sun Grid Engine v. 5.3. Crystallographic atomic coordinates were used as input where appropriate. Optimized geometries and molecular orbitals were visualized with the ADFView graphical routine of the ADF- GUI37 and the Gaussview 3 program.
In ADF program suite calculations, the triple-ζ Slater-type orbital TZ2P ADF basis set was utilized without frozen cores for all atoms. Relativistic effects were included by use of the zero-order regular approximation (ZORA).38 To ensure consistency over a range of exchange/correlation profiles, the molecular geometries and energies were evaluated with both the BP86 and OLYP functionals.
In BP86 calculations, the local density approximation (LDA) of Vosko et al.39 (VWN) was coupled with the generalized gradient approximation (GGA) corrections described by Becke40 and Perdew41, 42 for electron exchange and correlation, respectively. In OLYP calculations, the parameterized (X = 0.67) exchange-only LDA was coupled with the GGA corrections described by Handy and Cohen (OTPX)43 and Lee, Yang and Parr (LYP)44 for electron exchange and correlation, respectively.
Supplementary Material
Acknowledgments
This work was supported by NSF (CHE-0908115, protein design, modification, unfolding studies), DOE BES (DE-FG02-10ER46677, metal binding studies), a traineeship under an NIH Heme and Blood Training Grant (T32DK007233) (R.J.R), and Beckman Young Investigator and Hellman Faculty Scholar awards (F.A.T.).
Footnotes
Supporting Information Available: Additional experimental details, figures, and tables. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bertini I, Gray HB, Stiefel EI, Valentine JS. Biological Inorganic Chemistry, Structure & Reactivity. University Science Books; Sausalito: 2007. [Google Scholar]
- 2.Rosenzweig AC. Acc Chem Res. 2001;34:119–128. doi: 10.1021/ar000012p. [DOI] [PubMed] [Google Scholar]
- 3.Bush AI, Pettingell WH, Multhaup G, Paradis MD, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 4.Arnold FH, Haymore BL. Science. 1991;252:1796–1797. doi: 10.1126/science.1648261. [DOI] [PubMed] [Google Scholar]
- 5.Winkler JR, Gray HB. Chem Rev. 1992;92:369–379. [Google Scholar]
- 6.Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. J Am Chem Soc. 2002;124:6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 7.(a) Salgado EN, Faraone-Mennella J, Tezcan FA. J Am Chem Soc. 2007;129:13374–13375. doi: 10.1021/ja075261o. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Salgado EN, Lewis RA, Faraone-Mennella J, Tezcan FA. J Am Chem Soc. 2008;130:6082–6084. doi: 10.1021/ja8012177. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Salgado EN, Lewis RA, Mossin S, Rheingold AL, Tezcan FA. Inorg Chem. 2009;48:2726–2728. doi: 10.1021/ic9001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radford RJ, Tezcan FA. J Am Chem Soc. 2009;131:9136–9137. doi: 10.1021/ja9000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Radford RJ, Nguyen PC, Ditri TB, Figueroa JS, Tezcan FA. Inorg Chem. 2010;49:4362–4369. doi: 10.1021/ic100534y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martell AE, Smith RM. Critical Stability Constants. Plenum Press; New York: pp. 1974–1989. [Google Scholar]
- 11.Ghadiri MR, Choi C. J Am Chem Soc. 1990;112:1630–1632. [Google Scholar]
- 12.Ruan FQ, Chen YQ, Hopkins PB. J Am Chem Soc. 1990;112:9403–9404. [Google Scholar]
- 13.Blackwell HE, Grubbs RH. Angew Chem Int Ed. 1998;37:3281–3284. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 14.Schafmeister CE, Po J, Verdine GL. J Am Chem Soc. 2000;122:5891–5892. [Google Scholar]
- 15.Zhang FZ, Sadovski O, Xin SJ, Woolley GA. J Am Chem Soc. 2007;129:14154–14155. doi: 10.1021/ja075829t. [DOI] [PubMed] [Google Scholar]
- 16.Verdine GL, Walensky LD. Clin Cancer Res. 2007;13:7264–7270. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 17.Kortemme T, Baker D. Curr Opin Chem Biol. 2004;8:91–97. doi: 10.1016/j.cbpa.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Shoemaker BA, Panchenko AR. PLos Comput Biol. 2007;3:595–601. doi: 10.1371/journal.pcbi.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohler JJ, Metallo SJ, Schneider TL, Schepartz A. Proc Natl Acad Sci USA. 1999;96:11735–11739. doi: 10.1073/pnas.96.21.11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Due to the inherent chirality of the α-helical scaffolds, the Δ configuration is not possible.
- 21.Crane BR, Di Bilio AJ, Winkler JR, Gray HB. J Am Chem Soc. 2001;123:11623–11631. doi: 10.1021/ja0115870. [DOI] [PubMed] [Google Scholar]
- 22.Castellano FN, Dattelbaum JD, Lakowicz JR. Anal Biochem. 1998;255:165–170. doi: 10.1006/abio.1997.2468. [DOI] [PubMed] [Google Scholar]
- 23.Singh SK, Trivedi M, Chandra M, Sahay AN, Pandey DS. Inorg Chem. 2004;43:8600–8608. doi: 10.1021/ic049256m. [DOI] [PubMed] [Google Scholar]
- 24.For a previous study, where a Ru-polypyridyl complex was used to crosslink two loops on cytochrome c, see Muheim A, Todd RJ, Casimiro DR, Gray HB, Arnold FH. J Am Chem Soc. 1993;115:5312–5313.
- 25.Alber T. Ann Rev Biochem. 1989;58:765–798. doi: 10.1146/annurev.bi.58.070189.004001. [DOI] [PubMed] [Google Scholar]
- 26.Faraone-Mennella J, Tezcan FA, Gray HB, Winkler JR. Biochemistry. 2006;45:10504–10511. doi: 10.1021/bi060242x. [DOI] [PubMed] [Google Scholar]
- 27.Braun M, Thony-Meyer L. Proc Natl Acad Sci USA. 2004;101:12830–12835. doi: 10.1073/pnas.0402435101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou ZG, Sarova GH, Zhang S, Ou ZP, Tat FT, Kadish KM, Echegoyen L, Guldi DM, Schuster DI, Wilson SR. Chem Eur J. 2006;12:4241–4248. doi: 10.1002/chem.200600021. [DOI] [PubMed] [Google Scholar]
- 29.Linse S. In: Calcium-Binding Protein Protocols, Vol. 2: Methods and Techniques. Vogel HJ, editor. Totowa: Humana Press; 2002. [Google Scholar]
- 30.Nozaki Y. Methods Enzymol. 1972;26:43–50. doi: 10.1016/s0076-6879(72)26005-0. [DOI] [PubMed] [Google Scholar]
- 31.Pace NC, Shirley BA, Thomson JA. In: Protein Structure: A Practical Approach, Creighton. TF, editor. IRL Press; Oxford: 1990. pp. 311–330. [Google Scholar]
- 32.John DM, Weeks KM. Prot Sci. 2000;9:1416–1419. doi: 10.1110/ps.9.7.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuck P. Biophys Chem. 2004;108:187–200. doi: 10.1016/j.bpc.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 34.Te Velde G, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, van Gisbergen SJA, Snijders JG, Ziegler T. J Comput Chem. 2001;22:931–67. [Google Scholar]
- 35.Guerra CF, Snijders JG, te Velde G, Baerends EJ. J Theor Chem Acc. 1998;99:391–403. [Google Scholar]
- 36.ADF2007.01, SCM, Theoretical Chemistry. Vrije Universiteit; Amsterdam, The Netherlands: www.scm.com. [Google Scholar]
- 37.ADF-GUI 2007.01, SCM. Amsterdam, The Netherlands: [Access date, February, 2008.]. www.scm.com. [Google Scholar]
- 38.van Lenthe E, Baerends EJ, Snijders JG. J Chem Phys. 1993;99:4597–4610. [Google Scholar]
- 39.Vosko SH, Wilk L, Nusair M. Can J Phys. 1980;58:1200–1211. [Google Scholar]
- 40.Becke AD. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 41.Perdew JP, Yue W. Phys Rev B. 1986;33:8800–8802. doi: 10.1103/physrevb.33.8800. [DOI] [PubMed] [Google Scholar]
- 42.Perdew JP. Phys Rev B. 1986;34:7406–7406. [PubMed] [Google Scholar]
- 43.Handy NC, Cohen AJ. Mol Phys. 2001;99:403–412. [Google Scholar]
- 44.Lee CT, Yang WT, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.